

Abstract
In health, the pancreatic islet cells work as a network with highly coordinated signals over time to balance glycaemia within a narrow range. In type 1 diabetes (T1DM), with autoimmune destruction of the β-cells, lack of insulin is considered the primary abnormality and is the primary therapy target. However, replacing insulin alone does not achieve adequate glucose control and recent studies have focused on controlling the endogenous glucagon release as well. In T1DM, glucagon secretion is disordered but not absolutely deficient; it may be excessive postprandially yet it is characteristically insufficient and delayed in response to hypoglycaemia. We review our system-level analysis of the pancreatic endocrine network mechanisms of glucagon counterregulation (GCR) and their dysregulation in T1DM and focus on possible use of α-cell inhibitors (ACI) to manipulate the glucagon axis to repair the defective GCR. Our results indicate that the GCR abnormalities are of “network origin”. The lack of β-cell signalling is the primary deficiency which contributes to two separate network abnormalities: (i) absence of a β-cell switch-off trigger and (ii) increase intraislet basal glucagon. A strategy to repair these abnormalities with ACI is proposed which could achieve better control of glycaemia with reduced hypoglycaemia risk.
Keywords: glucagon, counterregulation, diabetes mellitus, feedback, hypoglycaemia, intrapancreatic network, mathematical model
Introduction
Standard insulin therapies aiming to achieve tight blood glucose (BG) control in diabetes mellitus result in up to 3 fold excess of severe hypoglycaemia [1–3] generally caused by insulin overdosing (basal or meal insulin) and delayed and deficient hormonal counterregulation. In health, early defences against hypoglycaemia include reduction in β-cell insulin secretion (not possible in β-cell deficient patients), pancreatic glucagon release, epinephrine secretion by the adrenal medulla and sympathetic nervous system activation [4,5]. Later defences include cortisol and GH secretion as well as hepatic auto-regulation which may also help to restore euglycemia. The first of the hormone defences lost during the course of T1DM [6–8] is glucagon, which occurs despite the normal number and histological appearance of α-cells in the pancreatic islets. GCR is also defective in advanced type 2 diabetes [9]. The abnormal GCR impedes the safe treatment of the disease [10,11]. Failure of glucagon secretion in response to hypoglycaemia may occur within the first few years of T1DM and is a hypoglycaemia–specific abnormality, as responses to exercise or amino acids administration are preserved or even sometimes exaggerated. No subsequent recovery of glucagon response to hypoglycaemia appears later in the course of the disease. In this regard, glucagon counterregulation differs from other aspects of the Hypoglycaemia Associated Autonomic Failure (HAAF) syndrome described by Cryer and colleagues [9,12] in that cessation of hypoglycaemia allows for recovery of the other hormone defences (cortisol, epinephrine, sympathetic nervous system activity and GH) within a few days.
To understand the mechanisms of GCR impairment in insulin deficiency we used a system-level approach, combining in vivo and in silico studies, to reconstruct the network control of glucagon release and GCR. Our premise is that the temporal relationships between the pancreatic peptides and the relative contribution of each interaction to the system behaviour cannot be properly assessed if its components are studied in isolation, such as looking for a specific single missing signal. In particular, we believe that important properties of the time-dependent glucagon release are a by-product of the action of a (feedback) network within the endocrine pancreas. Our approach provides a different perspective on the mechanism that controls GCR and on the nature of its impairment in T1DM. The models developed during the course of the implementation of this approach are a powerful tool to predict the response of the endocrine pancreas to various interventions and to design strategies to repair the defective GCR in insulin deficiency – a task which up to now seemed difficult to approach.
The mechanisms of GCR dysregulation in diabetes are largely unknown
The mechanism by which hypoglycaemia stimulates GCR and how it is impaired in insulin deficient diabetes is still unclear [13] and may include impaired BG-sensing in the α-cells [7], autonomic dysfunction [14,15], or/and loss of an insulin “switch-off” signal from the β-cells [16]. Recent studies in support of the “switch-off” hypothesis show that in STZ-treated rats GCR is impaired, but can be restored if the intrapancreatic insulin is re-established and switched off during hypoglycaemia [17] (see also [18,19]). Whether insulin is the specific trigger is unclear since the same group showed later that the switch-off of zinc ions rather than switch-off of insulin initiates the GCR [20]. Shortly after the first reports describing the in vivo repair of GCR by intrapancreatic infusion and switch-off of insulin (or zinc) [18–20], we applied mathematical modelling to analyze and reconstruct the GCR control network and predicted that the GCR develops as a general disinhibition phenomenon of a feedback network rather than as a response to a specific switch-off signal (insulin or zinc). Importantly, we have confirmed this prediction in vivo by showing that a defective GCR can be repaired by two different ACI signals (insulin and somatostatin) which upon switch-off trigger pulsatile GCR [21] (below).
Glucagon secretion abnormalities and treatment of T1DM
Lack of insulin classically defines T1DM - both in its pathophysiology and its therapy. Nonetheless, it has long been posited that glucagon abnormalities are also of crucial importance, as originally proposed by Unger and colleagues [22], and recent publications have highlighted anew the focus on this hormone [22–24]. However, the focus on glucagon secretion in diabetes so far has been on pharmacological treatment to reduce its excessive secretion. Thus, high glucagon levels with poor control of type 1 or type 2 diabetes (e.g., DKA) or its failure to suppress normally after meals (in both types of diabetes) have thought to represent potential opportunities to use drugs such as GLP-1, leptin or amylin agonists to reduce hyperglycaemia.
Unger and colleagues have found that treatment of an insulin deficient animal model of uncontrolled T1DM with α-cell inhibition (leptin analog or leptin overexpression), reverses most of the glycaemic instability even without provision of insulin [24]. Similarly, a knockout of glucagon receptors in an animal model also seems to stabilize insulin deficient diabetes [23]. Such studies indicate that suppression of glucagon secretion or action may be a critical intervention to stabilize insulin deficient diabetes although no human data is yet available. In this regard, our in vivo and in silico results support the concept that the abnormalities in glucagon secretion and GCR result at least in part from defects in the pancreatic endocrine network relationships specific for insulin deficiency and can be repaired by manipulating the secretion of glucagon (but not its action) by ACI.
Primary pancreatic endocrine relationships involved in glucagon secretion
Figure 1 summarizes the primary, largely consensus, interactions between blood glucose and the α- and β-cells to which we refer as a Minimal Control Network (MCN) of glucagon secretion. The top diagram represents the network connections in the healthy pancreas, the middle panel exemplifies the remaining interactions after autoimmune destruction of the β-cells, and the third panel shows the expected action of an ACI on the pancreatic network. An extensive background supporting each of these interactions can be found elsewhere [21,25–27]. Here, we only point out that the α-cell auto-feedback which is key to our system-level analysis could be either direct [28], or indirect, by which we mean that it is mediated by δ-cell somatostatin (glucagon stimulates somatostatin [29–33] and, in turn, somatostatin inhibits glucagon [31,34–42]).
Figure 1.
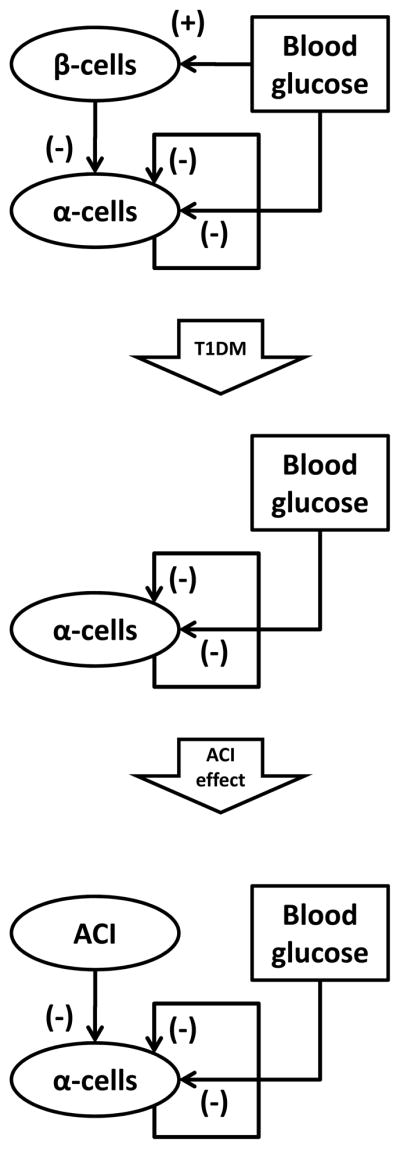
Minimal Control Network (MCN) of glucagon secretion in normal physiology (top), T1DM (middle) and in T1DM under the control of exogenous ACI (bottom).
The network in Figure 1 is clearly an oversimplification of the existing signals and interactions that control the secretion of glucagon, which are influenced by numerous extrapancreatic factors, some of which have important impacts on glucagon secretion and GCR. These include the autonomic input, catecholamines, growth hormone, ghrelin, and incretins [13,43–45]. The three major autonomic influences on the α-cell include: sympathetic nerves, parasympathetic nerves and circulating epinephrine, all of which are activated by hypoglycaemia, and are capable of stimulating glucagon and suppressing insulin [46–48]. These signals have no explicit terms in our model. However, they are not omitted or considered unimportant and when we describe the network their impact is taken into account (implicitly) even though they have no individual terms in the model.
The pancreatic network control of glucagon secretion and network abnormalities in T1DM
In support of our idea that each of the relationships shown in Figure 1 are important for the dynamic of the secretion of glucagon in vivo we note the following. In health, glucagon, like insulin, is secreted in a basal and pulsatile manner, with pulses produced in apparently linked anti-phasic manner to those of insulin and δ-cell somatostatin [49–52]. Such pulsatile behaviour and linked secretion implies a network within the endocrine pancreas that is highly regulated through a series of controlling influences, some from within and others possibly from outside the endocrine pancreas. The anti-phasic link between insulin and glucagon, their reciprocal dynamics during hyper and hypoglycaemia strongly suggests that the α-cells are under constant tonic inhibition by the β-cells which is therefore an important component of the α-cells regulation. We ([21,25–27]), and many others (e.g., [22]), have argued that this characteristic of the normal pancreas mediates the proper timing and amount of the glucagon responses to meals and hypoglycaemia and maintains a proper balance between insulin and glucagon in their roles of key regulators of the liver and the hepatic glucose production (HGP). Indeed, postprandial increase in insulin could explain the observed glucagon suppression and insulin decrease during hypoglycaemia could account in part for the GCR. The fact that the α-cell glucagon secretion in response to insulin decrease appears glucose dependent supports the notion that the BG->α-cell interaction shown in Figure 1 (direct or indirect) is operational in both the healthy and the β-cell deficient pancreas. Finally, we [21] and others [17–20] have observed that intrapancreatic infusion of ACI followed by their switch-off during hypoglycaemia triggers a rebound-like GCR which exceeds the typical (unsuppressed) basal glucagon levels: Figure 2.
Figure 2.

Glucagon (MEAN±SEM) from t=−10 to t=0 min before the switch-off (BASAL GLUCAGON) and from t=25 to t=45 min after the switch-off (SWITCH-OFF TRIGGERED GCR) in the saline, somatostatin and insulin groups. *p<0.05
Using deconvolution analysis [21] we detected a low-frequency pulsatility of glucagon oscillations triggered by the switch-off (e.g., cessation of intrapancreatically infused insulin or somatostatin) which matches the frequency in the glucagon pulsatile response to hypoglycaemia in health [49]. This frequency is markedly different from the high-frequency glucagon pulsatility typical for the portal circulation which could be attributed to the direct control of the α-cells by the β-cells [50,52].
Such behaviour suggests that the α-cells are also auto-feedback regulated [53] as shown in Figure 1 with the auto-feedback amplifying the pulsatile GCR and mediating the parameters of the glucagon oscillations during hypoglycaemia.
In summary, there are direct and indirect lines of evidence supporting the concept that all of the relationships shown in Figure 1 contribute significantly to the observed in vivo glucagon responses to hypoglycaemia of the normal and the β-cell deficient pancreas.
In silico analysis of the glucagon control network
To support the concept that the MCN shown in Figure 1 unifies the primary interactions responsible for the control of glucagon secretion and GCR we applied mathematical modelling to approximate the network and demonstrate that it can explain the in vivo behaviour of the system. In particular, we formalized the MCN with a set of coupled nonlinear delayed differential equations that describe the rate of change of the system components and their interaction following the methodology described in [53]. The model equations are shown below.
| (A) |
| (B) |
Here, GL(t), BG(t), and INS(t) denote the time-dependent concentrations of glucagon, blood glucose, and insulin (or exogenous ACI in the insulin-deficient model), respectively; the derivative is the rate of change with respect to the time t. Pulse denotes a β-cell-specific pulse generator superimposed to mimic the in vivo physiology of the β-cells and published data on insulin pulsatility in the portal circulation. The meaning of the parameters and the way they have been determined is explained in detail elsewhere [21,25–27]. In brief, the half-life of glucagon and insulin determines the elimination constants kGL and kINS. The delay DGL, the ED50-s, tBG, tGL and the slopes nBG and nGL in the auto-feedback guarantee glucagon pulsatility with a frequency similar to the experimentally observed [21]. The secretion rates rINS, rINS,basal, the ID50, tBG,2, the slope nBG, and the amplitude of the pulse generator, Pulse, are determined so that a glucose stimulus leads to an increase in insulin similar to that of published data (in the insulin sufficient model). The value of the ED50, tINS guarantees that hypoglycaemia-induced insulin withdrawal will trigger a GCR. The secretion rates rGL and rGL,basal control the amplitude of the GCR. The determination of the nominal values of these parameters is discussed in [25–27].
We have used the mathematical approximation outlined above of the glucagon axis to demonstrate that the MCN (Figure 1) explains the transition from a normal physiology to a β-cell deficiency in terms of gradual disappearance of GCR (Figure 3, left panels) and postprandial glucagon suppression (Figure 3, right panel) in response to decline in β-cell activity. We note however, that this minimal model cannot explain why hyperglycaemia may paradoxically increase glucagon levels as seen in some animal models.
Figure 3.

Transition from a normal physiology to a β-cell deficiency in terms of gradual disappearance of GCR (left panels) and postprandial glucagon suppression (right panel) in response to decline in β-cell activity.
We have also shown that the MCN could account for the observed in vivo pulsatile GCR response to hypoglycaemia triggered by switch-off signals in insulin deficiency as detailed in Figure 2 in [27] (see also [21]). These results exemplify the excellent agreement of the model predictions with the experimental observations.
Our system-level in silico analysis confirms that the MCN (Figure 1) is an excellent approximation of the glucagon control axis and can explain most of its in vivo behaviour. The postulated MCN interactions are both necessary and sufficient in terms of replicating the key features of the glucagon control axis.
Specific network deficiency behind the defects in GCR in T1DM
Once the MCN-based model has been validated as discussed in the previous section, it was used to establish the T1DM network deficiencies leading to defects in the GCR and to search for ways for their repair. One apparent difference between the MCN in health and in T1DM is the lack of β-cell signalling to the α-cells (Figure 1). Our in silico analysis shows that this key network deficiency mediates at least in part the severely damaged ability of the pancreas to properly control the secretion of glucagon in patients with T1DM: Figure 3. In particular, absence of a β-cell switch-off trigger during hypoglycaemia could be implicated in the GCR deficiencies in T1DM and lack of β-cell suppression of the α-cells could contribute to the impaired postprandial decline in glucagon. It is important to note that the increase in glucagon triggered by the switch-off of the β-cells is a rebound-like effect and takes advantage of the auto-feedback control of the α-cells, rather than solely being a product of the β/α-cell interactions. Indeed, from a control theory point of view, if a node of a system that is auto-feedback regulated is suppressed (hence, the system is removed from its steady state), a removal of the suppression forces the system to initiate a waning sequence of pulses. The effect is exemplified in Figure 4 which illustrates the impact of a suppression of the secretion of glucagon by an ACI followed by its disinhibition (switch-off); the secretion of glucagon is assumed to be auto-feedback regulated.
Figure 4.

Illustration of the impact of a brief infusion of a small (left) and large (right) dose of an ACI on the glucagon secretion under (delayed) auto-feedback control assuming lack of other influences.
Figure 4 also illustrates that theoretically a more profound suppression is expected to provide more energy to the system and its removal will lead to a higher rebound response (Figure 4; left vs. right panel). One important implication of interpreting the GCR as a rebound is that that magnitude of the post switch-off pulses would depend on the availability of sufficiently low nadirs immediately preceding these pulses [53]. Going back to the design of our mathematical model we note that the experimental data cannot be explained without separating the glucagon secretion into auto-feedback regulated (AFR) [or pulsatile intraislet glucagon release component] and auto-feedback independent (AFI) [or basal intraislet glucagon release component] as assumed in Eq. A. Therefore, an increase in the AFI component of glucagon secretion will effectively dampen the amplitude of the pulses, reduce the difference between the basal and maximal post switch-off levels, and also increase the basal concentration. The simulations in Figure 3 show the link between increase in basal glucagon (primarily due to increase in AFI) and GCR reduction.
The in silico analysis of the MCN predicts that lack of β-cell signalling to the α-cells is a key network deficiency which impairs the GCR by contributing to two separate network abnormalities: (1) lack of a β-cell switch-off as hypothesized by us [21,25–27] and others [16–20] (in a more narrow sense) and (2) increase in intraislet AFI glucagon as discussed by us [27].
Our analysis shows that both mechanisms can be targeted by ACI to repair the defective GCR in insulin deficiency. Previously, we have predicted and confirmed experimentally the possibility to use ACI switch-off signals to repair the GCR [21,25,26] which illustrates how the modelling and the experimental data are intertwined in these studies. More recently, we have focused on the second mechanism analyzing and designing strategies for its modulation with ACI to amplify the defective GCR [27]. These results, along with some new analysis, are presented in the next section.
Note. In our terminology, the basal intraislet glucagon release component (or AFI glucagon) is linked to the basal glucagon release understood as the secretion of glucagon during euglycaemia. However, they are not exactly the same since the basal glucagon secretion during euglycaemia is the sum of AFI and AFR glucagon.
Optimizing glucagon suppression to repair defective GCR
Identification of the intraislet hyperglucagonaemia as a possible network abnormality leading to defects in GCR in response to hypoglycaemia fostered the investigation of the possibility to use ACI to enhance either the absolute or the relative (with respect to basal levels) increase in GCR. We have used a model-based analysis to show that α-cell inhibition may repair defective GCR in insulin deficiency even without a switch-off. Using our MCN model we simulated the response to hypoglycaemia assuming different strategies of glucagon reduction: gradual suppression of AFI or AFR glucagon, or both. Reduction in AFI glucagon improved (Figure 4 [27], top curve), while reduction in AFR secretion reduced the GCR (Figure 4 [27], lower curve). However, if inhibition of both AFI and AFR glucagon is assumed (as expected during an ACI infusion) the model predicts initial GCR enhancement followed by GCR gradual decline: Figure 4 [27], middle curve.
The in silico analysis suggests that some ACI could be more suitable for use for GCR repair than others since if they are not switched-off or their switch-off is delayed (e.g., as a result of failure to detect hypoglycaemia on time) the risk associated with such behaviour will be minimal. The ACI infusion rate is critical and may require careful adjustment. It is also possible that the ACI infusion may need to be dynamically modulated to enhance GCR.
We then estimated the improvement in defective GCR caused by constant infusions of ACI depending on the extent to which they inhibit the AFI and AFR glucagon (AFIG and AFRG, respectively). To this end, we tabulated the predicted hypoglycaemia-stimulated glucagon release assuming graded levels of AFIG and AFRG inhibition by ACI. Restoring GCR requires more than 40% reduction of AFIG, but less than 80% suppression of AFRG. 40% reduction in both AFIG and AFRG restores the GCR response to hypoglycaemia, but glucagon remains inappropriately elevated during eu- and hyperglycaemia. Normalization of glucagon requires reduction of AFIG accompanied by only minor AFRG suppression. Table 1 summarizes the impact of ACI on the fold increase in glucagon in response to hypoglycaemia assuming 0% to 100% reduction in AFIG (by rows) and AFRG (by columns). Values >7 (bold) indicate restoration to levels similar to the normal pancreas (vs. 1.6 in insulin deficiency) and values that are also underlined correspond to a combination of AFIG and AFRG suppressions that have repaired the absolute GCR as well.
Table 1.
Simulated impact of a constant ACI infusion on the fold increase in glucagon in response to hypoglycaemia assuming 0% to 100% reduction in AFIG (by rows) and AFRG (by columns). Values in bold indicate restoration to levels similar to the normal pancreas; additional underlining mark repair of the absolute GCR as well.
| AFIGl↓/ARIG→ | 0% | 20% | 40% | 60% | 80% | 100% |
|---|---|---|---|---|---|---|
| 0% | 1.6 | 1.5 | 1.4 | 1.4 | 1.3 | 1 |
| 20% | 4.3 | 2.8 | 2 | 1.6 | 1.5 | 1 |
| 40% | 8.6 | 7.5 | 6 | 3 | 1.9 | 1 |
| 60% | 13.3 | 11.6 | 9.8 | 7.7 | 3.1 | 1 |
| 80% | 18.9 | 17.3 | 15.6 | 13.1 | 7.2 | 1 |
| 100% | 28.8 | 28.8 | 28.8 | 27.2 | 28.3 |
These results show that the most effective repair of defective GCR by constant infusion of ACI is achieved by signals that suppress preferentially the AFI rather than the AFR glucagon release. Most importantly, Table 1 can be used as a “screening criteria” to identify an ACI with the highest potential to improve GCR. Indeed, deconvolution analysis of hormone concentration time series can estimate separately these two secretory components (AFIG and AFRG) from frequently collected blood samples in vivo [54].
Summary of experimental, clinical and in silico data in support of the ACI-based approach to repair the defective GCR in β-cell deficiency
Based on the discussion in the previous two sections and on our earlier studies [21,25–27], GCR can be repaired by ACI. The effect is achieved either by suppressing glucagon release under normal conditions accompanied by a switch-off during hypoglycaemia to trigger a rebound pulsatile GCR or by permanent infusion of an ACI which preferentially suppresses the auto-feedback independent rather than the auto-feedback dependent component of glucagon secretion. Below we summarize experimental, clinical and in silico data in support of the applicability of these two approaches. Some of these data have already been discussed in the previous sections.
The concept that the switch-off mechanism can be applied to improve the pulsatile GCR is supported by in vivo (and in vitro) data showing that defective GCR can be significantly amplified by a switch-off of different ACIs during hypoglycaemia [17–21].
In silico results predict that a switch-off of an ACI during hypoglycaemia can trigger a pulsatile GCR [21,25–27].
In humans, infusions of certain ACIs not only do not disrupt, but may improve the GCR. For example, neither pramlintide (an amylin analog) [43] nor GLP-1 [55] impair the GCR response to hypoglycaemia in normal volunteers. In fact, exendin-4 (GLP-1 analog) improved the GCR response to hypoglycaemia in normal [56] subjects by both, reducing basal and amplifying hypoglycaemia-stimulated glucagon release. In addition, vildagliptin (which increases endogenous GLP-1) enhances GCR in type 2 DM primarily we believe by reducing basal hyperglucagonaemia and improving the fold response to hypoglycaemia [57]. In early, but not in later studies, pramlintide was associated with an increased risk of hypoglycaemia, probably related to an insufficient reduction of meal insulin when it was used with complex insulin regimens. In one analysis, we had found contrary to earlier data no increase in hypoglycaemia risk in T1DM with pramlintide added to insulin therapy [58]. Our theoretical platform suggests a potential to improve GCR with such an agent, but we note from our simulation studies, that different α-cell suppressing drugs may exert different effects on GCR at least partly based upon their mechanisms or effects upon AFI vs. AFR glucagon secretion [25–27].
Model-based simulations (above) suggest that (a) suppression of AFI glucagon secretion markedly amplifies the pulsatile GCR response to hypoglycaemia; (b) suppression of total glucagon may also amplify GCR and can present a strategy to repair defective GCR in β-cell deficient diabetes, but the ACI infusion rate may require careful adjustment.
In vivo preliminary experimental results in diabetic rats show that the GCR response to hypoglycaemia is enhanced by uninterrupted intrapancreatic infusion of GABA (a known α-cell inhibitor) [27].
A recent analysis of hypoglycaemia data from a study of GCR in T1DM subjects linked basal hyperglucagonaemia to defects in GCR ([59]: unpublished) and suggests that elevation of AFI glucagon (which leads to increase in basal glucagon) may be part of the mechanism of GCR impairment in T1DM as predicted by our model.
Suggested methodology for developing of a GCR repair strategies
Based on our results, an ACI that suppresses glucagon secretion can be used to improve GCR in several ways. First, maintaining constant α-cell suppression could be beneficial to GCR only if the used ACI suppresses preferentially the AFI glucagon release and does not markedly suppress the pulsatile (AFR) glucagon secretion. Currently, it is unclear whether such ACI exists even though some animal and clinical studies (above) suggest that this is potentially feasible. If such ACI can be identified its addition to a T1DM treatment strategy is straightforward and detection of hypoglycaemia will not be required. Therefore, such GCR improvement strategy can be added to a standard insulin therapy and is expected to significantly improve glycaemia control. Second, an ACI may enhance the GCR if it is permanently delivered most of the time (during eu- and hyperglycaemia) and is switched off during hypoglycaemia. This enhancement could be either only relative or relative and absolute. For example, if the ACI represses glucagon, which upon disinhibition (switch-off) returns back to basal levels the GCR improvement will be only relative. It may still be effective since even a small difference in glucagon levels could have a significant glucose raising effect if the increase in glucagon is accompanied with an appropriate timing of the decrease in insulin as shown by Shah et al [60]. However, if the ACI infusion represses glucagon which upon ACI switch-off rebounds at higher than basal levels the result will be essentially a network-based repair of defective GCR both in relative and absolute terms. Such an outcome which is based on our results appears to be feasible and would have strongest positive impact on improving glycaemia control and safety. We note however, that such a GCR repair strategy requires a detection of hypoglycaemia in order to switch-off the ACI signal at the appropriate time. We also point out that based on the in silico analysis summarised in Figure 5 a switch from a high suppression of glucagon during eu- and hyperglycaemia to a low suppression when BG declines from 110 to 80 mg/dL may restore to normal both basal and hypoglycaemia-stimulated glucagon. The model-based simulations show that if such ACI can be identified (which suppresses simultaneously the AFI and the AFR glucagon secretory components), the GCR repair will be relative and absolute and safe (note that failure to reduce the inhibition reduces only slightly the GCR).
Figure 5.

Plots 3 to 10: GCR response to different onset of a switch (black/gray bars) from 60% reduction to 40 % reduction of glucagon in order to restore both basal and hypoglycaemia-stimulated glucagon secretion: Plots 1–2: simulated stepwise BG decline into hypoglycaemia Left black arrow 80 mg/dl; Right grey arrow 60 mg/dl
Based on our findings, an approach for identifying an ACI for GCR repair and for the design of a GCR-repair strategy can be outlined. An initial screening will identify an ACI with a good potential to be part of a GCR repair strategy. According to the model-based predictions, the GCR response to insulin induced hypoglycaemia should be tested in the face of constant ACI delivery at a rate known to suppress basal glucagon at 30–50% on average (an infusion rate suggested by the in silico results). The following criteria listed in the order of their importance apply:
Improvement of relative GCR understood as the increase in glucagon above basal levels during hypoglycaemia. It is measured by the AUC of glucagon levels above basal and the relative % increase of maximal glucagon with respect to basal levels in the 90 min interval after BG drops below 70 mg/dL)
Improvement of absolute GCR understood as the absolute increase in glucagon during hypoglycaemia. It is measured by the AUC and the maximum glucagon levels reached in the 90 min interval after BG drops below 70 mg/dL)
Enhancement of glucagon pulsatility during hypoglycaemia in terms of increasing the zenith/nadir difference of the glucagon pulses, amplifying the pulsatile (AFR) and inhibition of AFI glucagon secretion. The determination of these parameters is based on analytical reconstruction of the glucagon secretion profile during hypoglycaemia via deconvolution hormone pulsatility analysis [54].
After an ACI is identified based on the above criteria, a more comprehensive study should evaluate the dose-response changes in the GCR caused by different modes of ACI delivery. In particular, the GCR response to hypoglycaemia induced by an insulin bolus (or by a hyperinsulinemic clamp) in the face of constant ACI delivery at different infusion rates with and without a switch-off should be estimated. This is a key step since our analysis shows that the repair of defective GCR requires a careful selection of the ACI infusion rate.
The analysis of the results of the comprehensive study will lead naturally to the design of a strategy for GCR repair with ACI based on three main principles:
Identification of an ACI infusion rate which provides a clinically significant improvement of the relative and/or absolute GCR response to hypoglycaemia.
Determination of the maximal ACI infusion rate (ACImax) which does not pose a significant risk of hypoglycaemia if there is a failure to switch off the ACI signal during a glucose decline (failure to detect hypoglycaemia).
Identification of an infusion rate to which ACImax should be reduced during hypoglycaemia (ACIhypo) such that it provides a maximal increase of the GCR. ACIhypo may or may not be zero.
Based on these principles an ACI-based GCR strategy can be designed and may consist of a simple constant rate ACI infusion or of a constant rate ACI infusion that is reduced (or switched-off) during hypoglycaemia. Note that different strategies will be needed to fully define the most effective and best tolerated treatment with the best reduction of highs and lows with meals and overnight fasting. We also point out that adding an ACI-based GCR repair strategy to an existing insulin therapy will likely require adaptation (decrease) of the exogenous insulin delivery rates since one of the anticipated effects of the use of ACI is a reduction of the hepatic glucose output. In terms of clinical implementation, an individualisation of the ACI infusion rates may be required.
Finally, we outline several arguments, which highlight additional advantages of an ACI-based GCR repair strategy to improve the safety of BG control.
Adding an α-cell inhibitor to an existing glucose control strategy may restore at least partially the balance between insulin and glucagon in their roles of key regulators of the liver and the HGP. Indeed, it has been repeatedly argued that the HGP in fasting and postprandial states depends on the net balance between the concentrations of insulin and glucagon rather than on their absolute levels (e.g., [61]). In T1DM, during euglycemia and hyperglycaemia, this balance is heavily biased towards glucagon. This follows easily from the observation that even in well controlled T1DM the absolute levels of insulin and glucagon in the circulation are not very different from the levels seen in health. Glucagon is either inappropriately elevated (postprandially) or is increased or unchanged during euglycemia and insulin levels required to control glycaemia are somewhat higher than those in health. Even with possibly higher insulin levels in T1DM the liver still “sees” by some estimates up to 50-fold lower insulin concentrations in the portal circulation and the same or higher glucagon concentrations as compared to normal physiology (e.g., see [22]). This is due to the fact that in health the circulating insulin levels reflect much higher portal concentrations and a significant clearance by the liver. Insulin replacement alone cannot restore the balance since it cannot be delivered at the appropriate levels to be effective at the liver.
In cases of pre-existing β-cell replacement therapies (e.g. islet transplantation) or in newly diagnosed diabetes the use of α-cell inhibitors may provide an additional tool (complementing insulin replacement) to allow reducing the contribution of endogenous β-cells and still maintain tight glycaemic control. This could be critical for β-cell survival.
Using α-cell inhibitors is expected to reduce the amount of insulin necessary to control glycaemia which may decrease the risk of hypoglycaemia related to possible failure to detect on time impending dangerous glucose declines.
Postprandial hyperglycaemia in patients with impaired glucose tolerance and diabetes has frequently been found to associate closely with cardiometabolic risk, post meal oxidant stress and endothelial dysfunction. For patients with T1DM, post meal control is the most common cause of hyperglycaemia extremes and practically winds up leading to ad hoc additional insulin doses to “catch up”, correction doses. Such dosing while needed with current strategies often presages overcorrection induced hypoglycaemia in subjects with greater glycaemic instability in an ever increasing spiral of highs and lows. The ACI-based strategy may help to minimize these lows and potentially some of the highs; for example pramlintide in our analysis of an insulin pump add-on study dramatically reduced supper hyperglycaemia [58].
-
Glucagon supplementation has been proposed and tested in an artificial pancreas closed loop strategy setting to help stabilize T1DM [62]. However, using exogenous glucagon as a controller to oppose the effect of insulin has the disadvantage that the liver may be forced to produce more glucose which should be then counterbalanced by more insulin. At this point, questions about exhausting liver glycogen stores are still not explored. In addition, glucagon replacement strategy has two other limits we perceive. The first is that while glucagon replacement is helpful in situations that engender heightened hypoglycaemia risk, it fails to lower glucagon at time when the risk for hyperglycaemia exists (e.g. postprandially). A second limitation is that raising both insulin and glucagon simultaneously is a strategy that theoretically might create a hormonal imbalance and inadvertently destabilize glycaemic control especially in rapidly changing circumstances, such as exercise, which if intense is notorious for causing hyperglycaemia with glucagon excess followed by hypoglycaemia risk with defective glucagon counterregulation. In addition:
Using ACI vs. supplementing glucagon may have the additional advantage of relying on endogenous rather than exogenous glucagon as a hypoglycaemia protection. Thereby, the circulating glucagon levels will be lower (since endogenous glucagon acts directly on the liver) thereby avoiding potentially dangerous glucagon overexposure. To achieve the same effect with exogenous glucagon, higher circulating levels will be required.
Using a pump to constantly infuse ACI rather than inject doses of glucagon may be advantageous in that there is more rapid absorption without a subcutaneous pool of medication and if the ACI needs to be downregulated (e.g, to trigger GCR), it can be done quite rapidly in comparison with achieving a similar glucose-stimulating effect with a subcutaneous glucagon injection.
-
To correct the impaired glucagon dynamics it is necessary to guarantee that glucagon increases during hypoglycaemia and decreases during hyperglycaemia relative to its euglycaemic levels. Theoretically, this can be achieved by using either glucagon inhibitors, or glucagon stimulators (or glucagon infusion). Figure 6 exemplifies the process of repair of glucagon dynamics by glucagon inhibition or stimulation/infusion.
However, while glucagon inhibitors reduce the overall glucagon levels, glucagon stimulators or glucagon infusions would have the opposite effect. Based on the clinical and in silico results (above) increase in glucagon may further impair the already defective GCR. In addition, higher insulin levels will be needed to balance the increase in glucagon. Therefore, a glucagon increase-based repair of glucagon dynamics is expected to augment the risk of hypoglycaemia, especially when the impending dangerous glucose declines are undetected.
Figure 6.

Illustration of the process of repair of glucagon dynamics by glucagon inhibition or glucagon stimulation/infusion.
Conclusions
In this review, we have presented a new approach to the study and understanding of glucagon secretion and its role in counterregulation. We have used traditional experimental physiological methods in lab animals, but we have added and intertwined a second component. That second component is a systems level understanding and a mathematical reconstruction, which brings an entirely different perspective on the normal physiology of GCR. By combining these two methods, it also permits a different type of analysis of the impairment of GCR in insulin deficient diabetes (whether type 1 or advanced type 2 diabetes) and allows us to propose the design of clinically feasible GCR repair strategies – a task which up to now appeared unapproachable. We show that lack of β-cell signalling to the α-cells can be viewed as a key network deficiency which impairs the GCR by contributing to two separate network abnormalities: (i) absence of a β-cell switch-off trigger and (ii) increase in intraislet auto-feedback independent glucagon release. Our in silico analysis of the glucagon axis shows that both abnormalities can be targeted in an effort to develop strategies to repair defective GCR. A key component in such strategies is the use of α-cell inhibitors to manipulate the glucagon levels by reduction of basal hyperglucagonaemia with or without disinhibition of the α-cells during hypoglycaemia to amplify the GCR. Such an α-cell inhibitor-based GCR repair treatment can be added to a variety of existing insulin replacement therapies to treat diabetes and is expected to stabilize blood glucose control and improve safety by reducing the risk of hypoglycemia.
Acknowledgments
Authors acknowledge support by NIH/NIDDK grant R01 DK082805.
Contributor Information
Leon S. Farhy, Email: leon@virginia.edu, Department of Medicine, PO Box 800735, University of Virginia, Charlottesville, Virginia, 22908, 434-924-2496, 434-982-3878 (fax).
Anthony L. McCall, Email: alm3j@virginia.edu, Departments of Medicine, PO Box 801407, University of Virginia, Charlottesville, Virginia, 22908, 434-243-9373, 434-982-3796 (fax).
References
- 1.The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 2.UK Prospective Diabetes Study Group (UKPDS) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes. Lancet. 1998;352:837–853. [PubMed] [Google Scholar]
- 3.The Action to Control Cardiovascular Risk in Diabetes Study Group. Effects of Intensive Glucose Lowering in Type 2 Diabetes. N Engl J Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cryer PE, Gerich JE. Relevance of glucose counterregulatory systems to patients with diabetes: critical roles of glucagon and epinephrine. Diabetes Care. 1983;6:95–99. doi: 10.2337/diacare.6.1.95. [DOI] [PubMed] [Google Scholar]
- 5.Gerich JE. Lilly lecture 1988. Glucose counterregulation and its impact on diabetes mellitus. Diabetes. 1988;37:1608–1617. doi: 10.2337/diab.37.12.1608. [DOI] [PubMed] [Google Scholar]
- 6.Fukuda M, Tanaka A, Tahara Y, et al. Correlation between minimal secretory capacity of pancreatic beta-cells and stability of diabetic control. Diabetes. 1988;37:81–88. doi: 10.2337/diab.37.1.81. [DOI] [PubMed] [Google Scholar]
- 7.Gerich JE, Langlois M, Noacco C, Karam JH, Forsham PH. Lack of glucagon response to hypoglycemia in diabetes: evidence for an intrinsic pancreatic alpha cell defect. Science. 1973;182:171–173. doi: 10.1126/science.182.4108.171. [DOI] [PubMed] [Google Scholar]
- 8.Hoffman RP, Arslanian S, Drash AL, Becker DJ. Impaired counterregulatory hormone responses to hypoglycemia in children and adolescents with new onset IDDM. J Pediatr Endocrinol. 1994;7:235–244. doi: 10.1515/jpem.1994.7.3.235. [DOI] [PubMed] [Google Scholar]
- 9.Segel SA, Paramore DS, Cryer PE. Hypoglycemia-associated autonomic failure in advanced type 2 diabetes. Diabetes. 2002;51:724–733. doi: 10.2337/diabetes.51.3.724. [DOI] [PubMed] [Google Scholar]
- 10.Cryer PE. Hypoglycemia is the limiting factor in the management of diabetes. Diabetes Metab Res Rev. 1999;15:42–46. doi: 10.1002/(sici)1520-7560(199901/02)15:1<42::aid-dmrr1>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 11.Cryer PE. Hypoglycemia the limiting factor in the glycaemic management of type I and type II diabetes. Diabetologia. 2002;45:937–948. doi: 10.1007/s00125-002-0822-9. [DOI] [PubMed] [Google Scholar]
- 12.Dagogo-Jack SE, Craft S, Cryer PE. Hypoglycemia-associated autonomic failure in insulin-dependent diabetes mellitus. Recent antecedent hypoglycemia reduces autonomic responses to, symptoms of, and defense against subsequent hypoglycemia. J Clin Invest. 1993;91:819–828. doi: 10.1172/JCI116302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gromada J, Franklin I, Wollheim CB. α-Cells of the Endocrine Pancreas: 35 Years of Research but the Enigma Remains. Endocr Rev. 2007;28:84–116. doi: 10.1210/er.2006-0007. [DOI] [PubMed] [Google Scholar]
- 14.Hirsch BR, Shamoon H. Defective epinephrine and growth hormone responses in type I diabetes are stimulus specific. Diabetes. 1987;36:20–26. doi: 10.2337/diab.36.1.20. [DOI] [PubMed] [Google Scholar]
- 15.Taborsky GJ, Jr, Ahren B, Havel PJ. Autonomic mediation of glucagon secretion during hypoglycemia: implications for impaired alpha-cell responses in type 1 diabetes. Diabetes. 1998;47:995–1005. doi: 10.2337/diabetes.47.7.995. [DOI] [PubMed] [Google Scholar]
- 16.Banarer S, McGregor VP, Cryer PE. Intraislet hyperinsulinemia prevents the glucagon response to hypoglycemia despite an intact autonomic response. Diabetes. 2002;51:958–965. doi: 10.2337/diabetes.51.4.958. [DOI] [PubMed] [Google Scholar]
- 17.Zhou H, Tran PO, Yang S, Zhang T, LeRoy E, Oseid E, Robertson RP. Regulation of alpha-cell function by the beta-cell during hypoglycemia in Wistar rats: the “switch-off“ hypothesis. Diabetes. 2004;53:1482–1487. doi: 10.2337/diabetes.53.6.1482. [DOI] [PubMed] [Google Scholar]
- 18.Hope KM, Tran PO, Zhou H, Oseid E, Leroy E, Robertson RP. Regulation of alpha-cell function by the beta-cell in isolated human and rat islets deprived of glucose: the “switch-off“ hypothesis. Diabetes. 2004;53:1488–1495. doi: 10.2337/diabetes.53.6.1488. [DOI] [PubMed] [Google Scholar]
- 19.Zhou H, Zhang T, Oseid E, Harmon J, Tonooka N, Robertson RP. Reversal of Defective Glucagon Responses to Hypoglycemia in Insulin-Dependent Autoimmune Diabetic BB Rats. Endocrinology. 2007;148:2863–2869. doi: 10.1210/en.2006-1375. [DOI] [PubMed] [Google Scholar]
- 20.Zhou H, Zhang T, Harmon JS, Bryan J, Robertson RP. Zinc, Not Insulin, Regulates the Rat α-Cell Response to Hypoglycemia In Vivo. Diabetes. 2007;56:1107–1112. doi: 10.2337/db06-1454. [DOI] [PubMed] [Google Scholar]
- 21.Farhy LS, Du Z, Zeng Q, et al. Amplification of pulsatile glucagon secretion by switch-off of α-cell suppressing signals in Streptozotocin (STZ)-treated rats. Am J Physiol Endocrinol Metab. 2008;295:E575–E585. doi: 10.1152/ajpendo.90372.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Unger RH, Orci L. Paracrinology of islets and the paracrinopathy of diabetes. Proc Natl Acad Sci USA. 2010;107:16009–16012. doi: 10.1073/pnas.1006639107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee Y, Wang MY, Du XQ, Charron MJ, Unger RH. Glucagon receptor knockout prevents insulin-deficient type 1 diabetes in mice. Diabetes. 2011;60:391–397. doi: 10.2337/db10-0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang MY, Chen L, Clark GO, et al. Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci U S A. 2010;107:4813–4819. doi: 10.1073/pnas.0909422107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farhy LS, McCall AL. System-Level Control to Optimize Glucagon Counterregulation by Switch-Off of alpha-Cell Suppressing Signals in beta-Cell Deficiency. J Diabetes Sci Technol. 2009;3:21–33. doi: 10.1177/193229680900300104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farhy LS, McCall AL. Pancreatic Network Control of Glucagon Secretion and Counterregulation. Methods Enzymol, Academic Press. 2009;467:547–581. doi: 10.1016/S0076-6879(09)67021-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farhy LS, McCall AL. Models of Glucagon Secretion, Their Application to the Analysis of the Defects in Glucagon Counterregulation and Potential Extension to Approximate Glucagon Action. J Diabetes Sci Technol. 2010;4:1345–1356. doi: 10.1177/193229681000400608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawai K, Unger RH. Inhibition of glucagon secretion by exogenous glucagon in the isolated, perfused dog pancreas. Diabetes. 1982;31:512–515. doi: 10.2337/diab.31.6.512. [DOI] [PubMed] [Google Scholar]
- 29.Stagner JI, Samols E, Bonner-Weir S. beta----alpha----delta pancreatic islet cellular perfusion in dogs. Diabetes. 1988;37:1715–1721. doi: 10.2337/diab.37.12.1715. [DOI] [PubMed] [Google Scholar]
- 30.Stagner JI, Samols E, Marks V. The anterograde and retrograde infusion of glucagon antibodies suggests that A cells are vascularly perfused before D cells within the rat islet. Diabetologia. 1989;32:203–206. doi: 10.1007/BF00265095. [DOI] [PubMed] [Google Scholar]
- 31.Brunicardi FC, Kleinman R, Moldovan S, et al. Immunoneutralization of somatostatin, insulin, and glucagon causes alterations in islet cell secretion in the isolated per fused human pancreas. Pancreas. 2001;23:302–308. doi: 10.1097/00006676-200110000-00012. [DOI] [PubMed] [Google Scholar]
- 32.Epstein S, Berelowitz M, Bell NH. Pentagastrin and glucagon stimulate serum somatostatin-like immunoreactivity in man. J Clin Endocrinol Metab. 1980;51:1227–1231. doi: 10.1210/jcem-51-6-1227. [DOI] [PubMed] [Google Scholar]
- 33.Utsumi M, Makimura H, Ishihara K, Morita S, Baba S. Determination of immunoreactive somatostatin in rat plasma and responses to arginine, glucose and glucagon infusion. Diabetologia. 1979;17:319–323. doi: 10.1007/BF01235888. [DOI] [PubMed] [Google Scholar]
- 34.Cejvan K, Coy DH, Efendic S. Intra-islet somatostatin regulates glucagon release via type 2 somatostatin receptors in rats. Diabetes. 2003;52:1176–1181. doi: 10.2337/diabetes.52.5.1176. [DOI] [PubMed] [Google Scholar]
- 35.Klaff LJ, Taborsky GJ., Jr Pancreatic somatostatin is a mediator of glucagon inhibition by hyperglycemia. Diabetes. 1987;36:592–596. doi: 10.2337/diab.36.5.592. [DOI] [PubMed] [Google Scholar]
- 36.Ludvigsen E, Olsson R, Stridsberg M, Janson ET, Sandler S. Expression and distribution of somatostatin receptor subtypes in the pancreatic islets of mice and rats. J Histochem Cytochem. 2004;52:391–400. doi: 10.1177/002215540405200310. [DOI] [PubMed] [Google Scholar]
- 37.Sumida Y, Shima T, Shirayama K, Misaki M, Miyaji K. Effects of hexoses and their derivatives on glucagon secretion from isolated perfused rat pancreas. Horm Metab Res. 1994;26:222–225. doi: 10.1055/s-2007-1001669. [DOI] [PubMed] [Google Scholar]
- 38.Portela-Gomes GM, Stridsberg M, Grimelius L, Oberg K, Janson ET. Expression of the five different somatostatin receptor subtypes in endocrine cells of the pancreas. Appl Immunohistochem Mol Morphol. 2000;8:126–132. doi: 10.1097/00129039-200006000-00007. [DOI] [PubMed] [Google Scholar]
- 39.Strowski MZ, Parmar RM, Blake AD, Schaeffer JM. Somatostatin inhibits insulin and glucagon secretion via two receptors subtypes: an in vitro study of pancreatic islets from somatostatin receptor 2 knockout mice. Endocrinology. 2000;141:111–117. doi: 10.1210/endo.141.1.7263. [DOI] [PubMed] [Google Scholar]
- 40.Schuit FC, Derde MP, Pipeleers DG. Sensitivity of rat pancreatic A and B cells to somatostatin. Diabetologia. 1989;32:207–212. doi: 10.1007/BF00265096. [DOI] [PubMed] [Google Scholar]
- 41.Kleinman R, Gingerich R, Ohning G, et al. The influence of somatostatin on glucagon and pancreatic polypeptide secretion in the isolated perfused human pancreas. Int J Pancreatol. 1995;18:51–57. doi: 10.1007/BF02825421. [DOI] [PubMed] [Google Scholar]
- 42.Huypens P, Ling Z, Pipeleers D, Schuit F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia. 2000;43:1012–1019. doi: 10.1007/s001250051484. [DOI] [PubMed] [Google Scholar]
- 43.Heise T, Heinemann T, Heller S, et al. Effect of Pramlintide on Symptom, Catecholamine, and Glucagon Responses to Hypoglycemia in Healthy Subjects. Metabolism. 2004;53:1227–1232. doi: 10.1016/j.metabol.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 44.Havel PJ, Ahren B. Activation of autonomic nerves and the adrenal medulla contributes to increased glucagon secretion during moderate insulin-induced hypoglycemia in women. Diabetes. 1997;46:801–807. doi: 10.2337/diab.46.5.801. [DOI] [PubMed] [Google Scholar]
- 45.Havel PJ, Taborsky GJ., Jr The contribution of the autonomic nervous system to changes of glucagon and insulin secretion during hypoglycemic stress. Endocr Rev. 1989;10:332–350. doi: 10.1210/edrv-10-3-332. [DOI] [PubMed] [Google Scholar]
- 46.Brelje TC, Scharp DW, Sorenson RL. Three-dimensional imaging of intact isolated islets of Langerhans with confocal microscopy. Diabetes. 1989;38:808–814. doi: 10.2337/diab.38.6.808. [DOI] [PubMed] [Google Scholar]
- 47.Bolli GB, Fanelli CG. Physiology of glucose counterregulation to hypoglycemia. Endocr Metab Clin North Am. 1999;28:467–493. doi: 10.1016/s0889-8529(05)70083-9. [DOI] [PubMed] [Google Scholar]
- 48.Taborsky GJ, Ahren B, Havel PJ. Autonomic mediation of glucagon secretion during hypoglycemia: implications for impaired alpha-cell responses in type 1 diabetes. Diabetes. 1998;47:995–1005. doi: 10.2337/diabetes.47.7.995. [DOI] [PubMed] [Google Scholar]
- 49.Genter P, Berman N, Jacob M, Ipp E. Counterregulatory hormones oscillate during steady-state hypoglycemia. Am J Physiol. 1998;275:E821–E829. doi: 10.1152/ajpendo.1998.275.5.E821. [DOI] [PubMed] [Google Scholar]
- 50.Grapengiesser E, Salehi A, Quader SS, Hellman B. Glucose Induces Glucagon Release Pulses Antisynchronous with Insulin and Sensitive to Purinoceptors Inhibition. Endocrinology. 2006;147:3472–3477. doi: 10.1210/en.2005-1431. [DOI] [PubMed] [Google Scholar]
- 51.Jaspan JB, Lever E, Polonsky KS, Van Cauter E. In vivo pulsatility of pancreatic islet peptides. Am J Physiol. 1986;251:E215–E226. doi: 10.1152/ajpendo.1986.251.2.E215. [DOI] [PubMed] [Google Scholar]
- 52.Salehi A, Quader SS, Grapengiesser E, Hellman B. Pulses of somatostatin release are slightly delayed compared with insulin and antisynchronous to glucagon. Regul Pept. 2007;144:43–49. doi: 10.1016/j.regpep.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 53.Farhy LS. Modeling of oscillations in endocrine networks with feedback. Methods Enzymol. 2004;384:54–81. doi: 10.1016/S0076-6879(04)84005-9. [DOI] [PubMed] [Google Scholar]
- 54.Johnson ML, Pipes L, Veldhuis PP, Farhy LS, Boyd DG, Evans WS. AutoDecon, a Deconvolution Algorithm for Identification and Characterization of Luteinizing Hormone Secretory Bursts: Description and Validation Using Synthetic Data. Anal Biochem. 2008;381:8–17. doi: 10.1016/j.ab.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nauck MA, Heimesaat MM, Behle K, et al. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab. 2002;87:1239–1246. doi: 10.1210/jcem.87.3.8355. [DOI] [PubMed] [Google Scholar]
- 56.Degn KB, Brock B, Juhl CB, et al. Effect of intravenous infusion of exenatide (synthetic exendin-4) on glucose-dependent insulin secretion and counterregulation during hypoglycemia. Diabetes. 2004;53:2397–2403. doi: 10.2337/diabetes.53.9.2397. [DOI] [PubMed] [Google Scholar]
- 57.Ahrén B, Schweizer A, Dejager S, et al. Vildagliptin enhances islet responsiveness to both hyper- and hypoglycemia in patients with type 2 diabetes. J Clin Endocrinol Metab. 2009;94:1236–1243. doi: 10.1210/jc.2008-2152. [DOI] [PubMed] [Google Scholar]
- 58.McCall AL, Cox DJ, Crean J, Gloster MA, Kovatchev BP. A novel analytical method for assessing glucose variability: using CGMS in type 1 diabetes mellitus. Diabetes Technol Ther. 2006;8:644–653. doi: 10.1089/dia.2006.8.644. [DOI] [PubMed] [Google Scholar]
- 59.Chan A, Breton MD, Anderson SM, Kovatchev BP, McCall AL, Farhy LS. Basal hyperglucagonaemia impairs the glucagon response to hypoglycemia in type 1 diabetes. Presented at the 12TH SERVIER-IGIS SYMPOSIUM; St.-Jean-Cap-Ferrat France. 31 March – 3 April 2011.. [Google Scholar]
- 60.Shah P, Basu A, Basu R, Rizza R. Impact of lack of suppression of glucagon on glucose tolerance in humans. Am J Physiol. 1999;277:E283–E290. doi: 10.1152/ajpendo.1999.277.2.E283. [DOI] [PubMed] [Google Scholar]
- 61.Unger RH. Glucagon and the insulin: glucagon ratio in diabetes and other catabolic illnesses. Diabetes. 1971;20:834–838. doi: 10.2337/diab.20.12.834. [DOI] [PubMed] [Google Scholar]
- 62.El-Khatib FH, Russell SJ, Nathan DM, Sutherlin RG, Damiano ER. A bihormonal closed-loop artificial pancreas for type 1 diabetes. Sci Transl Med. 2010;2:27ra27. doi: 10.1126/scitranslmed.3000619. [DOI] [PMC free article] [PubMed] [Google Scholar]