

Abstract
One of the outstanding questions concerning the early Earth is how ancient phototrophs made the evolutionary transition from anoxygenic to oxygenic photosynthesis, which resulted in a substantial increase in the amount of oxygen in the atmosphere. We have previously demonstrated that reaction centers from anoxygenic photosynthetic bacteria can be modified to bind a redox-active Mn cofactor, thus gaining a key functional feature of photosystem II, which contains the site for water oxidation in cyanobacteria, algae, and plants [Thielges M, et al. (2005) Biochemistry 44:7389–7394]. In this paper, the Mn-binding reaction centers are shown to have a light-driven enzymatic function; namely, the ability to convert superoxide into molecular oxygen. This activity has a relatively high efficiency with a kcat of approximately 1 s-1 that is significantly larger than typically observed for designed enzymes, and a Km of 35–40 μM that is comparable to the value of 50 μM for Mn-superoxide dismutase, which catalyzes a similar reaction. Unlike wild-type reaction centers, the highly oxidizing reaction centers are not stable in the light unless they have a bound Mn. The stability and enzymatic ability of this type of Mn-binding reaction centers would have provided primitive phototrophs with an environmental advantage before the evolution of organisms with a more complex Mn4Ca cluster needed to perform the multielectron reactions required to oxidize water.
In photosynthesis, the primary conversion of light energy into chemical energy is performed by the evolutionarily related pigment-protein complexes, reaction centers in purple and green bacteria and photosystems I and II in cyanobacteria, algae, and plants (1, 2). In the purple bacterium Rhodobacter sphaeroides, the reaction center is composed of two core protein subunits, identified as the L and M subunits, which each have five transmembrane helices and are related by an approximate twofold symmetry axis. These protein subunits surround the cofactors, which are arranged in two branches with the same twofold symmetry axis. In addition, reaction centers have a third subunit, identified as the H subunit, which has a single transmembrane helix and a large extramembrane domain. Light excitation of the primary electron donor P, a bacteriochlorophyll dimer, is followed by electron transfer steps predominately along one branch of cofactors to QA, the primary quinone, and then to QB, the secondary quinone, forming the charge-separated state P+·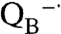 . In the cell, the oxidized bacteriochlorophyll dimer is reduced by a water-soluble cytochrome c2 allowing a second electron transfer to QB in a proton-coupled process. The electrons and protons associated with the oxidized cytochrome c2 and reduced quinone (quinol) are coupled to the cytochrome bc1 complex resulting in a net proton transfer across the cell membrane that is used to create energy-rich compounds. Photosystem II has a significant structural homology to this type of reaction center, with a core of two protein subunits surrounding two branches of cofactors and a similar electron transfer pathway from the primary electron donor to the quinones. However, the Mn4Ca cluster and a redox-active tyrosine serve as the secondary electron donors rather than a water-soluble electron carrier. The ability of the Mn4Ca cluster to collect four electron equivalents provides photosystem II with the unique ability to oxidize water.
. In the cell, the oxidized bacteriochlorophyll dimer is reduced by a water-soluble cytochrome c2 allowing a second electron transfer to QB in a proton-coupled process. The electrons and protons associated with the oxidized cytochrome c2 and reduced quinone (quinol) are coupled to the cytochrome bc1 complex resulting in a net proton transfer across the cell membrane that is used to create energy-rich compounds. Photosystem II has a significant structural homology to this type of reaction center, with a core of two protein subunits surrounding two branches of cofactors and a similar electron transfer pathway from the primary electron donor to the quinones. However, the Mn4Ca cluster and a redox-active tyrosine serve as the secondary electron donors rather than a water-soluble electron carrier. The ability of the Mn4Ca cluster to collect four electron equivalents provides photosystem II with the unique ability to oxidize water.
To experimentally address the molecular evolution of photosynthesis, in particular the development of the Mn4Ca cluster that performs water oxidation and played a critical role in the creation of an oxygen-rich atmosphere (3), reaction centers from Rb. sphaeroides are being modified to simulate the functional properties of photosystem II, specifically the binding of a Mn cofactor (4). Protein engineering of novel metal centers that can carry out specific reactions is a challenging area of rational structure-based design (5–8). One of the metals commonly found in proteins, Mn, is usually coordinated in an approximate octahedral geometry by aspartate, glutamate, and histidine side chains (7–10), as is true for the manganese cluster of photosystem II (2). Previously, reaction centers from a mutant identified as M2 were shown to exhibit tight binding of Mn with a dissociation constant of 1 μM (4). The three-dimensional structure of the M2 mutant was determined by X-ray diffraction revealing a mononuclear manganese ion bound at the designed Mn-binding site to carboxylate and histidine side chains (4). Also studied here is the M8 mutant that has the additional substitutions of Val M192 to Glu and Glu M173 to His, which are designed to provide a binuclear Mn-binding site as found in enzymes such as catalase. In both mutants, light excitation results initially in the charge-separated state Mn2+ P+·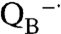 followed by the bound Mn2+ rapidly becoming oxidized as it transfers an electron to P+· in a first-order reaction. To drive this electron transfer, the Mn-binding reaction centers have additional mutations designed to enhance the oxidation capability, as wild-type reaction centers have too low an oxidation/reduction midpoint potential to oxidize Mn (11, 12).
followed by the bound Mn2+ rapidly becoming oxidized as it transfers an electron to P+· in a first-order reaction. To drive this electron transfer, the Mn-binding reaction centers have additional mutations designed to enhance the oxidation capability, as wild-type reaction centers have too low an oxidation/reduction midpoint potential to oxidize Mn (11, 12).
Redox-active Mn cofactors, such as found in the Mn-binding reaction centers, are found in several metalloenzymes, in particular Mn-superoxide dismutase. Superoxide is formed through a variety of processes, and its presence can result in cellular damage. To minimize such damage, virtually all aerobic organisms from bacteria to humans possess the enzyme superoxide dismutase that reacts with superoxide in the environment. In Mn-superoxide dismutase, three histidines, an aspartate, and a bound hydroxide molecule coordinate a mononuclear Mn cofactor that cycles between the Mn2+ and Mn3+ states as it converts superoxide into molecular oxygen and hydrogen peroxide (9, 10). As a test of whether the Mn cofactor in the modified reaction centers has the ability to react with superoxide, thus functionally mimicking the Mn cofactor of Mn-superoxide dismutase, the possible loss of superoxide was determined using an optical assay while the production of molecular oxygen was measured using an oxygen-sensing microelectrode. The implications of these results on the evolutionary development of photosystem II are discussed.
Results and Discussion
To access the possible reactivity of the Mn-binding reaction centers with superoxide, the concentration of superoxide in the presence of the reaction centers was measured using an optical assay. Superoxide and uric acid are produced by xanthine oxidase from oxygen and xanthine, and activity is identified by the reduction of the optical signal associated with a dye that is formed by reacting with superoxide. In the presence of light, Mn, and the Mn-binding reaction centers, essentially all of the generated superoxide was removed (Fig. 1). However, the amount of superoxide was unchanged, within error, when no Mn was present or when the samples were kept in the dark. The loss of superoxide was observed only for the Mn-binding reaction centers, and no activity was observed for wild-type reaction centers or a control mutant, which has the Mn-binding site but a wild-type potential that is too low to oxidize manganese. Thus, the Mn-binding reaction centers have the enzymatic ability of superoxide dismutase to react with superoxide with the exception that light is required for oxidation of the bound manganese.
Fig. 1.

Assay showing the ability of the Mn-binding reaction centers to react with superoxide. Spectra are shown for control mutant reaction centers, Mn-binding reaction centers, and wild-type reaction centers (solid spectra). Activity is observed only for the Mn-binding reaction centers in the presence of light and manganese. No change within experimental error is observed for wild type or the control mutant. The control mutant has the Mn-binding site but cannot oxidize Mn because it has an oxidation/reduction midpoint potential similar to wild type. In this assay, superoxide is generated by the reaction of xanthine with xanthine oxidase. The superoxide converts an indicator reagent (see Materials and Methods) into a formazan dye that has an absorption maximum at approximately 440 nm. The maximum absorption under each condition is shown (dashed spectra). If superoxide dismutase activity is present, the dye is not formed and a low absorption is observed (dotted spectra). Shown are the data for the M8 mutant; the data for the M2 mutant are provided in Fig. S1.
The reduction of Mn and the loss of superoxide should result in production of molecular oxygen by the Mn-binding reaction centers as occurs in the reaction mechanism of superoxide dismutase. Using an oxygen-sensing microelectrode, the concentration of molecular oxygen was measured with superoxide being generated using xanthine oxidase. For all measurements, the baseline level of oxygen showed a steady decrease due to the conversion of molecular oxygen into superoxide by xanthine oxidase (Fig. 2). In the light, the Mn-binding reaction centers with Mn showed a pronounced increase in oxygen compared to the measured level without Mn (Fig. 2). No measurable difference in oxygen concentration was observed when the Mn-binding reaction centers were in the dark. The oxygen increase was observed only for the Mn-binding reaction centers and not for wild-type reaction centers or the control mutant. Thus, the Mn-binding reaction centers are capable of the production of molecular oxygen in the light and with bound Mn.
Fig. 2.

Assay showing the production of oxygen from the Mn-binding reaction centers. (A) At -200 s (i.e., 200 s before illumination), xanthine oxidase is introduced into the solution, initiating the production of superoxide from molecular oxygen and resulting in a steady decrease in the oxygen concentration. From 0 to +300 s, the samples are illuminated. Only the Mn-binding reaction centers show a measurable increase in oxygen with Mn present compared to the absence of Mn. Both the Mn-binding reaction centers and reaction centers from the triple mutant are highly oxidizing and show a pronounced decrease in oxygen in the light, presumably due to photodamage. (B) The difference in the O2 concentration for samples with and without Mn. When Mn is bound to the Mn-binding reaction centers, P+· is rapidly reduced by the bound metal forming Mn3+ that subsequently can react with superoxide to form O2. For wild-type reaction centers, the triple mutant, the control mutant, solutions with no reaction centers, and solutions without superoxide, no measurable difference is observed. Shown are the data for the M8 mutant; the data for the M2 mutant are provided in Fig. S2.
The enzymatic mechanism was elucidated by systematically varying each experimental observable (namely, the light intensity and concentrations of reaction centers, manganese, and superoxide) while monitoring the difference in oxygen production with and without manganese. Evident in the oxygen production is a fast initial phase, with a time constant of 5 s, followed by a slow phase, with a time constant of 110 s (Fig. 2). The extent of oxygen production associated with the fast phase is directly proportional to the reaction center concentration as expected if the reaction centers are acting as an enzyme (Fig. 3A). The slow phase increases with protein concentration, although with a weaker dependence. For both phases, oxygen production requires light with the extent increasing as the light intensity increases (Fig. 3B). These results demonstrate that the oxygen production has two components that both arise from the light-induced activity of the Mn-binding reaction centers.
Fig. 3.

Oxygen production under different experimental conditions. (A) Increasing the reaction center concentration increases the extent of both the fast phase (measured at 30 s, closed circles) and slow phase (measured at 300 s, open circles) of the oxygen production when comparing the samples with and without manganese. A protein concentration of 10 μM is used for measurements shown in B–D. (B) Increasing the light intensity increases the extent of oxygen production. For most measurements, a subsaturating intensity of 1 mW/cm2 is used. (C) The addition of manganese proportionally increases the amount of oxygen production associated with the fast phase until a maximal amplitude of approximately 3∶1 for the ΔO2/protein ratio is reached. When electron transfer to QB is blocked by terbutryne, this ratio decreases to approximately 1∶1. (D) The extent of oxygen production associated with the fast phase was directly related to the concentration of superoxide according to a Lineweaver–Burke dependence yielding average values of 35 μM, 1.3 s-1, and 4 × 104 M-1 s-1 for Km, kcat, and Km/kcat, respectively, when averaged for three independent sets of measurements for the M8 mutant. For the M2 mutant, these values are 40 μM, 1.1 s-1, and 3 × 104 M-1 s-1 for Km, kcat, and Km/kcat, respectively.
As the amount of manganese increased, the oxygen production associated with the fast phase increased to a maximum O2/protein ratio of approximately 3∶1 corresponding to three rapid electron transfers that form the state 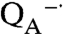
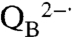 , which is the fully reduced state until QB exchanges with a quinone pool (Fig. 3C). In the absence of exogeneous quinone, additional charge-separation reactions are not possible and the fast phase ends. The average value of 2.7 for the O2/protein ratio at high Mn concentration is lower because of incomplete occupancy of the QB site; the addition of ubiquinone prior to the measurements results in a 15% increase of this ratio consistent with full occupancy. To verify this assignment of the fast phase, terbutryne was added to block electron transfer from
, which is the fully reduced state until QB exchanges with a quinone pool (Fig. 3C). In the absence of exogeneous quinone, additional charge-separation reactions are not possible and the fast phase ends. The average value of 2.7 for the O2/protein ratio at high Mn concentration is lower because of incomplete occupancy of the QB site; the addition of ubiquinone prior to the measurements results in a 15% increase of this ratio consistent with full occupancy. To verify this assignment of the fast phase, terbutryne was added to block electron transfer from 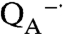 to QB. In this case, a maximal value of approximately 1 for the O2/protein ratio was observed that is consistent with the fast phase stopping after one electron transfer and formation of the
to QB. In this case, a maximal value of approximately 1 for the O2/protein ratio was observed that is consistent with the fast phase stopping after one electron transfer and formation of the  QB state. Thus, the initial phase corresponds to the rapid electron transfer within the reaction center until the inability to further reduce the quinones results in the slow phase, which presumably is limited by charge recombination that requires several minutes (4).
QB state. Thus, the initial phase corresponds to the rapid electron transfer within the reaction center until the inability to further reduce the quinones results in the slow phase, which presumably is limited by charge recombination that requires several minutes (4).
The dependence of the amount of the substrate, superoxide, on the oxygen release was obtained by adjusting the delay time between the injection of xanthine oxidase and the start of the illumination. Increasing this time delay allows the xanthine oxidase to convert more molecular oxygen into superoxide, as evidenced by the steady decrease of molecular oxygen with time in the dark (Fig. 2A). Increasing the amount of superoxide resulted in an increase in the amount of molecular oxygen produced associated with the fast phase (Fig. 3D). A fit according to the Lineweaver–Burke relationship yielded Km values of 40 and 35 μM and kcat values of 1.1 and 1.3 s-1, for the M2 and M8 mutants, respectively, with estimated errors of 5 μM and 0.5 s-1 for Km and kcat, respectively, based upon averages for three independent sets of measurements. The similarity of the parameters for the two mutants shows that the values are independent of the specific nature of the Mn ligands. Although the value of kcat is small compared to many enzymes [for example, Mn-superoxide dismutase has a value of 4,000 s-1 (13)], the value of 1 s-1 is larger than the 10-5 to 10-4 s-1 values reported for artificial enzymes (14–16) and the 0.01 to 0.22 s-1 rates obtained for an artificial oxo-protein with phenol oxidase activity (17). Comparable values of 0.02 to 1.4 s-1 are observed only for designed catalysts that have been modified using directed evolution (18). The value of 35–40 μM for Km for the Mn-binding mutants is comparable to the measured value of 50 μM for Mn-superoxide dismutase (13), indicating that the Mn-binding reaction centers can tightly bind superoxide. The kcat/Km ratio of 3–4 × 104 M-1 s-1 is much smaller than the 8 × 108 M-1 s-1 value observed for Mn-superoxide dismutase where the reaction has been proposed to be diffusion limited with superoxide binding being facilitated by favorable electrostatic interactions between superoxide and the entry channel to the Mn cofactor (13).
The superoxide activity measured for the Mn-binding reaction centers shares functional features with the enzymatic conversion process found in superoxide dismutase (Table 1). In Mn-superoxide dismutase, the oxidation state of the metal cycles between Mn3+ and Mn2+, converting superoxide into molecular oxygen and hydrogen peroxide (19, 20). For the Mn-binding reaction center, the Mn reduction reaction also produces molecular oxygen from superoxide. However, the Mn oxidation reaction differs between these two proteins. In Mn-superoxide dismutase, the metal-oxidation reaction converts superoxide into hydrogen peroxide and is coupled to the transfer of two protons. The Mn-binding reaction center does not perform this proton-coupled reaction because Mn is oxidized using the energy from the absorbed light. For any Mn-enzyme to catalyze both superoxide reduction and oxidation, the manganese must have a midpoint potential between -160 and +890 mV (19, 20). Because the Mn-binding reaction centers have an oxidation/reduction potential of 625 mV for the Mn cofactor (11), both reactions are energetically favorable. Thus, this functional difference must arise from another aspect, most likely the inability of the Mn-binding reaction center to couple the reaction to proton transfer as required for the reaction in Mn-superoxide dismutase (19, 20).
Table 1.
Comparison of the reaction with superoxide for Mn-superoxide dismutase and the Mn-binding reaction center
| Mn-superoxide dismutase | Mn-binding mutant | |
| Mn reduction |  |
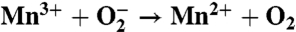 |
| Mn oxidation | 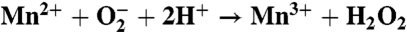 |
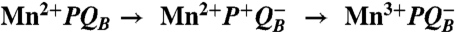 |
To determine if the light-induced loss of oxygen evident for the Mn-binding reaction centers without Mn (Fig. 2A) is due to photodamage, the impact of light and superoxide on the reaction centers was determined using an optical activity assay (Fig. 4). For wild type, a brief illumination at 865 nm results in a light-minus-dark spectrum characteristic of the P+·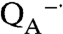 state that increases slightly in amplitude after a 2-min illumination due to the use of subsaturating light. No further change is observed after additional exposure up to 5 min, indicating that the wild-type reaction centers are stable under prolonged illumination. Similar spectra are obtained for the control mutant that can bind Mn but has a wild-type potential that is too low to oxidize Mn. In the absence of Mn, the Mn-binding mutants show a similar spectrum after a brief illumination, although the amplitude is reduced 10-fold because of the low quantum yield for these mutants (21, 22). However, longer light exposures result in pronounced loss of activity, as evident by absorption losses centered near 760 and 800 nm that correspond to loss of the bacteriopheophytin and bacteriochlorophyll pigments, respectively. This absorption loss becomes very pronounced if the samples are exposed to superoxide or illuminated for time periods beyond 2 min. Because the triple mutant also shows absorption loss near 760 and 800 nm, this pigment loss is likely associated with the highly oxidizing P+·. In striking contrast, only small absorption losses are observed for the Mn-binding reaction centers in the presence of Mn, which has the light-minus-dark spectral features of the P
state that increases slightly in amplitude after a 2-min illumination due to the use of subsaturating light. No further change is observed after additional exposure up to 5 min, indicating that the wild-type reaction centers are stable under prolonged illumination. Similar spectra are obtained for the control mutant that can bind Mn but has a wild-type potential that is too low to oxidize Mn. In the absence of Mn, the Mn-binding mutants show a similar spectrum after a brief illumination, although the amplitude is reduced 10-fold because of the low quantum yield for these mutants (21, 22). However, longer light exposures result in pronounced loss of activity, as evident by absorption losses centered near 760 and 800 nm that correspond to loss of the bacteriopheophytin and bacteriochlorophyll pigments, respectively. This absorption loss becomes very pronounced if the samples are exposed to superoxide or illuminated for time periods beyond 2 min. Because the triple mutant also shows absorption loss near 760 and 800 nm, this pigment loss is likely associated with the highly oxidizing P+·. In striking contrast, only small absorption losses are observed for the Mn-binding reaction centers in the presence of Mn, which has the light-minus-dark spectral features of the P 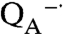 state because P+· is reduced by the bound manganese. Thus, the binding of the manganese cofactor not only serves a protective role against reactive oxygen species but also minimizes photooxidation damage in highly oxidizing reaction centers.
state because P+· is reduced by the bound manganese. Thus, the binding of the manganese cofactor not only serves a protective role against reactive oxygen species but also minimizes photooxidation damage in highly oxidizing reaction centers.
Fig. 4.

Stability of reaction centers under illumination. The ability of the reaction center to perform charge separation after different times of illumination was determined by measurement of the light-induced absorption changes. Shown are the light-minus-dark spectra after illuminations of 20 s (solid) and 2 min (dashed). Reaction centers from wild type and the control mutant, which has the Mn-binding site but a potential similar to wild type, show spectra characteristic of the P+·
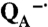 state. In the absence of bound Mn, the Mn-binding reaction centers have the same spectral features as wild type for 20-s illumination, but longer exposure results in significant loss of absorption near 760 and 800 nm associated with loss of the bacteriopheophytin and bacteriochlorophyll pigments. A similar behavior is observed for the reaction centers from the triple mutant that is highly oxidizing but does not have a Mn-binding site. The light-induced pigment loss of the Mn-binding mutant is increased in the presence of superoxide. However, when manganese was present, the Mn-binding reaction centers showed a relatively stable spectrum characteristic of the P
state. In the absence of bound Mn, the Mn-binding reaction centers have the same spectral features as wild type for 20-s illumination, but longer exposure results in significant loss of absorption near 760 and 800 nm associated with loss of the bacteriopheophytin and bacteriochlorophyll pigments. A similar behavior is observed for the reaction centers from the triple mutant that is highly oxidizing but does not have a Mn-binding site. The light-induced pigment loss of the Mn-binding mutant is increased in the presence of superoxide. However, when manganese was present, the Mn-binding reaction centers showed a relatively stable spectrum characteristic of the P 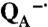 state, with P+· being reduced by the manganese. Shown are the data for the M8 mutant; the data for the M2 mutant are provided in Fig. S3.
state, with P+· being reduced by the manganese. Shown are the data for the M8 mutant; the data for the M2 mutant are provided in Fig. S3.
These results provide insight into the evolution from anaerobic to oxygenic photosynthesis. The core cofactors and subunits of photosystem II and the bacterial reaction center have similar three-dimensional structures (4, 23), with the D1 and D2 subunits of photosystem II and the L and M subunits of bacterial reaction centers being derived from a common ancestor. The evolutionary transition from primitive anaerobic phototrophs to organisms capable of oxygenic photosynthesis is thought to have triggered the great oxidation event approximately 2.4 Gyr ago, in which molecular oxygen emerged as a significant constituent of Earth’s atmosphere (24–29). This transition would have required the development of a highly oxidizing complex with a Mn cluster capable of water oxidation. Creation of a highly oxidizing protein complex could have been achieved through a combination of altered interactions between the bacteriochlorophyll dimer and the surrounding protein as well as the incorporation of more highly oxidizing tetrapyrroles, such as chlorophyll d (29–31). The incorporation of appropriate metal ligands through a few amino acid substitutions that were similar to those in the Mn-binding reaction centers would have provided a metal-binding site and consequently resulted in the production of molecular oxygen from any superoxide that was present at that time. The ability of two different mutants, the M2 and M8 mutants, to bind Mn and produce molecular oxygen with very similar reaction parameters suggests a flexibility in the amino acid substitutions that gave rise to metal binding and activity.
Given the complex nature of the four-electron water oxidation process, the development from anoxygenic to oxygenic photosynthesis has been proposed to be enabled by a catalytic intermediate containing a Mn cofactor (30–37). A complex mechanism has been developed in cyanobacteria and plants to recover from the damage that occurs due to the presence of the highly oxidizing primary donor (38). The bound Mn cofactor would have stabilized the highly oxidizing reaction centers against damage from both photooxidation and superoxide. Such reaction centers would have been capable of converting superoxide into molecular oxygen, and so provide protection against reactive oxygen species. At that time, the oxygen level of the atmosphere has been estimated to be decreased by 10-5 compared to the present atmospheric level (24, 28, 29), yielding a concentration of dissolved oxygen of 3 nM. Essentially all of the dissolved oxygen would have been in radical forms due to the intense UV radiation at the Earth’s surface in the absence of an ozone layer. The presence of reactive oxygen species could have been mitigated by the enzymatic activity of highly oxidizing reaction centers with a Mn cofactor, and so such complexes could represent an intermediary in the evolutionary transitional period.
The pathway used by primitive anaerobic phototrophs is unknown, but the versatility of pathways exhibited by purple and green bacteria may have been exploited during evolution. The metabolic incorporation of an external electron donor to reduce the bound Mn cofactor would have required involvement of a suitable electron acceptor, such as NAD or NADP, as utilized by some purple bacteria when ferrous iron is the donor or in the noncyclic pathway with sulfide donors in green bacteria (36). In a model of the transition from a single photosystem complex to two coupled complexes (35), the ancient phototrophs have been proposed to be initially capable of expressing either a photosystem I or a photosystem II equivalent complex depending upon local environmental factors. In this case, the involvement of superoxide as a new electron donor with the resulting oxygen production may have triggered the simultaneous production of both complexes giving rise to the Z-scheme of oxygenic photosynthesis.
In summary, our results demonstrate that modifications of bacterial reaction centers can produce a highly oxidizing protein with a tight Mn binding site that is redox active. After light-induced electron transfer from the primary donor to the electron acceptors, the bound Mn is oxidized and can react with superoxide to produce molecular oxygen. The reaction has similarities to the metal-reduction step in superoxide dismutase but a novel light-driven process replaces the metal-oxidation step. This newly created light-driven enzyme serves as a useful model for understanding the involvement of intermediates in the evolutionary development of photosystem II.
Materials and Methods
Protein Isolation.
Mutants were constructed and purified as previously described (4, 21, 22). The strains were grown semiaerobically in the dark for 3 to 4 d at 30 °C. The Mn-binding mutant M8 has three changes near P to increase the oxidation/reduction potential, Leu L131 to His, Leu M160 to His, and Phe M197 to His, and three changes that create the Mn-binding site, Arg M164 to Tyr, Met M168 to Glu, and Gly M188 to Asp. The highly oxidizing triple mutant has only the three changes near P, Leu L131 to His, Leu M160 to His, and Phe M197 to His. The Mn-binding control mutant has only the three changes that create the Mn-binding site, Arg M164 to Tyr, Met M168 to Glu, and Gly M188 to Asp. For spectroscopic measurements, after solubilization, the reaction centers were purified using a DEAE Sephacryl ion exchange column equilibrated with 0.05% Triton X-100. After the chromatography, the reaction centers were dialyzed against 15 mM Tris-HCl pH 8 and 0.05% Triton X-100 to remove EDTA.
Enzyme Activity.
The enzymatic activity with superoxide was measured using an optical assay that is commercially available (Sigma-Aldrich) and developed for superoxide dismutase (39). Briefly, oxygen and xanthine are converted into superoxide and uric acid by xanthine oxidase. After the addition of the protein of interest, either the modified reaction centers or superoxide dismutase, the amount of superoxide remaining in solution is determined using an indicator, 2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium, monosodium salt, which upon reduction with superoxide is converted into a water-soluble formazan dye with a maximum absorbance at 440 nm after a 20-min incubation period. Thus, the extent of dye formation is inversely related to the amount of superoxide lost and correspondingly the enzymatic activity. Excitation was using 865- or 550-nm steady-state light; no difference was observed for these two wavelengths. Oxygen measurements were performed by use of an oxygen-sensing microelectrode (Ocean Optics) inserted into a sealed 0.5-mL glass vial containing the protein solution with superoxide generated as for the other assay. Steady-state optical spectra were obtained using a Cary 5 spectrometer (Varian) with light excitation using an Oriel tungsten lamp with an 860-nm interference filter.
Supplementary Material
Acknowledgments.
For assistance with the oxygen measurements we thank Sara Bowen and Wilson Francisco, and for the protein preparations we thank Alicia Kishpaugh and Camille Black. This work was supported by Grant CHE 1158552 from the National Science Foundation.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1115364109/-/DCSupplemental.
References
- 1.Hunter N, Daldal F, Thurnauer M, Beatty JT, editors. The Purple Phototrophic Bacteria. Dordrecht, The Netherlands: Springer; 2008. [Google Scholar]
- 2.Wydrzynski T, Satoh K, editors. Photosystem II: The Light-Driven Water: Plastoquinone Oxidoreductase. Dordrecht, The Netherlands: Springer; 2005. [Google Scholar]
- 3.Leslie M. On the origin of photosynthesis. Science. 2009;323:1286–1287. doi: 10.1126/science.323.5919.1286. [DOI] [PubMed] [Google Scholar]
- 4.Thielges M, et al. Design of a redox-linked active metal site: Manganese bound to bacterial reaction centers at a site resembling that of photosystem II. Biochemistry. 2005;44:7389–7394. doi: 10.1021/bi050377n. [DOI] [PubMed] [Google Scholar]
- 5.Barker PD. Designing redox metalloproteins from bottom-up and top-down perspectives. Curr Opin Struct Biol. 2003;13:490–499. doi: 10.1016/s0959-440x(03)00108-8. [DOI] [PubMed] [Google Scholar]
- 6.Kaplan J, DeGrado WF. De novo design of catalytic proteins. Proc Natl Acad Sci USA. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu Y, Yeung N, Sieracki N, Marshall NM. Design of functional metalloproteins. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeung N, et al. Rational design of a structural and functional nitric oxide reductase. Nature. 2009;462:1079–1085. doi: 10.1038/nature08620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller AF. Superoxide dismutases: Active sites that save, but a protein that kills. Curr Opin Chem Biol. 2004;8:162–168. doi: 10.1016/j.cbpa.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 10.Perry JJP, Shin DS, Getzoff ED, Tainer JA. The structural biochemistry of the superoxide dismutases. Biochim Biophys Acta. 2010;1804:245–262. doi: 10.1016/j.bbapap.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kálmán L, Williams JC, Allen JP. Energetics for oxidation of a bound manganese cofactor in modified bacterial reaction centers. Biochemistry. 2011;50:3310–3320. doi: 10.1021/bi1017478. [DOI] [PubMed] [Google Scholar]
- 12.Allen JP, Williams JC. The evolutionary pathway from anoxygenic to oxygenic photosynthesis examined by comparison of the properties of photosystem II and bacterial reaction centers. Photosynth Res. 2011;107:59–69. doi: 10.1007/s11120-010-9552-x. [DOI] [PubMed] [Google Scholar]
- 13.Hsu JL, Hsieh Y, Tu C, O’Connor CD, Nick HS, Silverman DN. Catalytic properties of human manganese superoxide dismutase. J Biol Chem. 1996;271:17687–17691. doi: 10.1074/jbc.271.30.17687. [DOI] [PubMed] [Google Scholar]
- 14.Jiang L, et al. De novo computational design of retro-aldo enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siegel JB, et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bolon DN, Mayo SL. Enzyme-like proteins by computational design. Proc Natl Acad Sci USA. 2001;98:14274–14279. doi: 10.1073/pnas.251555398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faielle M, et al. An artificial di-iron oxo-protein with phenol oxidase activity. Nat Chem Biol. 2009;5:882–884. doi: 10.1038/nchembio.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rothlisberger D, et al. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 19.Miller AF. Fe superoxide dismutase. In: Messerschmidt A, Huber R, Poulos TL, Weinhardt K, editors. Handbook of Metalloproteins. New York: Wiley; 2001. pp. 668–682. [Google Scholar]
- 20.Leveque VJP, Vance CK, Nick HS, Silverman DN. Redox properties of human manganese superoxide dismutase and active-site mutants. Biochemistry. 2001;40:10586–10591. doi: 10.1021/bi010792p. [DOI] [PubMed] [Google Scholar]
- 21.Lin X, et al. Specific alteration of the oxidation potential of the electron donor in reaction centers from Rhodobacter sphaeroides. Proc Natl Acad Sci USA. 1994;91:10265–10269. doi: 10.1073/pnas.91.22.10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kálmán L, LoBrutto R, Allen JP, Williams JC. Modified reaction centres oxidize tyrosine in reactions that mirror photosystem II. Nature. 1999;402:696–699. [Google Scholar]
- 23.Umena Y, Kawakami K, Shen JR, Kamiya N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature. 2011;473:55–61. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 24.Bekker A, et al. Dating the rise of atmospheric oxygen. Nature. 2004;427:117–120. doi: 10.1038/nature02260. [DOI] [PubMed] [Google Scholar]
- 25.Kaufman AJ, et al. Late Archean biosphere oxygenation and atmospheric evolution. Science. 2007;317:1900–1903. doi: 10.1126/science.1138700. [DOI] [PubMed] [Google Scholar]
- 26.Buick R. When did oxygenic photosynthesis evolve? Philos Trans R Soc Lond B Biol Sci. 2008;B363:2731–2743. doi: 10.1098/rstb.2008.0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Falkowski PG, Isozaki Y. The story of O2. Science. 2008;322:540–545. doi: 10.1126/science.1162641. [DOI] [PubMed] [Google Scholar]
- 28.Kump LR. The rise of atmospheric oxygen. Nature. 2008;451:277–278. doi: 10.1038/nature06587. [DOI] [PubMed] [Google Scholar]
- 29.Sessions AL, Doughty DM, Welander PV, Summons RE, Newman DK. The continuing puzzle of the great oxidation event. Curr Biol. 2009;19:R567–R574. doi: 10.1016/j.cub.2009.05.054. [DOI] [PubMed] [Google Scholar]
- 30.Blankenship RE, Hartman H. The origin and evolution of oxygenic photosynthesis. Trends Biochem Sci. 1998;23:94–97. doi: 10.1016/s0968-0004(98)01186-4. [DOI] [PubMed] [Google Scholar]
- 31.Larkum AWD. The evolution of chlorophylls and photosynthesis. In: Grimm B, Porra RJ, Rudinger W, Scheer H, editors. Chlorophylls and Bacteriochlorophylls: Biochemistry, Biophysics, Functions, and Applications. Dordrecht, The Netherlands: Springer; 2006. pp. 261–282. [Google Scholar]
- 32.Olson JM, Pierson BK. Origin and evolution of photosynthetic reaction centers. Origins Life. 1987;17:419–430. [Google Scholar]
- 33.Ananyev GM, Zaltsman L, Vasko C, Dismukes GC. The inorganic biochemistry of photosynthetic oxygen evolution/water oxidation. Biochim Biophys Acta. 2001;1503:52–68. doi: 10.1016/s0005-2728(00)00215-2. [DOI] [PubMed] [Google Scholar]
- 34.Sauer K, Yachandra VK. A possible evolutionary origin for the Mn4 cluster of the photosynthetic water oxidation complex from natural MnO2 precipitates in the early ocean. Proc Natl Acad Sci USA. 2002;99:8631–8636. doi: 10.1073/pnas.132266199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen JF, Martin W. Out of thin air. Nature. 2007;445:610–612. doi: 10.1038/445610a. [DOI] [PubMed] [Google Scholar]
- 36.Hohmann-Marriott MF, Blankenship RE. Evolution of photosynthesis. Annu Rev Plant Biol. 2011;62:515–548. doi: 10.1146/annurev-arplant-042110-103811. [DOI] [PubMed] [Google Scholar]
- 37.Williamson A, Conlan B, Hillier W, Wydrzynski T. The evolution of photosystem II: Insights into the past and future. Photosynth Res. 2011;107:71–86. doi: 10.1007/s11120-010-9559-3. [DOI] [PubMed] [Google Scholar]
- 38.Chow WS, Aro EM. Photoinactivation and mechanisms of recovery. In: Wydrzynski T, Satoh K, editors. Photosystem II: The Light Driven Water Plastoquinone Oxidireductase. Dordecht, The Netherlands: Springer; 2005. pp. 627–648. [Google Scholar]
- 39.Bull C, Niederhoffer EC, Yoshida T, Fee JA. Kinetic studies of superoxide dismutases: Properties of manganese-containing protein from Thermus thermophilus. J Am Chem Soc. 1991;113:4069–4076. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.