

Abstract
Autism spectrum disorder (ASD) and schizophrenia (SCZ) are two common neurodevelopmental syndromes that result from the combined effects of environmental and genetic factors. We set out to test the hypothesis that rare variants in many different genes, including de novo variants, could predispose to these conditions in a fraction of cases. In addition, for both disorders, males are either more significantly or more severely affected than females, which may be explained in part by X-linked genetic factors. Therefore, we directly sequenced 111 X-linked synaptic genes in individuals with ASD (n = 142; 122 males and 20 females) or SCZ (n = 143; 95 males and 48 females). We identified > 200 non-synonymous variants, with an excess of rare damaging variants, which suggest the presence of disease-causing mutations. Truncating mutations in genes encoding the calcium-related protein IL1RAPL1 (already described in Piton et al. Hum Mol Genet 2008) and the monoamine degradation enzyme monoamine oxidase B were found in ASD and SCZ, respectively. Moreover, several promising non-synonymous rare variants were identified in genes encoding proteins involved in regulation of neurite outgrowth and other various synaptic functions (MECP2, TM4SF2/TSPAN7, PPP1R3F, PSMD10, MCF2, SLITRK2, GPRASP2, and OPHN1).
Keywords: autism spectrum disorder, schizophrenia, X chromosome, synaptic genes, rare variants
Introduction
Autism spectrum disorder (ASD) and schizophrenia (SCZ) are two common neurodevelopmental disorders that typically appear during childhood, adolescence, or early adulthood. Twin and epidemiological studies strongly support the function of genetic factors in these diseases.1–3 Several linkage and association studies have been performed over the past decades, with limited success.3–7 Positive linkage signals were obtained in numerous genomic regions and several genes or markers have been associated with ASD or SCZ; however, many of these results failed to be replicated in subsequent studies. The hypothesis implicating genetic heterogeneity, in which rare highly penetrant mutations (some of which may be de novo) in different genes specific to single families would predispose to these disorders, is supported by the recent findings that rare and de novo mutations in the NLGN4X, NLGN3, and SHANK3 synaptic genes cause ASD in a small number of families.8–12 Thus, direct resequencing of the complete coding regions of candidate genes is the best approach to identify such rare variants.
A significant body ofwork suggests thatASDand SCZ are synaptic disorders. Reduced dendritic spines have been observed in neurons of these patients and nearly all the genes associated with these disorders are involved in the formation, regulation, or normal function of the synapse.13–19 Therefore, we screened our ASD and SCZ cohorts for variants in candidate genes that encode proteins involved in various synaptic processes, as part of the Synapse-to-Disease project (www.synapse2disease.ca). Here, we focused on X-linked synaptic genes, as several evidences have implicated the X chromosome in ASD and SCZ.20,21 The proportion of X-linked genes involved in brain development and cognition is high when compared with autosomal genes.22–24 Numerous X-linked genes have been implicated in non-syndromic mental retardation (NS-MR) and in syndromes associated with autistic features.23,25,26 In addition, linkage and association studies have identified several ASD and SCZ candidate regions on the X chromosome.27–36 Moreover, the prevalence of ASD or SCZ is increased in individuals with sex-chromosome aneuploidy, as is the case in Turner and Klinefelter syndromes.23,37–39 Finally, the prevalence of ASD is four times higher in males than in females.40 This sex-related difference in disease prevalence does not hold for SCZ; however, schizophrenic males seem to develop more severe symptoms at an earlier age than females.41
On the basis of the evidences presented above, we screened X-linked synaptic genes for rare damaging variants in a cohort of 142 ASD and 143 SCZ individuals (Figure 1a). The cohort of ASD includes patients with classic autism, Asperger Syndrome, and pervasive developmental disorder not otherwise specified, whereas the SCZ cohort includes patients with SCZ, schizoaffective disorder (SCZaff), and childhood onset schizophrenia (COS).
Figure 1.

Strategy used for the selection and screening of synaptic candidate genes. (a) Schematic overview of the whole project: from the selection of candidate genes to the identification of genes probably involved in ASD and SCZ. (b) Representation of the number of synaptic genes identified according to the three different sources: proteomic studies, SynDB (Synapse Database), and manual query of PubMed (see Materials and methods; Supplementary Methods). The list of genes selected according to the different sources is provided in Supplementary Table 1. (c) List of the criteria used for the ranking and scoring of synaptic genes. The global score for each gene was the sum of all the points obtained using the various criteria. Further details are provided in Supplementary Tables 2 and 3.
Materials and methods
Creation of the synaptic X-linked gene list
From a list of 1125 X-linked genes annotated in the NCBI Mapviewer Build 35.1, we performed a quick search on each gene using information obtained from NCBI Gene and PubMed, as detailed in the Supplementary Information. Separately, we used different lists from proteomic studies42–44 and recovered 51 proteins encoded by X-linked genes. In parallel, we used a list extracted from Synapse DataBase (SynDB) (http://syndb.cbi.pku.edu.cn/), a new database dedicated to synaptic proteins that was created by the center for Bioinformatics of Peking University.45 They found ~3000 human synaptic proteins, of which 130 are encoded by X-chromosome genes.
Selection of synaptic X-linked candidate genes
To select the best candidate genes, we built a scoring system based on different criteria. The first one was the involvement in a human disease: genes known to cause ASD, SCZ, or related syndromes (for example Rett syndrome RTTor NS-MR) were directly selected for the study, whereas genes known to cause other non-psychiatric diseases were excluded from the selection. Genes not clearly involved in any disease were ranked according to five different criteria relevant for ASD or SCZ: nature of synaptic function, evidence of synaptic localization, expression in brain tissues, cognitive impairment in animal models, genetic data, and involvement in a relevant pathway for ASD or SCZ. A score was calculated that corresponded to the sum of all the points attributed using the five criteria described above (Supplementary Table 3). Additional details for each criterion are provided in the Supplementary Information. We selected 113 synaptic genes for the variant screening; 25 based on their involvement in related diseases and 88 that had a score superior to zero. Two out of the 113 genes were not screened: SYN1, which had been sequenced and analyzed before the beginning of this project (Cossette et al., unpublished data), and NXF5, because we were unable to design specific primers for this gene.
Patient and control cohorts
The 142 unrelated ASD patients (122 males and 20 females) were as described.46 The 143 SCZ subjects (95 males and 48 females) were collected from five different centers and included 28 cases of COS and 115 sporadic or familial cases of adult onset SCZ or SCZaff. They were evaluated by experienced investigators using the Diagnostic Interview for Genetic Studies or Kiddie Schedule for Affective Disorders and Schizophrenia, and multidimensional neurological, psychological, psychiatric, and pharmacological assessments. Family history for psychiatric disorders was also collected using the Family Interview for Genetic Studies. All Diagnostic Interview for Genetic Studies and Family Interview for Genetic Studies have been reviewed by two or more psychiatrists for a final consensus diagnosis based on DSM-IIIR or DSM-IV at each center. Exclusion criteria included patients with psychotic symptoms mainly caused by alcohol, drug abuse, or other clinical diagnosis including major cytogenetic abnormalities. Our population control group included 277 X chromosomes (from 103 males and 87 females) collected as part of the Quebec Newborn Twin Study. Ethnic origins of the grandparents were self-reported by the parents of probands and population control subjects. The ASD cohort is composed of French Canadians (85), of other European Caucasians (47), and of non-Caucasians (10). The SCZ cohort is composed of European Caucasians (133) and Pakistani or non-Caucasians (10). The control population is composed of French Canadians (129), of other European Caucasians (37), of South Americans (4), of non-Caucasians (3), and of individuals of mixed origin (16). Paternity and maternity of each individual of all families were confirmed using a panel of six highly informative unlinked microsatellite markers (D2S1327, D3S1043, D4S3351, D6S1043, D8S1179, D10S677). These markers were also used to confirm that cell line and blood samples corresponded to the same individual in the case of de novo mutations. An additional set of 285 ASD patients (247 males and 38 females) was used for the screening of IL1RAPL1 and several exons of TM4SF2/TSPAN7 and an additional set of 190 SCZ patients (118 males and 72 females) was used for the screening of monoamine oxidase B (MAOB) and several exons of PLXNB3 and GRPR.
Gene resequencing and variant identification
Genomic DNA was extracted from blood samples or lymphoblastoid cell lines using the Puregene DNA kit (Gentra System, USA). The amplification of the coding regions of all isoforms of the 111 genes required the design of > 1200 primer pairs, which was carried out using the Primers3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi), through Exonprimer (http://ihg2.helmholtz-muenchen.de/ihg/ExonPrimer.html), when possible. PCR reactions were performed in 384-well plates using 5 ng of genomic DNA, according to standard procedures. PCR products were sequenced at the McGill University and Genome Quebec Innovation Centre (Montreal, Canada) on a 3730XL DNA Analyzer. A fragment was considered successfully sequenced if the analysis of over 90% of the traces was possible. PolyPhred (v.5.04), PolySCAN (v.3.0), and Mutation Surveyor (v. 3.10, Soft Genetics) were used for variant detection analyses. In each case, unique novel exonic variants were confirmed manually by reamplifying the fragment and resequencing the proband and both parents using reverse and forward primers. For all de novo transmission variants, the specific exon was sequenced a second time using DNA extracted from blood instead of from cell lines, when possible. Variants found more than once or already described (in dbSNP http://www.ncbi.nlm.nih.gov/projects/SNP/, Exoseq http://www.sanger.ac.uk/cgi-bin/humgen/exoseq/exoseqview or in the literature) were considered as automatically confirmed (CA).
Genetic and statistical analyses
Rare variant follow-up
For each rare variant, the mode of transmission and the cosegregation between the variant and the disease were evaluated by sequencing the corresponding exon in the parents and other available relatives. For the most relevant variants, we resequenced the corresponding exon in 190 individuals from our control population and/or in an additional 285 ASD and 190 SCZ patients.
Prediction of potential functional effect
The potential consequence of each missense variant was evaluated using the PolyPhen (http://genetics.bwh.harvard.edu/pph/), SIFT (http://blocks.fhcrc.org/sift/SIFT.html), and PANTHER (http://www.pantherdb.org/tools/csnpScoreForm.jsp) software.47–49
Statistical analyses
The number of coding base pairs screened was calculated as follows: 3×number of codons successfully sequenced×(number of males+2×number of females). The Ka of each gene was calculated as the number of non-synonymous (NS) variants identified divided by the number of NS sites screened. The Ks of each gene was calculated as the number of synonymous (S) variants identified divided by the number of S sites screened. Ka was also calculated for polymorphisms and for rare variants, ASD rare variants, or SCZ rare variants only. For the McDonald–Kreitman test, NS and S changes between human and macacque sequences were calculated using DNASP as described earlier by Tarpey et al.50 The significance of differences between S and NS rare variants and human/macacque S and NS changes was evaluated for each gene using a Fisher’s exact test. Significance was set at P < 0.01. The proportion of novel vs known (or rare variants vs polymorphisms) for S and NS variants was compared using a χ2 test (1 df). The proportion of damaging vs benign predictions for rare variants and polymorphisms was also analyzed using a χ2 test (1 df).
Results
Selection and screening of candidate genes
From the 1125 X-linked annotated genes, we established a list of 188 genes encoding synaptic or potentially synaptic proteins using PubMed searches, published proteomic studies, and the SynDB (see Figure 1b; Material and methods; Supplementary Table 1). This corresponds to 17% of the total number of X-linked genes, with all the categories of synaptic functions being represented. We narrowed this list down to 111 genes by applying selective criteria and a prioritization scoring scheme (see Figure 1c; Material and methods; Supplementary Tables 2 and 3). To amplify the exons and the intronic boundaries of all available isoforms of each gene, we designed 1324 PCR amplicons and successfully optimized PCR conditions for 1263 fragments, which cover 96% of the coding regions. Fragments were amplified in the 285 patient samples and PCR products were sequenced (one strand).
We identified and confirmed 533 coding and splice variants (Table 1; Supplementary Table 4), which included 526 single-nucleotide changes and seven insertions/deletions (indels). A large number of silent (S) and missense (NS) variants as well as two nonsense variants, one splicing alteration, and one substitution of the first methionine were found. Five of the seven indels (in the CACNA1F, PAK3, GPR50, GABRQ, and ATP6AP1 genes) were in frame and added/removed one or several amino acids. These indels did not target conserved amino acids predicted to be implicated in protein function, and, therefore, were unlikely to be pathogenic (Table 2). The two other indels caused a frameshift and are described in the next section.
Table 1.
Distribution of the exonic variants, annotated by type of variant
| Variant type | Total | Known vs novel | Pol vs rare | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Indel | |||||||||
| Indel frameshifta | 2 | 0 | 2 | 0 | 2 | ||||
| Indel inframe | 5 | 1 | 4 | 4 | 1 | ||||
| Single nucleotide | |||||||||
| Nonsensea | 2 | 1 | 1 | 1 | 1 | ||||
| Splicea | 1 | 0 | 1 | 0 | 1 | ||||
| First methioninea | 1 | 0 | 1 | 0 | 1 | ||||
| Missense (NS) | 245 | 60 | 185 | | | P = 0.0004 | 86 | 159 | | | P = 0.0004 |
| Silent (S) | 277 | 108 | 169 | 140 | 137 | ||||
The ‘known’ variants were reported earlier in SNP public databases or in the literature, whereas other variants were ‘novel’. The ‘rare’ variant group included all the novel variants identified in only one individual or in individuals sharing the same disease, either ASD or SCZ. In contrast, the ‘pol’ (polymorphism) group comprised all variants known as polymorphisms or that were not specific to one disease (variants found in both the ASD and SCZ cohorts). indel, insertion, or deletion of one or more nucleotides; single-nucleotide, single-nucleotide changes; NS, single-nucleotide non-synonymous missense variants; S, single-nucleotide synonymous variants; rare, variant found only once or in one cohort; pol, polymorphism, variant found in both cohorts or already described.
Truncating variants.
Table 2.
List of the six truncating and five indel inframe variants identified during the screening of the 111 X-linked synaptic genes in 285 ASD and SCZ patients
| Gene | Variant | Variant type | Already described | Frequencies
|
Comments | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Disease | Sex | ASD | SCZ | CONT | Inheritance | Familial info | |||||
| MAOB | c.1342C > T p.R448X |
Nonsens | No | SCZ | M | 0/142 | 1/332 | 0/190 | Mother | 1 SCZ brother does not have the variant | Removes the TM, IM and one part of the cytoplasmic domain. |
| P2RY4 | c.1043G > A p.W348X |
Nonsens | rs41310667 | Both | M | 2/130 | 2/140 | 3/190 | n.a. | n.a. | Found in the three cohorts with similar frequencies. It might not affect protein function |
| PCDH11X | c.3034-1G > A | Splice | No | SCZ | F | 0/139 | 1/113 | 0/190 | n.a. | n.a. | Affects the end of the shortest isoform. Nonsense polymorphisms are described in dbSNP in the last exon, which is removed |
| PLXNB3 | c.1A > G p.M1? |
Unknown | No | SCZ | M M |
0/142 | 2/325 | 0/190 mother |
Mother | 1 SCZ sister has the variant 1 SCZ sister does not have the variant |
This first methionine codon is unlikely to be the real initiator codon. It is not conserved between species and does not match the Kozak consensus sequence. |
| HS6ST2 | c.23delT p.V8AfsX26 |
Indel (fshift) | No | SCZ | F | 0/142 | 1/143 | 1/190 | Mother | The SCZ half-brother has the variant | Found in a girl from the CONT cohort. The variant is inherited from her unaffected father. It might be a polymorphism. |
| IL1RAPL1 | c.1100-1106 delTACTCTT p.I367fsX6 |
Indel (fshift) | No | ASD | F | 1/422 | 0/143 | 0/189 | de novo | No familial history | Affects protein function (Piton et al.).51 |
| CACNA1F | c.2440 delGAGGAAGAA p.E814-816del |
Indel (inf) | No | Both | Both | 3/142 | 9/142 | n.a. | n.a. | n.a. | Deletion/insertion of one or several Glu in a region with Glu repetition. |
| PAK3 | c.525delGAT p.D175del |
Indel (inf) | No | SCZ | M | n.a. | 1/94 | n.a. | n.a. | n.a. | Deletion of one Asp in a region with Glu repetition. It might not have a damaging effect on protein function. |
| GABRQ | c.1432delATT p. I478del |
Indel (inf) | No | Both | M | 1/141 | 1/143 | n.a. | n.a. | n.a. | One polymorphism I478F was earlier described at this position. It might not be a critical amino acid for the protein. |
| GPR50 | c.1505-1516 del12bp p. 502-505del |
Indel (inf) | No | Both | Both | 57/142 | 66/143 | n.a. | n.a. | n.a. | Already described with no association found with SCZ. |
| ATP6AP1 | c.97insGCGGCG p.A33_A34insAA |
Indel (inf) | No | Both | Both | 2/142 | 1/143 | n.a. | n.a. | n.a. | Insertion of 2 Ala found in the two cohorts. It might be a polymorphism. |
Abbreviations: ASD, autism spectrum disorder; CONT, control population; F, female; fshift, frameshift; IM, intermembrane domain; inf, in frame; M, male; n.a., non available; SCZ, schizophrenia; TM, transmembrane domain.
Variants that may cause protein truncation
In total, six potentially truncating variants, four single-nucleotide substitutions and two indels were identified (Table 2). The nonsense variant found in P2RY4 (c.1043G > A, p.W348X) was clearly a polymorphism, referenced as rs41310667. The frameshift variant detected in HS6ST2 (female (F): c.23delT, p.V8AfsX26), which was predicted to truncate a large portion of the protein, was also found in one control female and her father and was presumed to be a polymorphism. Variants affecting the consensus splice site (F: c.3034-1G > A) and initiation methionine (2 males (M): c.1A > G, 2 M) of PCDH11X and PLXNB3, respectively, were not predicted to have a strong damaging effect because of their relative position in the protein (see comments in Table 2). In contrast, the 7 bp deletion (F: c.1100-1106del- TACTCTT) in the IL1RAPL1 gene, which caused a frameshift p.I367fsX6 in one girl with Asperger Syndrome, led to a loss of function of IL1RAPL1, as described earlier.51 The screening of IL1RAPL1 in additional ASD patients led to the identification of a new missense variant (M: 59A > G; p.K20R), which was not present in the control population. Finally, a nucleotide substitution (M: c.1342C > T) leading to a premature stop codon (p.R448X) was identified in one boy in the gene encoding MAOB (Figure 2a). This patient had a SCZaff disorder with an early age of onset and the R448X variant was inherited from his mother, who had a history of major depression. Moreover, there was some familial history of SCZ on the mother’s side. The patient’s brother, who had a diagnosis of SCZ, did not carry this variant. However, the proband displayed a different diagnosis (SCZaff vs SCZ) and an earlier age of onset (15 vs 22 years) than his elder brother. This R448X variant was predicted to delete the last 73 amino acids of the MAOB protein, which encompass the mitochondrial anchor to the outer membrane and the intermembrane domains; this truncation could affect the targeting of the protein to mitochondria. Sequencing of 190 additional SCZ patients led to the identification of three novel missense variants, c.326C >T (M: p.P109L), c.958G >A (M+F: p.E320K), and c.1003G >A (M: p.G335S), in three boys, all transmitted by a heterozygous unaffected mother (Figure 2b; Supplementary Table 6). Moreover, the E320K variant was also found in one other girl with SCZ. These variants all affected evolutionarily conserved amino acids located in the cytoplasmic domain of the protein and may affect protein function, particularly the P109L variant, which modifies an amino acid involved in binding to the mitochondrial membrane. 52 The exons of MAOB that contain the variants described were sequenced in the 190 control individuals and no NS changes were identified.
Figure 2.
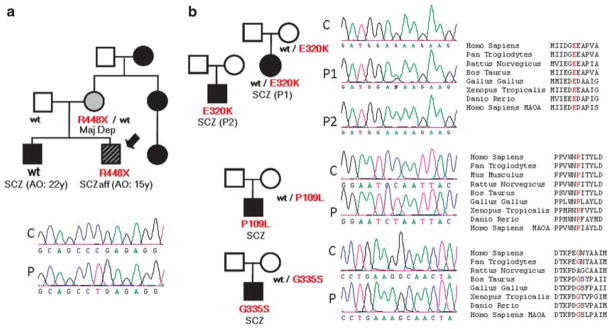
MAOB variants identified in SCZ individuals. The corresponding family pedigree (the proband is indicated by an arrow) and chromatograms for the proband (P) and control (C), as well as amino-acid conservation, are shown for each variant. (a) Nonsense variant identified during the first screening of 143 SCZ patients. (b) Missense variants identified during the screening of 190 additional SCZ patients.
Analysis of variants identified gene per gene
Taking into consideration the different RefSeq isoforms of all the genes sequenced, in this study, we screened an estimated 71,723 codons per patient (Supplementary Table 5), which corresponds to a total of 76Mb of coding sequence. S and NS variants were found in an approximately equal proportion, which is consistent with the findings of a recent analysis of the whole exome of one individual53. For each gene, we represented the number of S+NS variants as a function of the length of the coding sequence (Supplementary Table 5; Supplementary Figure 1a). Some genes (such as USP9X) had less variants than expected, according to their coding-sequence length, whereas others (for example SHROOM2 or HCFC1) exhibited an accumulation of variants; however, we could not establish a correlation between these findings and the chromosomal location of the different genes (data not shown). Genes such as HCFC1 had significantly less NS than S variants, which suggests that variation in their coding sequence is not well tolerated (Supplementary Figure 1b). In contrast, an excess of NS variants were observed in genes such as PLXNB3, GPR50, or HS6ST2. For each gene, we calculated the Ka, which is the number of NS variants divided by the number of NS sites. The analysis of genes with a high Ka in our cohorts did not allow the distinction between a highly polymorphic gene (in which greater coding-sequence variation is tolerated) from a gene enriched for disease-causing variants.
Excess of rare and damaging NS variants
We classified the variants identified during our study either as rare variants or polymorphisms. The ‘rare variant’ group included all novel (not reported in any database) variants identified in only one individual or in individuals sharing the same disease, either ASD or SCZ (as they could be rare variants shared by patients with a recent common ancestry). The ‘polymorphism’ group included all known variants or all variants not specific to one disease, which likely represent neutral polymorphisms.
Considering NS variants as more likely to have an effect on gene function, as they cause an amino-acid change, and S variants as a pool of generally neutral variants, we identified two parameters that were associated with an uneven distribution of NS and S variants. Only 30% of the variants identified were already known (either present in SNP databases (dbSNP and ExoSeq) or described in the literature). However, the distribution of novel vs known variants was significantly different between S and NS variants (P=0.0004), with an excess of NS variants being novel (Table 1). Perhaps more relevant, the grouping of variants into rare variants and polymorphisms revealed a significant difference in the distribution of S and NS variants (P=0.0004) toward the identification of more NS rare variants than expected. The analysis of the potential effect of NS missense variants using various prediction programs showed that, according to at least two out of the three algorithms tested, rare variants were significantly more damaging than polymorphisms (Supplementary Table 4; Figure 3), which suggests that they may include diseasecausing mutations for ASD or SCZ. Although we decided to focus on NS changes in our study, we are aware that, in some cases, certain S variants may have an effect on mRNA splicing or stability.54,55
Figure 3.

Potential effect on protein function of the different missense variants identified, according to three prediction programs: PolyPhen, PANTHER, and SIFT. The variants were divided into the ‘rare variant’ (rare) and ‘polymorphism’ (pol) groups. The percentage of variants associated with benign (PolyPhen) or tolerated (SIFT) predictions or with a score >–5 (PANTHER) are represented in green, whereas variants associated with a possibly or probably damaging effect (PolyPhen), affecting protein function prediction (SIFT), or with a score <–5 (PANTHER) are represented in red. The gray portion represents variants for which prediction was unknown (PolyPhen), given with low confidence (SIFT), or not available (PANTHER).
Genes that present an excess of NS rare variants
Some genes had a higher Ka for rare variants (Karare) than their Ka of polymorphisms (Kapol) (Supplementary Table 5; Supplementary Figure 2). Among them, some had a higher Karare in one cohort than in the other; these included genes such as TM4SF2/TSPAN7, HNRPH2, PSMD10, and CACNA1F for ASD and RBM3, PNCK, SLITRK2, or KCND1 for SCZ.
To identify genes that exhibited a greater number of rare NS variations in the patients than that expected from their rate of evolution, we performed a McDonald–Kreitman test (Supplementary Table 5). For the SCZ cohort, significant differences were obtained for SLITRK2 (P = 0.0006), a neuronal transmembrane protein family SLITRK, which affects neurite outgrowth. 56 We identified in this gene two novel missense variants c.265G >A and c.1646C >T (F: p.V89M and F: p.S549F) in girls with SCZ and in their affected siblings. Significant results were also obtained for SEPT6 (P = 0.004), and KCND1 (P = 0.004). Surprisingly, MECP2, the main Rett syndrome gene, showed an accumulation of NS rare variants (P = 0.004) in the SCZ cohort. A significant excess of NS rare variants in the ASD cohort was obtained for CACNA1F (P = 0.0011). This gene is involved in congenital stationary night blindness of type 2. A missense mutation (I745T) in CACNA1F was described in a large family with congenital stationary night blindness of type 2, in which five of the affected males had intellectual deficits and three of these five individuals were diagnosed with ASD.57 Interestingly, other members of this calcium channel family have been linked to ASD (CACNA1C,58 CACNA1H,59 and CACNA1G60). Although the two NS variants identified in two ASD patients (2 M: p.V635I and M+F: p.N746T) were also found in the control cohort, the four unique missense variants (M: p.L977F, M: p.D1966N, M: p.Q1625R, and M: p.G1684R) were absent in the control population; however, the analysis of cosegregation and functional effect predictions did not support their implication in ASD. CDKL5, which causes Rett syndrome,61 and PLXNB3, a plexin acting as a receptor for Semaphorin 5A, also exhibited an accumulation of NS rare variants (P = 0.0068 and P = 0.0012, respectively), but this was not significant for ASD or SCZ separately. To deal with the population structure issue, we performed again the analysis excluding rare variants from patients with a non-European-Caucasian origin (10 ASD and 10 SCZ), and we obtained similar results.
Transmission, gender, and disease distribution of rare variants
To evaluate the mode of transmission of each rare variant, we reamplified the corresponding genomic fragments and resequenced them in the proband and his/her parents. As the large majority of parents were non-affected, we can discern modes of transmission that favor the implication of rare variants in disease, that is de novo inheritance or transmission from a heterozygous mother to a hemizygous boy. We need to be particularly cautious with the de novo variants, as we found that a non-negligible proportion of the de novo variants identified here were artifacts associated with the use of DNA extracted from lymphoblastoid cell lines and could not be confirmed in corresponding blood DNA. Among the eight de novo variants initially identified in this study, only the deletion in IL1RAPL1 and the missense in MECP2 were confirmed as bona fide de novo variants. Two of these variants were cell-line variants and four were not tested because blood DNA was not available, but they are likely to be cell-line artifacts, as for most of them, they were found as heterozygous variants in male patients.
On the basis of the number of chromosomes tested, we observed no significant differences in the number of NS rare variants found in males and in females. Only one NS variant (F: c.175G > A; p.D59N in the TRO gene) was homozygous in one girl from a Pakistani family. This variant was predicted to have a benign effect on protein function and did not segregate with SCZ in the family. However, some of the NS rare variants identified in heterozygous females could also be causative. Indeed, in the case of variants transmitted by the unaffected mother, it could be a difference in the X-inactivation patterns that cause a phenotype in some carrier females and not in others. However, we decided not to test X-inactivation pattern in a systematic way for all the rare NS variants identified in females, given that extrapolation of X-inactivation pattern results in lymphoblastoid cell lines DNA, or even in blood, might be wrong to estimate what occurs in brain tissues. We did not observe any significant difference in the number of NS rare variants identified between the ASD and the SCZ cohorts. However, among the individuals of European-Caucasian origin, more SCZ than ASD (14 vs 7) patients accumulate several NS rare variants (Supplementary Figure 4).
Most promising NS rare variants
To identify the variants that are more likely to be disease-predisposing or -causing mutations, all the rare NS variants were ranked according to their potentially damaging effect on protein function and the mode of transmission and/or the cosegregation between the variants and the disease (Supplementary Table 7). The most promising variants are listed in Table 3 and several examples are described here.
Table 3.
List of the 30 most promising NS rare variants identified during the screening of the 111 X-linked synaptic genes in 285 ASD and SCZ patients
| Variant Info
|
Segregation analysis
|
Effect on protein functiona | ||||
|---|---|---|---|---|---|---|
| Gene | Variant | Gender | Pedigreea | |||
| Rare missense variants in ASD patients | ||||||
| PPP1R3F | T/C | Hemi | F245L | M | Mother + + | + + + |
| MECP2 | C/T | Hetero | R309W | F | de novo + + | + + |
| PSMD10 | G/A | Hemi | G129R | M | Mother + | + + |
| PDZD4 | G/A | Hemi | D326N | M | Mother + | + + |
| GLRA2 | G/T | Hetero | R350L | F | Mother | + + |
| TM4SF2/TSPAN7 | C/A | Hemi | P172H | M | Mother + | + |
| OPHN1 | A/G | Hemi | H705R | M | Mother + | + |
| CXCR3 | C/G | Hemi | V115L | M | Mother + | + |
| FRMPD4 | G/A | Hemi | G1147E | M | Mother + | + |
| MAOA | A/T | Hemi | N271I | M | Mother + | + |
| PCDH19 | C/G | Hemi | H146Q | M | Mother + | + |
| WNK3 | C/G | Hemi | S886C | M | Mother + | + |
| WNK3 | G/T | Hemi | G944V | M | Mother + | + |
| SH3KBP1 | A/G | Hemi | S458G | M | Mother + | + |
| HNRPH2 | G/C | Hemi | M396I | M | Mother + | + |
| Rare missense variants in SCZ patients | ||||||
| MCF2 | C/A | Hetero | F555L | F | Mother + + | + + |
| GPRASP2 | T/A | Hemi | I791K | M | Mother + + | + + |
| FRMPD4 | C/T | Hemi | R1189C | M | n.a. + | + + + |
| HCFC1 | C/T | Hetero | T142M | F | Mother + + | + + |
| GABRE | C/T | Hetero | R472H | F | Mother | + + + |
| OPHN1 | A/G | Hemi | M461V | M | Mother + | + + |
| BRS3 | C/T | Hemi | R352W | M | Mother + | + + |
| ATP2B3 | G/C | Hemi | D459H | M | Mother + | + + |
| BEX2 | G/A | Hemi | R53H | M | Mother + | + + |
| HTR2C | C/T | Hemi | A171V | M | Mother + | + + |
| SLITRK2 | G/A | Hetero | V89M | F | Mother + + | + |
| IDH3G | C/T | Hemi | R101C | M | Mother + + | + |
| PCDH19 | T/C | Hetero | V91A | F | Mother | + + |
| P2RY4 | C/T | Hetero | S130N | F | Mother | + + |
| SLC9A6 | G/A | Hetero | R536Q | F | Mother | + + |
Abbreviations: ASD, autism spectrum disorder; F, female; Hemi, hemizygous; Hetero, heterozygous; M, male; n.a., non available; SCZ, schizophrenia.
More details on pedigree (cosegregation analysis) or functional effect predictions are given in Supplementary Table 7.
We identified one potentially damaging variant c.733T >C (M: p.F245L) in PPP1R3F, which encodes a regulatory subunit of protein phosphatase 1, in one boy with an Asperger Syndrome diagnosis and seizures (Supplementary Figure 3a). This variant was transmitted from a mother who suffered from learning disabilities and seizures. Protein phosphatase 1 is a serine/threonine phosphatase that consists of a catalytic subunit that can interact with over 50 regulatory or scaffolding proteins and is involved in many cellular processes. In neurons, protein phosphatase 1 influences neurite outgrowth, synapse formation, and is critical for synaptic plasticity and learning and memory.62
We also identified a missense variant, c.385G >A (M: p.G129R), in the 26S proteasome non-ATPase regulatory subunit (PSMD10) gene in one boy with classical autism. This variant was inherited from his heterozygous mother (Supplementary Figure 3b). The Gly129 residue is perfectly conserved among species and is located in the ANK3 (ankyrin) domain of PSMD10 (which is also known as gankyrin). Numerous synaptic proteins, including scaffolding proteins or neurotransmitter receptors, are regulated by degradation mechanisms through the ubiquitin–proteasome pathway, a phenomenon that is particularly important for synaptic plasticity.63 Moreover, several proteins of this pathway, such as the ubiquitin ligase UBE3A, have been linked to autism-like disorders and MR.64,65 There is no direct evidence that PSMD10 is involved in the degradation of synaptic proteins; however, its synaptic localization is suggestive of this function. A non-conservative substitution of an amino acid in one ANK domain of this protein could alter its binding to protein partners and compromise its function.
The c.1845C >A (F+M: p.F555L) missense variant was identified in the gene encoding the MCF2/DBL protein, which is a member of the large family of GDP–GTP exchange factors for small GTPases of the Rho family (Supplementary Figure 3c). The F555L variant was found in one heterozygous SCZaff girl, her youngest SCZaff brother, and her unaffected mother. This variant affects a highly conserved amino acid and is predicted to be damaging to protein function, according to different programs (Supplementary Table 4). The Phe555 residue is located in the DBL motif, which is critical for the regulation of Rho-GTPases. Other rare variants or polymorphisms were observed in this gene, but did not involve highly conserved residues. Dbl knockout mice reportedly display shorter dendrites in distinct populations of Dbl-null cortical pyramidal neurons, which suggests a function for MCF2/Dbl in dendrite elongation.66 Reduced dendrite spines were observed in the prefrontal cortex of schizophrenic individuals, suggesting a possible involvement of the RhoGTPase pathway.18,19
Other promising variants were identified in genes encoding proteins involved in cell adhesion (SLITRK2, PCDH11X, PCDH19, TM4SF2/TSPAN7), serotonin pathway (MAOA, HTR2C), mRNA or protein transport (HNRPH2, GPRASP2), calcium homeostasis (ATP2B3), or RhoGTPase regulation (OPHN1). Variants in several neurotransmitter receptors (GLRA2, GABRE, HTR2C) were also identified. Finally, we identified variants in genes encoding proteins involved in various cell functions less specific of the synapse (WNK3, CXCR3, IDH3G, HCFC1 or SLC9A6) as well as proteins highly expressed in brain with unknown or unclear function (PDZD4, FRMPD4, BEX2).
Screening of MR genes
Sequence analysis of the TM4SF2/TSPAN7 gene led to the identification of the P172H missense variant, which was associated earlier with MR in two families.67,68 In the first study, the cosegregation of this variant was not perfect, with one non-affected male carrying the variant. This variant was also identified by another team who screened patients with MR and was absent in 77 ASD patients68 and 420 control chromosomes tested. We found the P172H variant in one ASD boy with MR; this variant was inherited from his mother (Supplementary Figure 3d). We sequenced an additional 285 ASD patients for the fragment encoding this amino acid and found the same variant in another boy diagnosed with pervasive developmental disorder not otherwise specified without MR (Supplementary Table 5). This variant was neither present in 143 SCZ nor in 190 ethnically matched control individuals. Taken together, ours and earlier reports revealed that the P172H variant has not been found in 697 control X chromosomes. Our results add evidence to the function of the P172H substitution in TM4SF2/TSPAN7 not only in MR but also in ASD. TM4SF2/TSPAN7 encodes a cell-surface glycoprotein from the tetraspanin family, a class of proteins that have a function in the regulation of cell development, activation, growth, and motility. The Pro172 residue is located in the extracellular loop, a region that may be involved in interaction with other adhesion proteins.
The screening of the RTT MECP2 gene revealed a de novo missense variant (F: p.R309W) in a girl with ASD. She presents with an important psychomotor retardation and had developed severe epileptic seizures from her adolescence. However, she does not satisfy the criteria for classical nor atypical RTT. This variant, already described in one boy with NS-MR, 69 affects a conserved amino acid located in a domain responsible for the interaction with the cyclin-dependent kinase-like CDKL5 and is predicted to be damaging.
We also identified two rare missense variants in OPHN1: c.1381A >G (M: p.M461V) in one COS patient and c.2114A >G (M: p.H705R) in one ASD patient (Supplementary Figure 3e). These two variants were predicted to affect protein function, according to different programs, and they target amino acids that are well conserved among species. In contrast, OPHN1 polymorphisms and variants found in the control cohort (Supplementary Table 5) were all predicted to have a benign effect on protein function. OPHN1 encodes a RhoGTPase-activating protein that interacts with RhoA, Rac, cdc42, actin filaments, and with the HOMER protein. It is expressed in both neuronal and glial cells and participates in the regulation of dendrite elongation.70 Nonsense, splice, frameshift, and missense mutations have been described for this gene in patients with MR and cerebellar hypoplasia.71 Interestingly, cerebellar abnormalities have also been described in COS and ASD patients.72,73
Among the other genes that are known to be involved in NS-MR, ASD, or ASD-related diseases, a subset showed no variation in their coding sequences, including AP1S2, ARHGEF9, DCX, GDI1, and SLC6A8. No NS variants were observed in CASK and SYP, two genes reported recently by Tarpey et al.50 to cause NS-MR. In addition, no novel NS variants were identified during the screening of ARHGEF6, ATP6AP2, ARX, JARID1C, and FMR1. This was also the case for RPL10, in which variants were described recently in ASD patients.74 Several novel NS variants were identified in CDKL5, DLG3, NLGN3, PAK3, AFF2, and RPS6KA3; however, the analysis of the pedigrees and of the predicted effect of these variants on protein function suggests that none of them are likely to be involved in ASD.
Discussion
Our study reports the systematic resequencing of the coding regions of 111 X-linked candidate genes in a cohort of 285 ASD and SCZ patients, aimed at the identification of genes potentially involved in these disorders. Using various methods, we prioritized these genes from a larger group of 1125 X-linked genes and ~5000 potentially synaptic genes. For the X chromosome, only 28 genes were shared between the gene list from SynDB and that derived from proteomic studies. This non-concordance may be explained by three major reasons: (1) some synaptic proteins, such as membrane receptors, are difficult to purify and could be missed by proteomic studies, (2) the presence of a higher number of false positives in SynDB because of inclusion of genes based on structural predictions, and (3) a fraction of the proteins found through proteomic studies may also represent false positives, as they could be due to contamination of the synaptic fraction during the preparation. These results convinced us to use the complementarity of these two types of sources. Altogether, the gene lists extracted from SynDB and from proteomic studies comprised 65% of the genes identified through our manual search of PubMed. The combination of these three resources resulted in a comprehensive list of X chromosome potentially synaptic genes. This list also provides good candidate genes for the screening of other psychiatric and neurological disorders, such as bipolar disorder or Tourette’s syndrome.
Here, we identified several hundred variants, the majority of which had not been described in any public database. Tarpey et al.,50 who recently published a list of X-chromosome variants found in patients with MR, identified 134 of the variants reported here, which corresponded mainly to the polymorphism category. However, their cohort is not a pertinent control for our study, as MR and ASD are genetically related and are often clinically comorbid. We screened three times less nucleotides (75 954 657 bp vs ~200 000 000 bp) than Tarpey et al. and found around three times less variants in coding regions (533 vs 1858). However, we identified significantly less truncating variants (6 vs 40). This result is not surprising, as their cohort included only families with X-linked MR. Consistent with their results, we observed that truncating variants in two genes (P2RY4 and HS6ST2) seemed to be well tolerated in males and to not lead to any obvious phenotype. Contrary to the variant in P2RY4, which removes only the C-terminal end of the protein, the variant in HS6ST2 removes almost all the protein. Possible functional redundancy with other heparan sulfate 6-O-sulfotransferase subtypes, such as HS6ST1 and HS6ST3, may explain the fact that HS6ST2 truncation is tolerated in male individuals.
Despite the probable function of rare variants with a recent origin in ASD and SCZ, we identified and confirmed only two de novo damaging variants, which were found in IL1RAPL1 and MECP2 in two ASD girls. However, because of the particularity of X-linked transmission, we are aware that X-linked genes are not ideal to test the de novo hypothesis. One nonsense and several missense variants were identified in the MAOB gene in SCZ patients. MAOB catalyzes the oxidative deamination of xenobiotic and biogenic amines, such as the neurotransmitter dopamine or the neuromodulator phenylethylamine.75 Phenylethylamine is involved in the modulation of mood and, as it is structurally close to amphetamine, it may cause, when expressed at high levels, a similar type of psychosis.76 Moreover, low MAOB activity and elevated phenylethylamine in urine were described earlier in SCZ patients.77 Several studies failed to identify any association between polymorphisms in MAOB and SCZ; however, one positive association was recently found in the Spanish population.78 Nonetheless, one would not expect positive associations if disease-predisposing mutations are different rare mutations. Although no evident neurological phenotype was found in two boys who carry a deletion of a fragment of MAOB and of the Norrie disease gene,79 MAOB knockout mice present elevated phenylethylamine in urine and an increased reactivity to stress.80 Regarding the possible implication of MAOB in SCZ, the identification of one nonsense mutation in this gene in one SCZaff patient is potentially relevant, even if the cosegregation was not perfect. Further studies are needed to determine the effect of the three missense variants found in the four additional families on MAOB function.
The NS rare variants identified in this study were predicted to be more damaging than the NS polymorphisms, which suggests that they may include disease-causing mutations responsible for ASD or SCZ. The genetic follow-up on these variants is challenging, as most of our patients are sporadic cases. Moreover, because of the genetic complexity of ASD and SCZ, we do not expect to find a perfect segregation of variants with the disease, which hampers the interpretation of the results. Several genes in which we identified potentially damaging variants have a function in structural modulation of the synapse. The involvement of the IL1RAPL1, OPHN1, and MCF2 proteins in neurite outgrowth is well documented66,70,81 and evidence suggests that SLITRK2 and TM4SF2/TSPAN7 may also be implicated in this process.56,67 We also highlighted variants in genes involved in serotonin function (HTR2C) and degradation (MAOB) in SCZ patients.
Our gene scoring system aimed at selecting the best candidate genes seems to have performed well. MAOB was selected with a score equal to 6, which was the maximum score. Several of the genes in which we found potentially relevant variants in ASD (for example IL1RAPL1, OPHN1, TM4SF2/TSPAN7, or MECP2) were selected because of their involvement in NS-MR. Interestingly, most of our ASD individuals who carried mutations in these genes did not have MR. These results support a genetic link between MR and ASD, suggesting that the same genes and mutations may predispose to these two diseases, as was shown earlier for NLGN4.12 Therefore, the screening of genes involved in MR in ASD patients is warranted, even if the patients do not have MR. The study of factors (environmental or genetic) that modulate these phenotypes will be critical to understand the molecular pathways that underlie MR, ASD, or both.
The identification of two damaging variants in OPHN1 in one male with ASD and in another with COS suggests a genetic link between these two diseases. The COS patient was also diagnosed with pervasive developmental disorder as one-third of the individuals with COS. The hypothesis of the existence of common genes between ASD and SCZ is emerging82 and has been shown by the discovery that copy number variants of the NRXN1 synaptic gene are associated with these two diseases.83 Similarly, the SCZ-associated DISC-1 gene was also found to be involved in ASD.84 Interestingly, in concordance with our results, a recent study showed that several copy number variants are common between MR, ASD, and SCZ, supporting the existence of shared biologic pathways in these neurodevelopmental disorders.85,86
In conclusion, our results indicate that large-scale direct resequencing of synaptic candidate genes constitutes a promising approach to dissect the genetic heterogeneity of SCZ and ASD and to explore the hypothesis that a number of distinct individually rare penetrant variants are involved in the pathogenesis of these two syndromes. Indeed, the identification of an excess of potentially damaging rare variants in ASD and SCZ patients validated the usefulness of this approach. However, with the exception of mutations in IL1RAPL1, MECP2 and maybe TM4SF2/TSPAN7 in ASD, it is difficult to make a definitive claim that the damaging variants identified here are disease causing. The truncating mutation in MAOB, as well as other missense variants in different genes are promising, but further work involving functional testing will be needed to confirm the implication of these rare variants in SCZ and ASD. If the hypothesis of rare variants with classical Mendelian inheritance does not seem to entirely explain the complexity of the genetic factors involved in ASD, we succeeded, at least, in the identification of causative mutations in some ASD patients. However, the story seems to be more complicated for SCZ. The excess of patients accumulating several NS rare variants in the SCZ cohort in comparison with the ASD cohort and the lack of identification of a clear segregating damaging mutation in SCZ in our study suggest that SCZ may involve more an interaction between rare variants with moderate effects in different synaptic genes rather than addition of Mendelian inheritances. Interestingly, a recent genome- wide association study on SCZ reported that common polygenic variants could also contribute to the risk of SCZ.7 These common variants may work in concert with rare variants to manifest SCZ. However, we are limited in this conclusion because we analyzed only variants on the X chromosome. A similar approach targeting autosomal genes is needed to examine whether autosomal rare variants provide a similar picture to that of our X-chromosome study. More generally, for these diseases, the ‘rare variant with penetrant effect’ vs the ‘common variant with low effect’ hypotheses should not be viewed as exclusive hypotheses, but more as a continuum including also variants with rare frequency and having moderate effect. That is why direct resequencing of candidate genes, as well as copy number variants or genome-wide association study analyses, could be viewed as complementary approaches to dissect the genetic susceptibilities to SCZ and ASD.
Acknowledgments
We thank the families involved in our study and the recruitment coordinators (Anne Desjarlais, Caroline Poulin, and Sabrina Diab). We thank Annie Levert, Judith St-Onge, and Isabelle Bachand for performing DNA extraction and paternity and identity testing. We are thankful for the efforts of the members of the McGill University and Genome Quebec Innovation Centre Sequencing (Pierre Lepage, Sébastien Brunet, and Hao Fan Yam) and Bioinformatic (Louis Létourneau and Louis Dumond Joseph) groups. This work was supported by a grant from Genome Canada and Génome Québec and was cofunded by Université de Montréal, for the ‘Synapse-to-disease’ (S2D) project.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Molecular Psychiatry website (http://www.nature.com/mp)
References
- 1.Cardno AG, Gottesman II. Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am J Med Genet. 2000;97:12–17. [PubMed] [Google Scholar]
- 2.Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, et al. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan PF. The genetics of schizophrenia. PLoS Med. 2005;2:e212. doi: 10.1371/journal.pmed.0020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. image 45. [DOI] [PubMed] [Google Scholar]
- 5.Yang MS, Gill M. A review of gene linkage, association and expression studies in autism and an assessment of convergent evidence. Int J Dev Neurosci. 2007;25:69–85. doi: 10.1016/j.ijdevneu.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 6.Ma D, Salyakina D, Jaworski JM, Konidari I, Whitehead PL, Andersen AN, et al. A genome-wide association study of autism reveals a common novel risk locus at 5p14. 1. Ann Hum Genet. 2009;73(Part 3):263–273. doi: 10.1111/j.1469-1809.2009.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gauthier J, Spiegelman D, Piton A, Lafreniere RG, Laurent S, St-Onge J, et al. Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:421–424. doi: 10.1002/ajmg.b.30822. [DOI] [PubMed] [Google Scholar]
- 9.Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74:552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 14.Garber K. Neuroscience. Autism’s cause may reside in abnormalities at the synapse. Science. 2007;317:190–191. doi: 10.1126/science.317.5835.190. [DOI] [PubMed] [Google Scholar]
- 15.Harrison PJ. The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology (Berl) 2004;174:151–162. doi: 10.1007/s00213-003-1761-y. [DOI] [PubMed] [Google Scholar]
- 16.Hashimoto R, Tankou S, Takeda M, Sawa A. Postsynaptic density: a key convergent site for schizophrenia susceptibility factors and possible target for drug development. Drugs Today (Barc) 2007;43:645–654. doi: 10.1358/dot.2007.43.9.1088821. [DOI] [PubMed] [Google Scholar]
- 17.Eastwood SL. The synaptic pathology of schizophrenia: is aberrant neurodevelopment and plasticity to blame? Int Rev Neurobiol. 2004;59:47–72. doi: 10.1016/S0074-7742(04)59003-7. [DOI] [PubMed] [Google Scholar]
- 18.Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, et al. Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry. 1998;65:446–453. doi: 10.1136/jnnp.65.4.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11:557–566. doi: 10.1038/sj.mp.4001792. [DOI] [PubMed] [Google Scholar]
- 20.Marco EJ, Skuse DH. Autism-lessons from the X chromosome. Soc Cogn Affect Neurosci. 2006;1:183–193. doi: 10.1093/scan/nsl028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Loat CS, Haworth CM, Plomin R, Craig IW. A model incorporating potential skewed X-inactivation in MZ girls suggests that X-linked QTLs exist for several social behaviours including autism spectrum disorder. Ann Hum Genet. 2008;72(Part 6):742–751. doi: 10.1111/j.1469-1809.2008.00470.x. [DOI] [PubMed] [Google Scholar]
- 22.Laumonnier F, Cuthbert PC, Grant SG. The role of neuronal complexes in human X-linked brain diseases. Am J Hum Genet. 2007;80:205–220. doi: 10.1086/511441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skuse DH. X-linked genes and mental functioning. Hum Mol Genet. 2005;4(Spec No 1):R27–R32. doi: 10.1093/hmg/ddi112. [DOI] [PubMed] [Google Scholar]
- 24.Skuse D. Genetic influences on the neural basis of social cognition. Philos Trans R Soc Lond B Biol Sci. 2006;361:2129–2141. doi: 10.1098/rstb.2006.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 26.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 27.Yonan AL, Alarcon M, Cheng R, Magnusson PK, Spence SJ, Palmer AA, et al. A genomewide screen of 345 families for autism-susceptibility loci. Am J Hum Genet. 2003;73:886–897. doi: 10.1086/378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petit E, Herault J, Raynaud M, Cherpi C, Perrot A, Barthelemy C, et al. X chromosome and infantile autism. Biol Psychiatry. 1996;40:457–464. doi: 10.1016/0006-3223(96)85270-X. [DOI] [PubMed] [Google Scholar]
- 29.Vincent JB, Melmer G, Bolton PF, Hodgkinson S, Holmes D, Curtis D, et al. Genetic linkage analysis of the X chromosome in autism, with emphasis on the fragile X region. Psychiatr Genet. 2005;15:83–90. doi: 10.1097/00041444-200506000-00004. [DOI] [PubMed] [Google Scholar]
- 30.Liu J, Nyholt DR, Magnussen P, Parano E, Pavone P, Geschwind D, et al. A genomewide screen for autism susceptibility loci. Am J Hum Genet. 2001;69:327–340. doi: 10.1086/321980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Auranen M, Vanhala R, Varilo T, Ayers K, Kempas E, Ylisaukko-Oja T, et al. A genomewide screen for autism-spectrum disorders: evidence for a major susceptibility locus on chromosome 3q25-27. Am J Hum Genet. 2002;71:777–790. doi: 10.1086/342720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gauthier J, Joober R, Dube MP, St-Onge J, Bonnel A, Gariepy D, et al. Autism spectrum disorders associated with X chromosome markers in French-Canadian males. Mol Psychiatry. 2006;11:206–213. doi: 10.1038/sj.mp.4001756. [DOI] [PubMed] [Google Scholar]
- 33.Dann J, DeLisi LE, Devoto M, Laval S, Nancarrow DJ, Shields G, et al. A linkage study of schizophrenia to markers within Xp11 near the MAOB gene. Psychiatry Res. 1997;70:131–143. doi: 10.1016/s0165-1781(97)03138-7. [DOI] [PubMed] [Google Scholar]
- 34.DeLisi LE, Wellman N, Stewart J, Smith AB, Churchman M, Crow TJ. Linkage disequilibrium study of markers within the pericentromeric region of the X chromosome. Am J Med Genet. 1999;88:588–589. [PubMed] [Google Scholar]
- 35.Hovatta I, Varilo T, Suvisaari J, Terwilliger JD, Ollikainen V, Arajarvi R, et al. A genomewide screen for schizophrenia genes in an isolated Finnish subpopulation, suggesting multiple susceptibility loci. Am J Hum Genet. 1999;65:1114–1124. doi: 10.1086/302567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei J, Hemmings GP. A further study of a possible locus for schizophrenia on the X chromosome. Biochem Biophys Res Commun. 2006;344:1241–1245. doi: 10.1016/j.bbrc.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 37.Roser P, Kawohl W. Turner syndrome and schizophrenia: a further hint for the role of the X-chromosome in the pathogenesis of schizophrenic disorders. World J Biol Psychiatry. 2008 Sep 11;:1–4. doi: 10.3109/15622970701599060. [DOI] [PubMed] [Google Scholar]
- 38.DeLisi LE, Friedrich U, Wahlstrom J, Boccio-Smith A, Forsman A, Eklund K, et al. Schizophrenia and sex chromosome anomalies. Schizophr Bull. 1994;20:495–505. doi: 10.1093/schbul/20.3.495. [DOI] [PubMed] [Google Scholar]
- 39.El Abd S, Patton MA, Turk J, Hoey H, Howlin P. Social, communicational, and behavioral deficits associated with ring X turner syndrome. Am J Med Genet. 1999;88:510–516. doi: 10.1002/(sici)1096-8628(19991015)88:5<510::aid-ajmg14>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 40.Fombonne E. Epidemiological trends in rates of autism. Mol Psychiatry. 2002;7 (Suppl 2):S4–S6. doi: 10.1038/sj.mp.4001162. [DOI] [PubMed] [Google Scholar]
- 41.Goldstein JM, Seidman LJ, Goodman JM, Koren D, Lee H, Weintraub S, et al. Are there sex differences in neuropsychological functions among patients with schizophrenia? Am J Psychiatry. 1998;155:1358–1364. doi: 10.1176/ajp.155.10.1358. [DOI] [PubMed] [Google Scholar]
- 42.Trinidad JC, Specht CG, Thalhammer A, Schoepfer R, Burlingame AL. Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Mol Cell Proteomics. 2006;5:914–922. doi: 10.1074/mcp.T500041-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 44.Collins MO, Husi H, Yu L, Brandon JM, Anderson CN, Blackstock WP, et al. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J Neurochem. 2006 Apr;97(Suppl 1):16–23. doi: 10.1111/j.1471-4159.2005.03507.x. [DOI] [PubMed] [Google Scholar]
- 45.Zhang W, Zhang Y, Zheng H, Zhang C, Xiong W, Olyarchuk JG, et al. SynDB: a Synapse protein DataBase based on synapse ontology. Nucleic Acids Res. 2007;35(Database issue):D737–D741. doi: 10.1093/nar/gkl876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gauthier J, Bonnel A, St-Onge J, Karemera L, Laurent S, Mottron L, et al. NLGN3/NLGN4 gene mutations are not responsible for autism in the Quebec population. Am J Med Genet B Neuropsychiatr Genet. 2005;132B:74–75. doi: 10.1002/ajmg.b.30066. [DOI] [PubMed] [Google Scholar]
- 47.Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, et al. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 2003;31:334–341. doi: 10.1093/nar/gkg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Piton A, Michaud JL, Peng H, Aradhya S, Gauthier J, Mottron L, et al. Mutations in the calcium-related gene IL1RAPL1 are associated with autism. Hum Mol Genet. 2008;17:3965–3974. doi: 10.1093/hmg/ddn300. [DOI] [PubMed] [Google Scholar]
- 52.Binda C, Newton-Vinson P, Hubalek F, Edmondson DE, Mattevi A. Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nat Struct Biol. 2002;9:22–26. doi: 10.1038/nsb732. [DOI] [PubMed] [Google Scholar]
- 53.Ng PC, Levy S, Huang J, Stockwell TB, Walenz BP, Li K, et al. Genetic variation in an individual human exome. PLoS Genet. 2008;4:e1000160. doi: 10.1371/journal.pgen.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet. 2002;3:285–298. doi: 10.1038/nrg775. [DOI] [PubMed] [Google Scholar]
- 55.Collin RW, de Heer AM, Oostrik J, Pauw RJ, Plantinga RF, Huygen PL, et al. Mid-frequency DFNA8/12 hearing loss caused by a synonymous TECTA mutation that affects an exonic splice enhancer. Eur J Hum Genet. 2008;16:1430–1436. doi: 10.1038/ejhg.2008.110. [DOI] [PubMed] [Google Scholar]
- 56.Aruga J, Mikoshiba K. Identification and characterization of Slitrk, a novel neuronal transmembrane protein family controlling neurite outgrowth. Mol Cell Neurosci. 2003;24:117–129. doi: 10.1016/s1044-7431(03)00129-5. [DOI] [PubMed] [Google Scholar]
- 57.Hemara-Wahanui A, Berjukow S, Hope CI, Dearden PK, Wu SB, Wilson-Wheeler J, et al. A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1. 4 channel activation. Proc Natl Acad Sci USA. 2005;102:7553–7558. doi: 10.1073/pnas.0501907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31. doi: 10.1016/j.cell.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 59.Splawski I, Yoo DS, Stotz SC, Cherry A, Clapham DE, Keating MT. CACNA1H mutations in autism spectrum disorders. J Biol Chem. 2006;281:22085–22091. doi: 10.1074/jbc.M603316200. [DOI] [PubMed] [Google Scholar]
- 60.Strom SP, Stone JL, Ten Bosch JR, Merriman B, Cantor RM, Geschwind DH, et al. High-density SNP association study of the 17q21 chromosomal region linked to autism identifies CACNA1G as a novel candidate gene. Mol Psychiatry. 2009 May 19; doi: 10.1038/mp.2009.41. e-pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Weaving LS, Christodoulou J, Williamson SL, Friend KL, McKenzie OL, Archer H, et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am J Hum Genet. 2004;75:1079–1093. doi: 10.1086/426462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Munton RP, Vizi S, Mansuy IM. The role of protein phosphatase-1 in the modulation of synaptic and structural plasticity. FEBS Lett. 2004;567:121–128. doi: 10.1016/j.febslet.2004.03.121. [DOI] [PubMed] [Google Scholar]
- 63.Bingol B, Schuman EM. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol. 2005;15:536–541. doi: 10.1016/j.conb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 64.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 65.Nurmi EL, Bradford Y, Chen Y, Hall J, Arnone B, Gardiner MB, et al. Linkage disequilibrium at the Angelman syndrome gene UBE3A in autism families. Genomics. 2001;77:105–113. doi: 10.1006/geno.2001.6617. [DOI] [PubMed] [Google Scholar]
- 66.Hirsch E, Pozzato M, Vercelli A, Barberis L, Azzolino O, Russo C, et al. Defective dendrite elongation but normal fertility in mice lacking the Rho-like GTPase activator Dbl. Mol Cell Biol. 2002;22:3140–3148. doi: 10.1128/MCB.22.9.3140-3148.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zemni R, Bienvenu T, Vinet MC, Sefiani A, Carrie A, Billuart P, et al. A new gene involved in X-linked mental retardation identified by analysis of an X;2 balanced translocation. Nat Genet. 2000;24:167–170. doi: 10.1038/72829. [DOI] [PubMed] [Google Scholar]
- 68.Maranduba CM, Sa Moreira E, Muller Orabona G, Pavanello RC, Vianna-Morgante AM, Passos-Bueno MR. Does the P172H mutation at the TM4SF2 gene cause X-linked mental retardation? Am J Med Genet A. 2004;124A:413–415. doi: 10.1002/ajmg.a.20401. [DOI] [PubMed] [Google Scholar]
- 69.Campos M, Jr, Abdalla CB, Santos-Reboucas CB, dos Santos AV, Pestana CP, Domingues ML, et al. Low significance of MECP2 mutations as a cause of mental retardation in Brazilian males. Brain Dev. 2007;29:293–297. doi: 10.1016/j.braindev.2006.09.012. [DOI] [PubMed] [Google Scholar]
- 70.Govek EE, Newey SE, Akerman CJ, Cross JR, Van der Veken L, Van Aelst L. The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci. 2004;7:364–372. doi: 10.1038/nn1210. [DOI] [PubMed] [Google Scholar]
- 71.Zanni G, Saillour Y, Nagara M, Billuart P, Castelnau L, Moraine C, et al. Oligophrenin 1 mutations frequently cause X-linked mental retardation with cerebellar hypoplasia. Neurology. 2005;65:1364–1369. doi: 10.1212/01.wnl.0000182813.94713.ee. [DOI] [PubMed] [Google Scholar]
- 72.Mittleman G, Goldowitz D, Heck DH, Blaha CD. Cerebellar modulation of frontal cortex dopamine efflux in mice: relevance to autism and schizophrenia. Synapse. 2008;62:544–550. doi: 10.1002/syn.20525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaufmann WE, Cooper KL, Mostofsky SH, Capone GT, Kates WR, Newschaffer CJ, et al. Specificity of cerebellar vermian abnormalities in autism: a quantitative magnetic resonance imaging study. J Child Neurol. 2003;18:463–470. doi: 10.1177/08830738030180070501. [DOI] [PubMed] [Google Scholar]
- 74.Klauck SM, Felder B, Kolb-Kokocinski A, Schuster C, Chiocchetti A, Schupp I, et al. Mutations in the ribosomal protein gene RPL10 suggest a novel modulating disease mechanism for autism. Mol Psychiatry. 2006;11:1073–1084. doi: 10.1038/sj.mp.4001883. [DOI] [PubMed] [Google Scholar]
- 75.Shih JC, Chen K, Ridd MJ. Role of MAO A and B in neurotransmitter metabolism and behavior. Pol J Pharmacol. 1999;51:25–29. [PubMed] [Google Scholar]
- 76.O’Reilly RL, Davis BA. Phenylethylamine and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 1994;18:63–75. doi: 10.1016/0278-5846(94)90024-8. [DOI] [PubMed] [Google Scholar]
- 77.Wyatt RJ, Potkin SG, Bridge TP, Phelps BH, Wise CD. Monoamine oxidase in schizophrenia: an overview. Schizophr Bull. 1980;6:199–207. doi: 10.1093/schbul/6.2.199. [DOI] [PubMed] [Google Scholar]
- 78.Gasso P, Bernardo M, Mas S, Crescenti A, Garcia C, Parellada E, et al. Association of A/G polymorphism in intron 13 of the monoamine oxidase B gene with schizophrenia in a Spanish population. Neuropsychobiology. 2008;58:65–70. doi: 10.1159/000159774. [DOI] [PubMed] [Google Scholar]
- 79.Lenders JW, Eisenhofer G, Abeling NG, Berger W, Murphy DL, Konings CH, et al. Specific genetic deficiencies of the A and B isoenzymes of monoamine oxidase are characterized by distinct neurochemical and clinical phenotypes. J Clin Invest. 1996;97:1010–1019. doi: 10.1172/JCI118492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Grimsby J, Toth M, Chen K, Kumazawa T, Klaidman L, Adams JD, et al. Increased stress response and beta-phenylethylamine in MAOB-deficient mice. Nat Genet. 1997;17:206–210. doi: 10.1038/ng1097-206. [DOI] [PubMed] [Google Scholar]
- 81.Gambino F, Pavlowsky A, Begle A, Dupont JL, Bahi N, Courjaret R, et al. IL1-receptor accessory protein-like 1 (IL1RAPL1), a protein involved in cognitive functions, regulates N-type Ca2+-channel and neurite elongation. Proc Natl Acad Sci USA. 2007;104:9063–9068. doi: 10.1073/pnas.0701133104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burbach JP, van der Zwaag B. Contact in the genetics of autism and schizophrenia. Trends Neurosci. 2009;32:69–72. doi: 10.1016/j.tins.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 83.Kirov G, Gumus D, Chen W, Norton N, Georgieva L, Sari M, et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet. 2008;17:458–465. doi: 10.1093/hmg/ddm323. [DOI] [PubMed] [Google Scholar]
- 84.Kilpinen H, Ylisaukko-Oja T, Hennah W, Palo OM, Varilo T, Vanhala R, et al. Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry. 2008;13:187–196. doi: 10.1038/sj.mp.4002031. [DOI] [PubMed] [Google Scholar]
- 85.Guilmatre A, Dubourg C, Mosca AL, Legallic S, Goldenberg A, Drouin-Garraud V, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sebat J, Levy DL, McCarthy SE. Rare structural variants in schizophrenia: one disorder, multiple mutations; one mutation, multiple disorders. Trends Genet. 2009;25:528–535. doi: 10.1016/j.tig.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]