

Abstract
A series of HIV integrase (HIV-1 IN) inhibitors were synthesized to evaluate the role of the metal-binding group (MBG) in this class of metalloenzyme inhibitors. A total of 21 different raltegravir-chelator derivative (RCD) compounds were prepared that differed only in the nature of the MBG. These IN strand-transfer inhibitors (INSTIs) were evaluated in vitro in cell-free enzyme activity assays, and the in vitro results were further validated in cell culture experiments. All of the active compounds showed selective inhibition of the strand-transfer reaction over 3′-processing, suggesting a common mode of action with raltegravir. The results of the in vitro activity suggest that the nature of the MBG donor atoms, the overall MBG structure, and the specific arrangement of the MBG donor atom triad are essential for obtaining maximal HIV-1 IN inhibition. At least two compounds (RCD-4, RCD-5) containing a hydroxypyrone MBG were found to display superior strand-transfer inhibition when compared to an abbreviated analogue of raltegravir (RCD-1). By isolating and examining the role of the MBG in a series of INSTIs, we have identified a scaffold (hydroxypyrones) that may provide access to a unique class of HIV-1 IN inhibitors, and may help overcome rising raltegravir resistance.
Human immunodeficiency virus (HIV) is a retrovirus that causes acquired immunodeficiency syndrome (AIDS) (1, 2). There is presently no cure for AIDS, although potent antiretroviral drugs have improved the management of the disease (3). HIV integrase (HIV-1 IN) is one of three essential enzymes for HIV replication (along with HIV reverse transcriptase and protease). HIV-1 IN performs two functions related to inserting the viral genome into the host DNA. In its first function, known as 3′-processing, HIV-1 IN generates reactive CpA 3′-hydroxyl ends (cytosine-adenosine 3′ recessed ends) by specifically cleaving a dinucleotide from the viral cDNA. The second function of HIV-1 IN, known as strand transfer, occurs upon translocation to the nucleus, where HIV-1 IN uses the hydroxyl ends to integrate the viral DNA into the host genome (4, 5).
The active site of HIV-1 IN is characterized by a dinuclear magnesium center, coordinated by three carboxylate ligands of a DDE amino acid motif (5–7). The metal-dependent activity of HIV-1 IN has proven to be exceptionally important in the development of inhibitors against this metalloenzyme. The US Federal and Drug Administration (FDA) approved the first HIV-1 IN inhibitor, raltegravir, in 2007. Raltegravir utilizes a 5-hydroxy-3-methylpyrimidin-4(3H)-one (HMPO) chelating group in combination with an amide carbonyl oxygen atom to bind the dinuclear Mg2+ metal site in HIV-1 IN. The HMPO metal-binding group (MBG) was discovered by high-throughput screening and was found to possess suitable pharmacokinetics (8–10). The HMPO chelator and the amide carbonyl oxygen atom provide three, essentially coplanar, oxygen atoms to bind and bridge the Mg2+ ions of HIV-1 IN (Fig. 1). Despite the success of raltegravir, resistant HIV strains have emerged with mutations in key active site residues (9, 11, 12). Importantly, the raltegravir-resistant mutants characterized do not alter the metal-binding motif of the enzyme (13). Indeed, substitution of any of the three metal-binding residues abolishes HIV-1 IN activity, suggesting that metal-binding is essential for HIV-1 IN (6).
Fig. 1.

Proposed mode of metal binding for the FDA-approved HIV-1 IN inhibitor raltegravir (in raised box, Left). Structure and strand-transfer IC50 values of advanced INSTIs, including raltegravir and its abbreviated analogue RCD-1. Proposed metal-binding atoms are shown in bold and red for each inhibitor. Raltegravir and RCD-1 are identical, except that RCD-1 lacks an oxadiazole substituent (highlighted in magenta).
This was further corroborated by a recent crystal structure of the prototype foamy virus (PFV) integrase bound to its cognate DNA (intasome) (14). Structures have also been determined in complex with several inhibitors, including raltegravir. The intasome structures show that these IN strand-transfer inhibitors (INSTIs) have two common features: (i) a heteroatom triad to bind the dinuclear metal center, and (ii) a halogenated benzene ring that serves to displace the 3′ adenine of the bound viral DNA (12). The structure of raltegravir bound to the PFV intasome reveals that both active site Mg2+ ions are coordinated by the inhibitor as shown schematically in Fig. 1. Other advanced HIV-1 IN inhibitors, such as elvitegravir, dolutegravir, MK2048, and MK0536 (11, 12, 14), were also shown to use similar heteroatom triads for binding the dinuclear Mg2+ center (Fig. 1). However, the metal-binding atoms in these compounds are not the same, using different combinations of carbonyl and phenolic oxygen atoms, or even endocyclic pyridyl-nitrogen atoms (12). In addition, the inhibitors do not have identical bond angles between the donor atoms. The difference in these MBGs indicates that diverse metal-binding atoms in various relative orientations can accommodate the HIV-1 IN active site (9, 12, 14); however, no systemic study that examines these features within a single chemical scaffold has been reported (15, 16).
In an attempt to better understand the key metal-ligand interactions involved in HIV-1 IN inhibition, a series of raltegravir-chelator derivatives (RCDs) have been synthesized and evaluated. These compounds were designed to specifically examine the inhibitory effect of each MBG by keeping the remainder of the inhibitor structure unaltered. This systematic study was achieved by appending various MBGs to the p-fluorobenzyl backbone via a carboxyamide linkage, the latter of which provides the first of the three donor atoms. These INSTIs were screened against HIV-1 IN to determine which MBGs produced inhibitors with comparable or better activity than an abbreviated raltegravir derivative (RCD-1). Several RCDs had comparable strand-transfer inhibitory activity to RCD-1 and two derivatives, containing a hydroxypyrone MBG, were more effective at inhibiting strand transfer. Computational docking studies of RCDs in the active site of PFV IN have been performed to elucidate key features that contribute to effective metal chelation to the HIV-1 IN active site. The findings presented here methodically investigate and rigorously analyze the importance of different MBGs in HIV-1 IN.
Results
Design and Synthesis of Inhibitors.
To isolate and examine the effect of the MBG in HIV-1 IN inhibitors, a series of RCDs were designed and synthesized. These INSTIs are identical to a core portion of raltegravir and vary only in the nature of the MBG. The RCDs that were prepared are shown in Table 1 (SI Appendix for synthetic details); all of the compounds contain the MBG attached to an amide-linked p-fluorobenzyl group. This shared structure makes all of these compounds analogues of a substructure of raltegravir, where only the oxadiazole substituent has been removed (Fig. 1). The omission of the oxadiazole substituent from the RCD compounds serves a dual purpose: (i) it greatly simplifies the synthesis of the desired compounds, and (ii) differences in potency can be more directly attributed to changes in the MBG, rather than substituent effects. The MBGs employed in the RCD compounds cover a wide range of chelators including hydroxypyridinones (RCD-2, -3, -7), hydroxypyrones (RCD-4, -5, -6), catechols (RCD-8, -9), p-dicarboxycatechols (RCD-10, -11), hydroxyquinolines (RCD-12, -13, 14), and several others. A total of 21 RCD compounds were prepared, each with a unique MBG, and covering approximately ten chemically distinct chelating motifs. To provide a suitable benchmark for comparison for these RCD compounds, the reported raltegravir derivative RCD-1 was prepared (Fig. 1). As with the other RCD compounds, RCD-1 is an abbreviated raltegravir derivative that lacks the oxadiazole substituent, but still shows good activity against HIV-1 IN (IC50 value ∼60 nM against the strand-transfer reaction of HIV-1 IN) (17). The reduced activity of RCD-1 when compared to raltegravir is attributed to the loss of interactions between the omitted oxadiazole substituent and the surrounding active site residues, specifically Tyr143 of HIV-1 IN or Tyr212 in PFV (13, 14, 18).
Table 1.
Assay results for RCD compounds against the 3P and ST reactions of HIV-1 IN, as well as inhibition of viral replication. The chelate ring sizes formed upon binding the active site metal ions is also indicated
| Compound | Structure (MBG)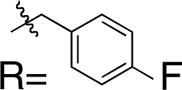
|
Chelate ring size(MgA, MgB) | 3′-processing IC50 (μM) | Strand-transfer IC50 (μM) | Antiviral activity IC50 (μM) |
| RCD-1 | 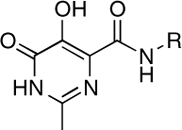 |
5-, 6- | > 100 | 1.0 ± 0.3 | 1.5 |
| RCD-2 | 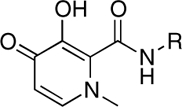 |
5-, 6- | > 100 | > 100 | ND |
| RCD-3 | 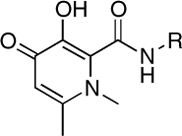 |
5-, 6- | > 100 | > 100 | ND |
| RCD-4 | 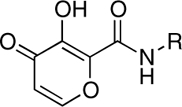 |
5-, 6- | > 100 | 0.96 ± 0.3 | ND |
| RCD-4S | 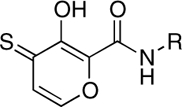 |
5-, 6- | > 100 | 11.5 ± 0.9 | ND |
| RCD-4S2 | 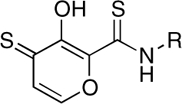 |
5-, 6- | 64 ± 6 | 7.3 ± 0.6 | ND |
| RCD-5 | 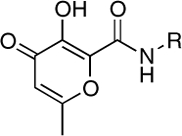 |
5-, 6- | 59.5 ± 1.4 | 0.55 ± 0.1 | 1.0 |
| RCD-6 | 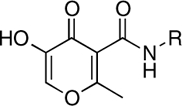 |
5-, 6- | > 100 | 56.0 ± 7.0 | ND |
| RCD-7 | 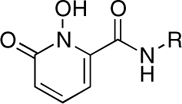 |
5-, 6- | > 100 | 19.7 ± 1.6 | ND |
| RCD-8 | 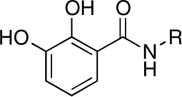 |
5-, 6- | > 100 | 39.4 ± 4.0 | ND |
| RCD-9 | 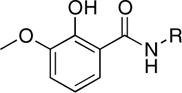 |
5-, 6- | > 100 | > 100 | ND |
| RCD-10 | 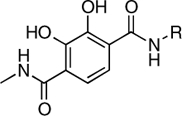 |
5-, 6- | > 200 | 1.5 ± 0.2 | 4.0 |
| RCD-11 | 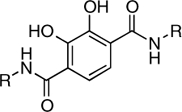 |
5-, 6- | > 300 | 1.7 ± 0.2 | ND |
| RCD-12 | 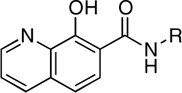 |
5-, 6- | > 100 | 14.5 ± 2.2 | 2.3* |
| RCD-13 | 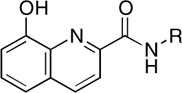 |
5-, 5- | > 100 | > 100 | > 100 |
| RCD-14 | 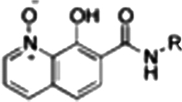 |
6-, 6- | 40.5 ± 2.0 | 3.8 ± 0.3 | 0.5* |
| RCD-15 | 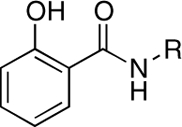 |
6-, NA | > 100 | > 100 | ND |
| RCD-16 | 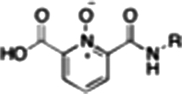 |
6-, 6- | 21.4 ± 3.0 | 9.2 ± 1.3 | ND |
| RCD-17 | 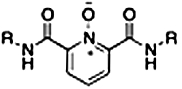 |
6-, 6- | > 300 | > 100 | > 100 |
| RCD-18 | 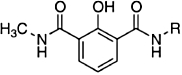 |
6-, 6- | > 300 | > 300 | > 100 |
| RCD-19 | 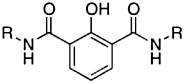 |
6-, 6- | > 300 | > 300 | ND |
NA, not applicable; ND, not determined
*Compound showed some cellular toxicity at 10 μM.
HIV-1 IN Activity Screen.
As described above, HIV-1 IN has two functions: 3′-processing (3P) and strand transfer (ST). Most HIV-1 IN inhibitors, including raltegravir, are targeted against the ST reaction of HIV-1 IN and hence are referred to as INSTIs. All 21 RCD compounds were screened for inhibitory activity against the 3P and ST reactions using published protocols (13, 19). Compounds were initially screened for activity at approximately 100 μM, and those compounds that showed ST inhibition were then further examined to assess inhibition of viral replication (SI Appendix). The results of the assays with the RCD compounds are listed in Table 1.
As expected, RCD-1 shows good activity against the ST reaction, with an IC50 value of approximately 1 μM. This value is higher than the reported value of 60 nM (17); however, under our assay conditions raltegravir also produces a higher IC50 value of approximately 50 nM (20). The difference in IC50 values results from differences in the assay. Some assays use preassembled HIV-1 IN on immobilized oligonucleotides (17), whereas our assay (SI Appendix) uses 32P-end-labeled oligonucleotides in solution and gel-based separation of the reaction products. RCD-1 also shows selectivity for the ST versus 3P reaction, consistent with previous findings (9). Indeed, examination of the in vitro assay results immediately reveals that all of the RCD compounds, with a few exceptions (RCD-14, -16), are highly selective for ST versus 3P, suggesting a common mode of action.
Of the compounds prepared, four RCD inhibitors showed activity comparable or better than RCD-1. RCD-4, -5, -10, and -11 gave ST inhibition IC50 values of 0.96, 0.55, 1.5, and 1.7 μM, respectively. Importantly, these compounds fall into only two distinct classes of MBG chelators: RCD-4 and RCD-5 contain hydroxypyrone chelators, whereas RCD-10 and RCD-11 contain p-dicarboxy catechol chelators. This result clearly highlights the role of the MBG for inhibitor efficacy, whereby only two of at least ten distinct metal-binding groups resulted in good ST inhibition. Other compounds showed modest activity, including RCD-4S, RCD-4S2, RCD-7, -12, -14, and -16 with IC50 values in the 4–20 μM range. Two compounds, RCD-6 and RCD-8, showed weaker activity with IC50 values > 40 μM. All of the remaining RCD compounds showed poor inhibition, with little or no activity at concentrations as high as 100 μM.
In addition to cell-free in vitro assays, eight RCD compounds were examined for inhibition of viral replication (Table 1) (21). The eight RCDs examined contained different MBGs and included both active (RCD-1, -5, -10, -12, -14) and inactive (RCD-13, -17, -18) compounds. Inhibition of viral replication by the selected RCDs was determined in P4R5 cells (21). Compounds with good ST activity were found to be the most effective at inhibiting P4R5 infection. RCD-1, -5, -10, -12, and -14, all of which have ST IC50 values below 15 μM, were shown to have IC50 values of < 4.0 μM (Table 1). RCD-13, -17, and -18, which perform poorly in vitro (ST IC50 > 100 μM), showed weak antiviral activity (IC50 > 100 μM). Toxicity assays showed that most of the compounds tested in the viral replication assay showed little affect on P4R5 cells at a concentration of 10 μM (22). Only RCD-12 and RCD-14 showed some toxicity at this concentration; therefore, follow up studies with these compounds or their derivatives will require greater consideration of their possible cytotoxicity. Overall, the cell-based infectivity assay was thus consistent with the in vitro ST activity, supporting the mechanism of action for the RCD compounds in HIV-1 IN inhibition.
Computational Docking Studies.
To elucidate the binding mode of the various RCD compounds, ligand-receptor docking studies were conducted. As previously described, the structure of the PFV IN in complex with raltegravir shows that the O, O, O donor triad binds to the active site Mg2+ ions, with the central oxygen atom acting as a bridge between the two metal centers. The coordinates for PFV IN [Protein Data Bank (PDB) ID code 3OYA] were used for computational docking of RCD compounds (23, 24). As a test of our docking procedure (SI Appendix), raltegravir was docked into the PFV IN structure, resulting in a pose consistent with that seen in the crystal structure complex (rmsd 0.19 Å). RCD-1 was docked into PFV IN using the same procedure and gave a binding pose identical to that found for raltegravir (rmsd 0.25 Å, Fig. 2).
Fig. 2.

Comparison of the computational docking of RCD-1 (magenta) in the PFV IN versus the reported crystal structure of raltegravir (green) bound in PFV IN (PDB ID code 3OYA). The rmsd between the inhibitors is 0.25 Å. MgA and MgB are shown as labeled, orange spheres.
The O, O, O donor atom triad of raltegravir and RCD-1 bind to the Mg2+ ions forming 5- and 6-membered chelate rings (Fig. 3). The hydroxyl oxygen and the amide-linked carbonyl oxygen together form the 6-membered ring whereas the same hydroxyl oxygen and the exocyclic carbonyl oxygen atom of the MBG make up the 5-membered ring. In both compounds, the deprotonated, anionic hydroxyl oxygen atom binds in a μ-bridging fashion between the two metal ions in the active site. The p-fluorobenzyl substituent of raltegravir and RCD-1 both rest in an identical pocket. It has been proposed that this pocket is formed by an induced fit mechanism upon displacement of an adenine residue (A17) from the nucleic acid substrate. The displacement of this nucleotide and the resulting pocket allow the p-fluorobenzyl group to interact with bases from the invariant CA dinucleotide, as well as residue Pro214 in the PFV intasome (equivalent to P145 in HIV-1 IN). The placement of this group is pivotal to the impairment of HIV-1 IN activity as it causes the viral DNA to be displaced from the active site (14). This docking exercise with raltegravir and RCD-1 validated our assumption that the only difference in binding between these compounds is the omitted oxadiazole moiety, and that the omission of this group has little or no effect on the binding of the MBG or p-fluorobenzyl components of the INSTI.
Fig. 3.

MBG numbering system and modes of metal coordination for raltegravir and select RCD compounds. Atoms in bold red are part of the heteroatom donor triad, which coordinate to the active site Mg2+ ions. Chelate rings with MgA and MgB are highlighted in blue and green, respectively.
Satisfied with the validity of the docking procedure and parameters, the remaining RCD compounds were docked in a similar manner (SI Appendix, Figs. S1–S19). Docking experiments showed that the other RCD compounds formed one of several chelate ring patterns (Fig. 3): (i) a 5-membered chelate ring with MgA and a 6-membered chelate ring with MgB (RCD-1 to RCD-12); (ii) two 5-membered chelate rings (RCD-13); (iii) two 6-membered chelate rings (RCD-14, -16, -17, -18, -19); or (iv) only a single 6-membered chelate ring with MgA (RCD-15). In addition, for all RCD compounds, the p-fluorobenzyl substituent was bound in the same pocket as described for raltegravir and RCD-1 (vide supra). The findings and interpretation of these docking studies are discussed in detail in the section below.
Discussion
Critical Features of MBGs.
Inspection of the in vitro ST inhibition data, in conjunction with the computational docking experiments, reveals several interesting trends about the MBG requirements for this series of HIV-1 IN inhibitors. One feature that may be important is the size of the chelate rings formed upon binding of the inhibitor (Fig. 3). Most of the active RCD compounds form a 5-membered chelate ring with MgA and a 6-membered chelate ring with MgB (RCD-1, -4, -5, -6, -7, -8, -10, -11, -12). Compounds that form two 5-membered chelate rings (RCD-13), two 6-membered chelate rings (RCD-17, -18, -19), or only a single chelate ring (RCD-15) were generally inactive. The preferred 5-,6-membered chelate ring binding arrangement found for most of the active RCD compounds is also formed by raltegravir (14) and several other second-generation INSTIs (12), including L-870,810, GS9160, and MK0536 (Fig. 1). However, there are exceptions to the observed trends. For example, RCD-14 and RCD-16 both form two 6-membered chelate rings upon binding (SI Appendix, Figs. S14 and S16) and still exhibit moderate inhibition. These compounds both possess highly Lewis acidic (vide infra) N-oxide donors and form dianionic (2-) chelators upon metal binding, which should result in a stronger electrostatic attraction between the inhibitors and active site Mg2+ ions. These features may explain the enhanced activity of RCD-14 and RCD-16 despite what may be a suboptimal coordination arrangement for this chemical scaffold.
Although the 5-,6-membered chelate ring appears to be favored by the RCD compounds and several other INSTIs, there are a number of examples in the literature indicating that other chelate ring motifs produce effective inhibitors. For example, dolutegravir reverses the size of the chelate rings, forming a 6-membered chelate ring with MgA and a 5-membered chelate ring with MgB (11). However, the chelate ring motifs of other INSTIs differ more substantially. Structures of the second-generation inhibitors MK2048 and PICA (Fig. 1) bound to the PFV intrasome show that these compounds form two 6-membered and two 4-membered chelate rings, respectively (12). Elvitegravir utilizes yet another motif, forming a 6-,4-membered chelate ring arrangement (14). Therefore, although the 5-,6-membered chelate ring arrangement appears to be most common among INSTIs, the numerous exceptions highlighted here clearly indicate that other productive binding modes are possible. Because of the intricate interplay between metal coordination and the positioning of the halogenated benzene group (12), it is likely that the metal chelate motif must be optimized in the context of different chemical scaffolds. Indeed, the RCD compounds also revealed an important trend concerning the relative positioning of the MBG to the p-fluorobenzyl backbone group.
A second observation from the RCD inhibition data shows the importance of the relative orientation of the amide-linked p-fluorobenzyl group on the MBG. Comparison of RCD-5 to RCD-6 clearly shows how a change in the position of this substituent has a dramatic effect on activity. Both RCD-5 and RCD-6 contain the same hydroxypyrone MBG and can provide O, O, O donor atom triads to the active site metal ions (Fig. 3). However, RCD-6 activity in vitro is found to be 100-fold less potent than RCD-5. Computational docking of RCD-5 and RCD-6 show that the molecules generally bind in a similar orientation, with little deviation (rmsd 0.30 Å) in the relative position of the p-fluorobenzyl group or in the scaffold of the MBG in the active site (SI Appendix, Figs. S5 and S6). However, the change in the point of attachment does affect the ordering of the oxygen atoms in the donor atom triad. The point of attachment of the p-fluorobenzyl group is the 2-position of the hydroxypyrone MBG ring in RCD-5, and the 5-position of the ring in RCD-6. As best illustrated in Fig. 3, RCD-5 bridges the two active site metal-ions through the 3-hydroxyl oxygen atom. In contrast, for RCD-6 the bridging donor atom is the 4-carbonyl oxygen atom. This subtle change in the donor atom triad arrangement contributes to the notable loss in activity between RCD-5 and RCD-6. The anionic hydroxyl group is a stronger Lewis base donor than the neutral carbonyl and will serve as a stronger bridging donor atom between the Mg2+ ions. This argument is supported by the activity of RCD-4, which also contains a hydroxypyrone MBG with a p-fluorobenzyl group on the 2-position of the ring (it lacks a 6-methyl group found in RCD-5 and RCD-6, vide infra). Like RCD-5, RCD-4 presents the anionic hydroxyl atom as the bridging donor atom (SI Appendix, Fig. S4) and similarly shows good ST inhibition (Table 1). Interestingly, essentially all of the lead INSTIs under investigation to date follow this motif, utilizing an anionic hydroxyl atom as the bridging atom (PICA is one notable exception) (11, 12, 14).
RCD-5 and RCD-6 both contain methyl groups at the 6-position of the MBG rings (Fig. 3). In addition to the change in the arrangement of the donor atom triads discussed above, the difference in the position of the amide-linked p-fluorobenzyl group results in these methyl groups occupying different locations in the protein active site (SI Appendix, Fig. S20). The orientation of the methyl group upon docking of RCD-5 in PFV IN does not result in any significant contacts with the protein. In contrast, the same methyl group, upon docking of RCD-6, results in a steric clash with Pro214 in the PFV IN active site (SI Appendix, Fig. S20). Pro214 is one of the few conserved residues in the IN active site loop that is directly involved in separating the viral DNA strands, and both raltegravir and elvitegravir make intimate van der Waals interactions with this residue (14). Therefore, the steric clash between Pro214 and the methyl group of RCD-6 also likely contributes to the loss of activity for this compound. The potential problems posed by the 6-methyl group in RCD-6 are further supported by the poor activity of hydroxypyridinones RCD-2 and RCD-3 (Table 1). The N-methyl group protruding from the MBGs in RCD-2 and RCD-3 is located in the same position as the 6-methyl group in RCD-6 (Fig. 3). Indeed, docking experiments confirm a steric clash with Pro214 (SI Appendix, Figs. S2 and S3), as observed for RCD-6. Importantly, unlike RCD-6, RCD-2, and RCD-3 contain the preferred bridging hydroxyl group found in RCD-4 and RCD-5, suggesting that the steric problems posed by the methyl substituent may be the more significant factor when considering the loss in activity of RCD-2, -3, and -6. The comparisons between RCD-2, -3, -4, -5, and -6 suggest that a combination of both the ordering of the donor triad as well as steric interactions can have a drastic affect on the potency of these inhibitors.
The dependence on the position of the amide p-fluorobenzyl substituent is also observed when comparing RCD-12 and RCD-13, both of which contain an 8-hydroxyquinoline MBG with identical O, O, N donor atom sets (Fig. 3). RCD-13, which contains the amide group at the 2-position, shows minimal (< 30%) inhibition at approximately 100 μM whereas RCD-12, which has the amide substituent attached at the 7-position, shows good activity with an IC50 value of approximately 14 μM. As with RCD-5 and RCD-6, RCD-12, and RCD-13 have the same molecular formula, overall composition, and MBG that provides an identical donor atom set (one hydroxyl oxygen atom, one amide oxygen atom, and one quinoline nitrogen atom). However, the position of the p-fluorobenzyl affects the overall arrangement of the donor atoms upon binding to the active site metal ions. As confirmed by docking studies (Fig. 4), the position of the p-fluorobenzyl amide substituent in RCD-12 versus RCD-13 results in a significant change in the arrangement of the donor atom triad for these two compounds. For RCD-13 the donor set will be arranged as O, N, O whereas for RCD-12 the arrangement will be O, O, N (Fig. 4), resulting in the donor atom arrangement for RCD-12 forming 6-membered and 5-membered chelate rings, with a bridging hydroxyl atom. The same arrangement is found in raltegravir and the other most active RCD compounds identified here. In contrast, when the p-fluorobenzyl amide group is attached to the 2-position of the scaffold as in RCD-13, the chelator is forced to adopt two 5-membered chelate rings, with the quinoline nitrogen atom serving as the bridging ligand. Such endocyclic nitrogen atoms do not readily engage in bridging modes of metal ion coordination (25). Furthermore, the quinoline nitrogen atom is positioned too far from the Mg2+ ions (> 3.7 Å) to form strong interactions. Despite the similar arrangement of the donor triad in RCD-12, this compound is still less potent than RCD-4 and RCD-5, which is likely due to the preference of the hard Mg2+ ions for the harder oxygen atom donor set found in the hydroxypyrone compounds. Hard Lewis base donors like anionic oxygen atoms are classically characterized by their small size, high charge state, and weak polarizability (26). Comparing these compounds clearly shows that having a heteroatom triad is not sufficient for good inhibition, but rather the correct or optimal atom arrangement of the triads is also essential along with the optimal matching of the Lewis acid character of the donor atoms.
Fig. 4.

Computational docking results for RCD-12 (Top) and RCD-13 (Bottom) in the PFV IN active site (PDB ID code 3OYA). Mg2+ ions are shown as orange spheres and bonding contacts between the inhibitor and metal ions are shown as dashed red lines.
The comparison between RCD-4/-5 and RCD-12 highlights a third trend related to the nature of the MBG donor atoms. The preference for certain donor atoms was explored by converting the O, O, O donor RCD-4 to two different sulfur analogues. As stated above, the catalytic Mg2+ ions are hard Lewis acids and hence should bind more tightly to harder Lewis base donor atoms. The introduction of softer, more polarizable Lewis base sulfur atoms to the donor triad were expected to lower the efficacy of the compounds. Isostructural hydroxypyrothione analogues, termed RCD-4S and RCD-4S2 (Table 1) provide O, O, S and S, O, S donor atom sets, respectively. Both RCD-4S and RCD-4S2 show a significant loss in activity when compared to RCD-4. The weaker ST inhibition by RCD-4S and RCD-4S2 is likely due to a hard-soft mismatch between the hard Lewis acid Mg2+ ions and the soft Lewis base sulfur donor atoms. This conclusion is consistent with the improved performance of sulfur compounds like RCD-4S2 against metalloenzymes that are dependent on the softer Lewis acid Zn2+ ion, such as the anthrax lethal factor (LF). In the case of anthrax LF, RCD-4S2 is a better inhibitor than RCD-4 (27, 28), precisely the opposite of what is observed for HIV-1 IN. Hence, the selection of the donor atoms with the appropriate Lewis acid character is important for obtaining optimal inhibition of HIV-1 IN.
MBG Scaffolds.
In this study, we have identified at least two, MBG types that appear to be promising scaffolds for the development of HIV-1 IN inhibitors. The first MBG is the hydroxypyrone group found in RCD-4 and RCD-5, both of which show good in vitro activity and RCD-5 also displayed good cell-based activity. The hydroxypyrone MBGs found in these compounds derive from the FDA-approved food additive maltol (3-hydroxy-2-methyl-4H-pyran-4-one) for which there has been extensive chemistry developed that should facilitate the preparation of even more potent inhibitors based on this scaffold (29–31). The second class of compounds that warrants additional investigation are those based on the p-dicarboxy catechol MBGs (RCD-10 and RCD-11). Four compounds were examined that are nominally based on a catechol MBG: RCD-8, RCD-9, RCD-10, and RCD-11. RCD-8 contains a catecholamide MBG and shows modest ST inhibition with an IC50 value of 39 μM. RCD-9 shows a complete loss of activity due to methylation of one of the phenol groups resulting in a reduced donor ability, whereas addition of a second carboxyamide group in RCD-10 and RCD-11 produces a significant improvement (> 20-fold) in activity with IC50 values < 2 μM. One possible explanation for the improved activity of RCD-10 and RCD-11 over RCD-8 would be additional interactions between the protein active site and the added carboxyamide substituents; however, RCD-10 and RCD-11 have very different substituents (methyl versus p-fluorobenzyl, Table 1), but essentially identical ST inhibition IC50 values (1.5 and 1.7 μM, respectively). With this observation in mind, we attribute the origin of the improved activity of RCD-10 and RCD-11 relative to RCD-8 to the reduced pKa of the MBG. To obtain optimal binding to the Mg2+ ions, the MBGs should be deprotonated upon metal binding. Catechol is a strong, hard Lewis donor, but it is also very basic (pKa1 = 9.2, pKa2 ∼ 13) (32) making deprotonation under physiological conditions more difficult. Addition of electron withdrawing groups, such as the carboxyamide groups in RCD-10 and RCD-11, are known to significantly reduce the pKa of the catechol ligand (32). Therefore, the addition of a second such carboxyamide group will result in an inhibitor that more readily achieves deprotontion of both phenolic groups in the catechol ligand, resulting in a dianionic (2-) ligand and a strong electrostatic attraction between the MBG and the active site metal ions.
Conclusions
To the best of our knowledge, this direct assessment of different MBGs on the activity of HIV-1 IN inhibitors is unique. Whereas numerous inhibitors have been prepared and studied (4, 9, 33), few or none have systematically dissected and evaluated the contribution and structure-activity relationship around the MBGs in these compounds (15). By preparing and evaluating the RCD compounds reported here, we have identified a number of important features of the MBG for use in INSTIs, including: (i) the heteroatom triad should consist of hard Lewis base donor atoms to match the hard Lewis acid character of the active site Mg2+ ions; (ii) the triad should possess a geometry that results in the formation of optimal chelate ring sizes [for RCDs adjacent 5- (MgA) and 6- (MgB) membered rings]; and (iii) the hardest, anionic donor atom should be located in the middle of the triad to provide a sufficiently electron-donating ligand in the μ-bridging position between the metal ions (16). These experiments also lead to the identification of at least two unique and distinct MBGs, hydroxypyrones (RCD-4 and RCD-5) and p-dicarboxy catechols (RCD-10 and RCD-11) that may prove to be promising scaffolds for next-generation HIV-1 IN inhibitors. Overall, these studies provide direct evidence that subtle variations in the MBG can substantially affect the activity of an HIV-1 IN inhibitor, and suggests that rational approaches to strengthening metal-ligand interactions can produce potent inhibitors to help mitigate the need for other active site interactions and hence overcome rising resistance against raltegravir.
Materials and Methods
All RCD compounds were prepared using standard synthetic methods, similar to those previously described (27). Computational docking was preformed using the Glide software package (Glide v5.5, Schrodinger, Inc.). Enzyme and cell-based assays were performed as previously described (13, 19, 21). Complete synthetic and experimental details are provided in the SI Appendix.
Supplementary Material
Acknowledgments.
This work was supported by National Institutes of Health Grant R01GM098435 (to S.M.C.), a developmental grant from the University of California at San Diego Center for AIDS Research (CFAR) (S.M.C.), the intramural program of the National Cancer Research, Center for Cancer Research, and an Intramural AIDS Targeted Program (K.M. and Y.P.). The authors thank the University of California at San Diego CFAR Translational Virology Core that is supported by National Institutes of Health Grants AI69432, AI043638, MH62512, MH083552, AI077304, AI36214, AI047745, AI74621, and AI080353 and the James B. Pendleton Charitable Trust.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. T.J.M. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1112389109/-/DCSupplemental.
References
- 1.Barre-Sinoussi F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune-deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.Schupbach J, et al. Serological analysis of a subgroup of human T-lymphotropic retroviruses (HTLV-III) associated with AIDS. Science. 1984;224:503–505. doi: 10.1126/science.6200937. [DOI] [PubMed] [Google Scholar]
- 3.Mehellou Y, De Clercq E. Twenty-six years of anti-HIV drug discovery: Where do we stand and where do we go? J Med Chem. 2010;53:521–538. doi: 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- 4.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov. 2005;4:236–248. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 5.Li X, Krishnan L, Cherepanov P, Engelman A. Structural biology of retroviral DNA integration. Virology. 2011;411:194–205. doi: 10.1016/j.virol.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiu TK, Davies DR. Structure and function of HIV-1 integrase. Curr Top Med Chem. 2004;4:965–977. doi: 10.2174/1568026043388547. [DOI] [PubMed] [Google Scholar]
- 7.Perryman AL, et al. A dynamic model of HIV integrase inhibition and drug resistance. J Mol Biol. 2010;397:600–615. doi: 10.1016/j.jmb.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwamoto M, et al. Safety, tolerability, and pharmacokinetics of raltegravir after single and multiple doses in healthy subjects. Clin Pharmacol Ther. 2008;83:293–299. doi: 10.1038/sj.clpt.6100281. [DOI] [PubMed] [Google Scholar]
- 9.Marchand C, Maddali K, Métifiot M, Pommier Y. HIV-1 IN inhibitors: 2010 update and perspectives. Curr Top Med Chem. 2009;9:1016–1037. doi: 10.2174/156802609789630910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Summa V, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51:5843–5855. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 11.Hare S, et al. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572) Mol Pharmacol. 2011;80:565–572. doi: 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hare S, et al. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Natl Acad Sci USA. 2010;107:20057–20062. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Metifiot M, et al. Biochemical and pharmacological analyses of HIV-1 integrase flexible loop mutants resistant to raltegravir. Biochemistry. 2010;49:3715–3722. doi: 10.1021/bi100130f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464:232–237. doi: 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bacchi A, et al. Investigating the role of metal chelation in HIV-1 integrase strand transfer inhibitors. J Med Chem. 2011;54:8407–8420. doi: 10.1021/jm200851g. [DOI] [PubMed] [Google Scholar]
- 16.Kirschberg T, Parrish J. Metal chelators as antiviral agents. Curr Opin Drug Discov Dev. 2007;10:460–472. [PubMed] [Google Scholar]
- 17.Pace P, et al. Dihydroxypyrimidine-4-carboxamides as novel potent and selective HIV integrase inhibitors. J Med Chem. 2007;50:2225–2239. doi: 10.1021/jm070027u. [DOI] [PubMed] [Google Scholar]
- 18.Metifiot M, Marchand C, Maddali K, Pommier Y. Resistance to integrase inhibitors. Viruses. 2010;2:1347–1366. doi: 10.3390/v2071347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchand C, Neamati N, Pommier Y. In vitro human immunodeficiency virus type 1 integrase assays. Methods Enzymol. 2001;340:624–633. doi: 10.1016/s0076-6879(01)40446-0. [DOI] [PubMed] [Google Scholar]
- 20.Marinello J, et al. Comparison of raltegravir and elvitegravir on HIV-1 integrase catalytic reactions and on a series of drug-resistant integrase mutants. Biochemistry. 2008;47:9345–9354. doi: 10.1021/bi800791q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Day JR, et al. A computer-based, image-analysis method to quantify HIV-1 infection in a single-cycle infectious center assay. J Virol Methods. 2006;137:125–133. doi: 10.1016/j.jviromet.2006.06.019. [DOI] [PubMed] [Google Scholar]
- 22.Hostetler KY, Aldern KA, Wan WB, Ciesla SL, Beadle JR. Alkoxyalkyl esters of (S)-9-[3-hydroxy-2-(phosphonomethoxy)propy-l]adenine are potent inhibitors of the replication of wild-type and drug-resistant human immunodeficiency virus type 1 in vitro. Antimicrob Agents Chemother. 2006;50:2857–2859. doi: 10.1128/AAC.01223-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hare S, et al. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Natl Acad Sci USA. 2010;107:20057–20062. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krishnan L, et al. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc Natl Acad Sci USA. 2010;107:15910–15915. doi: 10.1073/pnas.1002346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaes C, Katz A, Hosseini MW. Bipyridine: The most widely used ligand. A review of molecules comprising at least two 2,2′-bipyridine units. Chem Rev. 2000;100:3553–3590. doi: 10.1021/cr990376z. [DOI] [PubMed] [Google Scholar]
- 26.Ho T-L. Hard soft acids bases (HSAB) principle and organic chemistry. Chem Rev. 1975;75:1–20. [Google Scholar]
- 27.Agrawal A, et al. Thioamide hydroxypyrothiones supersede amide hydroxypyrothiones in potency against anthrax lethal factor. J Med Chem. 2009;52:1063–1074. doi: 10.1021/jm8013212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis JA, Mongan J, McCammon JA, Cohen SM. Evaluation and binding-mode prediction of thiopyrone-based inhibitors of Anthrax lethal factor. ChemMedChem. 2006;1:694–697. doi: 10.1002/cmdc.200600102. [DOI] [PubMed] [Google Scholar]
- 29.Finnegan MM, Rettig SJ, Orvig SJ. A neutral water-soluble aluminum complex of neurological interest. J Am Chem Soc. 1986;108:5033–5035. [Google Scholar]
- 30.Schugar H, et al. Combating Alzheimer’s disease with multifunctional molecules designed for metal passivation. Angew Chem Int Ed. 2007;46:1716–1718. doi: 10.1002/anie.200603866. [DOI] [PubMed] [Google Scholar]
- 31.Puerta DT, Mongan J, Tran BL, McCammon JA, Cohen SM. Potent, selective pyrone-based inhibitors of stromelysin-1. J Am Chem Soc. 2005;127:14148–14149. doi: 10.1021/ja054558o. [DOI] [PubMed] [Google Scholar]
- 32.Gorden AEV, Xu JD, Raymond KN, Durbin P. Rational design of sequestering agents for plutonium and other actinides. Chem Rev. 2003;103:4207–4282. doi: 10.1021/cr990114x. [DOI] [PubMed] [Google Scholar]
- 33.Serrao E, Odde S, Ramkumar K, Neamati N. Raltegravir, elvitegravir, and metoogravir: The birth of “me-too” HIV-1 integrase inhibitors. Retrovirology. 2009;6:25–39. doi: 10.1186/1742-4690-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.