

Abstract
Context:
Type IV collagen is a type of collagen found primarily in the skin within the basement membrane zone. The type IV collagen C4 domain at the C-terminus is not removed in post-translational processing, and the fibers are thus link head-to-head, rather than in a parallel fashion. Also, type IV collagen lacks a glycine in every third amino-acid residue necessary for the tight collagen helix. Thus, the overall collagen-IV conformation is structurally more pliable and kinked, relative to other collagen subtypes. These structural features allow collagen IV to form sheets, which is the primary structural form found in the cutaneous basal lamina. There are six human genes associated with collagen IV, specifically COL4A1, COL4A2, COL4A3, COL4A4, COL4A5 and COL4A6. The aim of this review is to highlight the significance of this protein in normal skin, and in selected diseases.
Results:
The alpha 3 protein constituent of type IV collagen is thought to be the antigen implicated in Goodpasture's syndrome, wherein the immune system attacks the basement membranes of the renal glomeruli and pulmonary alveoli. In addition, mutations to the genes coding for type IV collagen lead to the Alport syndrome. Furthermore, autoantibodies directed against denatured human type IV collagen have been described in rheumatoid arthritis, scleroderma, and SLE. Structural studies of collagen IV have been utilized to differentiate between subepidermal blistering diseases, including bullous pemphigoid, acquired epidermolysis bullosa, anti-epiligrin cicatricial pemphigoid, and bullous lupus erythematosus. Collagen IV is also of importance in wound healing and in embryogenesis.
Conclusions:
Pathological studies have demonstrated that minor structural differences in collagen IV can lead to distinct, clinically different diseases.
Keywords: Alport syndrome, Basement membrane zone, Collagen-type IV, Goodpasture's syndrome, Subepidermal blistering diseases
Introduction
Collagen IV is the primary collagen found in the extracellular basement membranes separating a variety of epithelial and endothelial cells. It is a major component of the dermal–epidermal junction, where it is mostly found in the lamina densa. Collagen IV is a heterotrimeric molecule containing two α1-like and one α2-like genes. Multiples diseases have been associated with the molecule, including Alport and Goodpasture's syndrome, as well as several rheumatological and dermatological diseases including, acquired epidermolysis bullosa.
Type IV Collagen
All the type IV collagen in mammals is derived from six genetically distinct α-chain polypeptides (α1–α6).[1] The type IV collagen α-chains have similar domain structures, demonstrating 50 – 70% homology at the amino acid level.[1] The α-chains can be separated into three domains: an amino-terminal 7S domain, a middle triple-helical domain, and a carboxy-terminal, globular, non-collagenous (NC)-1 domain. The triple-helical sector represents the longest domain, approximately 1,400 amino acids (aa) in length, with about 22 interruptions within its classical collagen Gly-X-Y sequence motif.[1,2] The NC1 domain of each α-chain is about 230 aa in length. As with all collagens, the NC1 domain is considered important for the assembly of the type IV collagen trimeric structure.[1,2] The assembly of a particular trimer begins when the three NC1 domains initiate a molecular interaction between three α-chains. Protomer trimerization then proceeds in a zipper-like format from the carboxy-terminal end, resulting in a fully assembled protomer. The assembled protomer is flexible, and can bend. The next step in the assembly is the type IV collagen dimer formation. Two type IV collagen protomers associate via their carboxy-terminal NC1 trimers to form an NC1 hexamer.[1,2] Next, four protomers interact at the glycosylated amino-terminal 7S region to form tetramers. These interactions form the nucleus for a type IV collagen scaffold. The scaffold evolves into a type IV collagen superstructure with the help of end-to-end associations and lateral associations between the type IV collagen protomers.[1,2] The COL4A3 gene provides instructions for making one component of a type IV collagen, which is a flexible protein. Specifically, this gene codes for the alpha3(IV) chain of the type IV collagen.[1,2] The alpha3(IV) chain combines with two other types of alpha(IV) chains (the alpha4 and alpha5 chains) to form a complete type IV collagen molecule. Type IV collagen molecules attach to each other to form complex protein networks.[1,2] These networks make up a large portion of the basement membrane zones (BMZs), which are thin, sheet-like structures that separate and support cells in many tissues. Type IV collagen, with the alpha3-4-5 networks, plays an important role in the BMZ of the kidney, inner ear, and eye.[1,2] Type IV collagen composed of alphal(IV) and alpha2(IV) networks is the major skeletal macromolecule of the basement membranes in selected tissues. These chains form heterotrimers through the association between their carboxy-terminal NC1 domains, associated with folding of the collagenous domains into triple helices. Such triple helices are also capable of forming networks through several types of intermolecular interactions.[1,2] These differential linkages between type IV collagen molecules produce a non-fibrillar polygonal assembly that serves as a scaffolding for the deposition of other matrix glycoproteins, as well as for cell attachment.[1,2]
Collagen IV as a Part of the Basement Membrane Zone
In recent years, the BMZ has been recognized as an important regulator of cell behavior, rather than just a structural feature of tissues.[3,4] The BMZ mediates tissue compartmentalization and sends signals to epithelial cells about the external microenvironment.[3,4] The BMZ also has important structural and functional effects on blood vessels, constituting an extracellular microenvironment sensor for endothelial cells and pericytes . Thus, BMZs are widely distributed extracellular matrices that interface the basilar portion of the epithelial and endothelial cells, and surround the muscle, adipose, and Schwann cells. These extracellular matrices, initially expressed in early embryogenesis, are self-assembled on competent cell surfaces through binding interactions among laminins, type IV collagens, nidogens, and proteoglycans. The BMZs play a role in tissue and organ morphogenesis, and help to maintain the functionality of these structures in adults. Mutations adversely affecting the expression of BMZ structural components are associated with developmental arrest at different stages, as well as postnatal diseases of the muscle, neural, ocular, cutaneous, vascular, and kidney tissues. Vascular BMZ components have recently been seen to be involved in the regulation of tumor angiogenesis, making them attractive candidate targets for potential oncological therapies.[3,4] In Figure 1, we show by direct immunofluorescence (DIF) and immunohistochemistry (IHC), the location of collagen IV in the skin within the BMZ, sweat glands, and blood vessels.[3,4]
Figure 1.
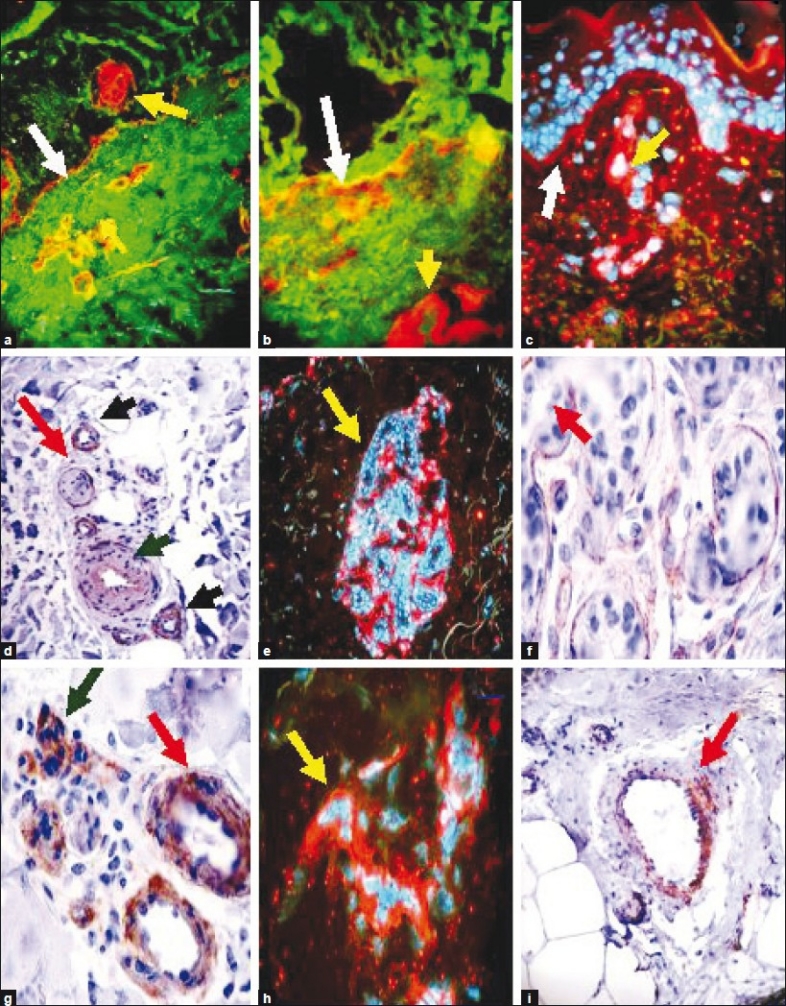
Examples of staining of collagen type IV antibody in the skin by direct immunofluorescence (DIF) (a,b,c,e, and h) and by immunohistochemistry (IHC) (d,f,g,i) under normal conditions, as well as in a patient with lupus erythematosus. All staining was performed as previously documented.[23–31] We utilized an antibody to collagen IV from Invitrogen (Carlsbad, California, USA). 1 a and b, Double positive DIF staining of the BMZ ‘lupus band’ in a lupus patient using an FITC conjugated antibody to human IgG, overlapping with the staining of Texas red conjugated collagen IV, resulting in orange staining (white arrows). The blood vessels also stained positive with the antibody to collagen IV (red staining; yellow arrows). c. Positive DIF staining of the cutaneous BMZ using the antibody to collagen IV alone (red staining; white arrow at the BMZ, yellow arrow showing positivity on a dermal blood vessel). d. Positive IHC staining for collagen IV antibodies around a nerve (brown staining; red arrow), blood vessels (green arrow), and eccrine sweat gland ducts (black arrows). e. Positive DIF staining of a sweat gland coil BMZ, utilizing Texas red conjugated collagen IV (red staining). The nuclei of the cells were counterstained with Dapi (blue staining; yellow arrow). f. Positive IHC staining of eccrine glands for collagen IV (brown staining; red arrow). g. Positive IHC staining of eccrine gland ducts (brown staining; red arrow) and of small blood vessels (green arrow). h. Positive DIF staining of skin blood vessels utilizing Texas red conjugated collagen IV (red staining, yellow arrow). The nuclei of the cells were counterstained with Dapi (blue). i. Positive IHC staining for collagen IV antibody on a large blood vessel in a subcutaneous adipose tissue septum (brown staining; red arrow)
Type IV Collagen and Embryogenesis
In the Drosophila system, it has been demonstrated that dorsal–ventral patterning in invertebrate embryos may be mediated by a conserved system of secreted proteins, which establishes a morphogenetic protein gradient. Although the Drosophila embryonic decapentaplegic gradient (Dpp) serves as a model to understand how morphogen gradients are established, no role of the extracellular matrix has been previously described.[5,6] Of late, some authors have shown that type IV collagen extracellular matrix proteins bind Dpp, and regulate its signaling in both the Drosophila embryo and in the ovary.[5,6] The researchers were able to identify a critical function of type IV collagens in modulating Dpp in the extracellular space during Drosophila development.[5,6]
Antibodies to Collagen IV in Selected Rheumatic Diseases
Some authors have demonstrated the presence of autoantibodies to collagen IV, in rheumatologic diseases.[7,8] These authors utilized a sensitive and specific enzyme-linked immunosorbent assay (ELISA) for antibodies to native and to denatured type IV collagen, derived from the basement membranes to collagen IV (1) bovine anterior lens capsules or (2) human placenta. No controls demonstrated human type IV collagen antibodies, and only 5.6% demonstrated an antibody to bovine type IV collagen. Antibodies to one or more of the four collagen antigens were observed in 20% of children with juvenile rheumatoid arthritis, 35% of patients with mixed connective tissue disease, 40% of children with juvenile dermatomyositis, 52% of adults with rheumatoid arthritis, 56% of patients with scleroderma, and 60% of patients with systemic lupus erythematosus (SLE).[7,8] Antibodies to native human type IV collagen were rare (0 – 10%), except in SLE (45%). Antibodies to denatured human type IV collagen were more common in RA, scleroderma, and SLE. Antibodies to native bovine type IV collagen occurred in 8 – 20% of the patient sera and to denatured bovine antigen in 25 – 26% of the scleroderma, mixed connective disease, and diabetes mellitus patients. Anti-bovine type IV collagen activity measured by ELISA could be detected from the positive sera following preincubation of the sera with bovine type IV collagen, but not bovine type I collagen or native human placental type IV collagen, indicating that the antibodies were specific for bovine type IV collagen.[7,8]
The authors also studied the presence of autoantibodies to collagen IV, using a murine system.[7,8] The authors examined the role that immunological sensitization to autologous connective tissue components might play in inducing an inflammatory response, resulting in pathological sequelae. Mice receiving a single subcutaneous injection of five micrograms of type IV collagen in a complete Freund's adjuvant mounted a delayed-type hypersensitivity response, characterized by a mononuclear cell infiltrate when challenged in the footpad with the sensitizing antigen.[7,8] Cell-mediated immunity to these connective tissue antigens could also be transferred to normal syngeneic mice with sensitized T lymphocytes. Furthermore, repeated immunizations with these homologous connective tissue components elicited antibody responses in mice. The authors found that the anti-type IV collagen autoantibodies were found to be primarily of an IgM subtype. The authors concluded that selective immunity to the BMZ may influence the clinical expression of diffuse connective tissue syndromes such as scleroderma (systemic sclerosis).[7,8]
Type IV Collagen and Alport Syndrome or Hereditary Nephritis
Alport syndrome (AS) is an inherited disorder of type IV collagen, the major collagenous constituent of the glomerular BMZ (GBMZ).[9–14] The hallmark of the disease is persistent microscopic hematuria, often associated with proteinuria, progressive renal failure, ocular abnormalities, and high-tone sensorineural hearing loss.[9–14] AS is a genetic disorder characterized by glomerulonephritis and end-stage kidney disease. AS can also affect the eyes (lenticonus). AS is caused by mutations in the COL4A3, COL4A4, and COL4A5 collagen biosynthesis genes.[9–14] Mutations in any of these genes prevent the proper production or assembly of the type IV collagen network, which is an important structural component of BMZ in the kidney, inner ear, and eye. When mutations prevent the formation of type IV collagen fibers, the basement membranes of the kidneys are not able to filter waste products from the blood, leading to pathological blood and protein detection in the urine. The abnormalities of type IV collagen in the kidney BMZ cause progressive scarring of the kidneys, leading to renal failure in many people with the disease. Progression of the disease leads to BMZ thickening and gives a pathological ‘basket-weave’ appearance from splitting of the lamina densa.[9–14] It is currently accepted that the diagnosis of AS is warranted when four out of ten of the following criteria are met: (1) Family history of nephritis or unexplained hematuria in a first degree relative of the index case, or in a male relative linked through any number of females. (2) Persistent hematuria without evidence of another possible inherited nephropathy, such as, thin GBM disease, polycystic kidney disease or IgA nephropathy. (3) Bilateral sensorineural hearing loss in the 2000 to 8000 Hz range. The hearing loss develops gradually, is not present in early infancy and commonly presents before the age of 30 years. (4) A mutation in COL4An, where n = 3, 4 or 5. (5) Immunohistochemistry evidence of complete or partial lack of the Alport epitope to BMZ in the skin, kidney or both. (6) Widespread glomerular BMZ ultrastructural abnormalities, in particular thickening, thinning, and / or splitting. (7) Ocular lesions, including anterior lenticonus, posterior subcapsular cataract, posterior polymorphous dystrophy, and retinal flecks. (8) Gradual progression to end-stage renal disease in the index case, or at least in two family members. (9) Macrothrombocytopenia or granulocytic inclusions. (10) Diffuse leiomyomatosis of the esophagus or female genitalia, or both.
The disease is genetically heterogeneous, but the majority of people affected by AS show an X-linked dominant inheritance and are affected by mutations in the COL4A5 gene located in the Chromosome Xq22 region.[9–14] More than 40 mutations in the COL4A3 gene have been found to cause the AS syndrome. Most of these mutations occur when single amino acid mutations occur in a region where the alpha3 (IV) collagen chain combines with other type IV collagen chains.[9–14] Other mutations in the COL4A3 gene severely decrease or prevent the production of alpha3(IV) chains. As a result of these mutations, there is a serious deficiency of the type IV collagen alpha3-4-5 network in the basement membranes of the kidney, inner ear, and eye.[9–14] In the kidney, other types of collagen accumulate in the BMZ, eventually leading to scarring of the kidneys and renal failure. Mutations of the COL4A5 gene result in a complete or segmental loss of the α5(IV) chain in the BMZ.[9–14] Interestingly, the glomerular BMZ of patients with the X-linked Alport syndrome (X-AS) displays characteristic ultrastructural features (diffuse thickening with splitting of the lamina densa into multiple interweaving strands), while no significant morphological alterations at the light or electron microscopic level have been demonstrated, to date, along the cutaneous BMZ.[9–14]
The reason for such a different reaction to the same genetic defect is not known, but it could be due to differences in the amino acid composition that exist between the two basement membranes. For example, while α1(IV) and α2(IV) chains are normally present in all BMZs, the GBM also contains α3(IV) and α4(IV) chains, but not the BMZ of the skin.[9–14] In addition, significant differences between the GBM and skin BMZ, different from those in the αx(IV) chains, also exist. Type VII collagen, for instance, is not found in normal glomeruli, being detectable only in sclerotic lesions.[9–14] However, it is present in large amounts in the normal skin BMZ, where it represents the predominant component of anchoring fibrils (attachment structures, which ensure the integrity of the skin BMZ), and maintains dermal-epidermal integrity.[9–14] A defective BMZ, as observed in X-AS, could also potentially induce significant effects on the epidermal extracellular matrix assembly in the dermis (e.g., due to the interaction between the skin basement membrane and the underlying dermis). Thus, IHC or electron microscopic study of the skin may be used as a screening method in patients suspected to have AS, as a skin biopsy is much less invasive than a kidney biopsy. Moreover, such skin biopsies will represent a useful adjunct to the conventional examination of biopsied renal tissue.
Goodpasture's Syndrome and Collagen Type IV
Goodpasture syndrome (GS) (also known as Goodpasture's disease and anti-glomerular basement membrane antibody disease) is a rare disease characterized by glomerulonephritis and hemorrhaging of the lungs.[10,15,16] Although many diseases can present with these symptoms, the name GS is usually reserved for the autoimmune disease triggered when the patient's antibodies attacks the Goodpasture antigen (a type II hypersensitivity reaction), which is found in the kidneys and lungs, causing damage to these organs over time.[10,15,16] Collagen type IV alpha3 represents the ‘Goodpasture antigen’; COL4A3 is the gene acronym. GS is a rare disease; in Caucasian populations, the incidence falls between 1 and 1,000,000 to 2,000,000. GS is less likely to present in non-Caucasian populations. Even though cases have occurred in patients between the ages of four and 80, presentation is most common between the ages of 18 and 30 and also between 50 and 65. Unlike many other autoimmune diseases, males are surprisingly affected six times more often than females.[10,15,16]
In GS, the primary affected organ is the kidney. The disease primarily affects the renal glomeruli, causing a form of nephritis. It is often not detected until a rapid advance of the disease occurs; the kidney function can be completely lost in a matter of days, a condition known as rapidly progressive glomerulonephritis. Hematuria is seen; the volume of urine output decreases, and the urea and other products normally excreted by the kidney are retained in the blood.[10,15,16] Classically, renal failure does not cause symptoms until more than 80% of the kidney function has been lost. Initial symptoms include loss of appetite and malaise; when the damage is more advanced, breathlessness, high blood pressure, and edema are appreciated. As previously noted, renal involvement usually presents as a nephritic syndrome (i.e., hematuria, a reduced glomerular filtration rate, and increased blood pressure). The nephritic presentation is in contrast to the nephrotic syndrome, a less common complication of GS, characterized by an abnormally large amount of protein in the urine, coupled with severe edema.[10,15,16] The diagnosis of GS is sometimes difficult, due to a vagueness of the early symptoms. Given the late, rapid progression of the clinically apparent disease, the diagnosis is often not established until very late in the pathological course. A renal biopsy for DIF will show linear IgG deposits along the renal glomerular basement membrane. Serologic tests for anti-GBM antibodies may also be useful, combined with tests for antibodies to neutrophil cytoplasmic antigens, which are also directed against the patient's own proteins. The second most commonly affected organ in the GS is the lung. Pulmonary symptoms may present as nothing more than a dry cough and minor breathlessness; such mild symptoms may last for many years. In a severe GS presentation, lung damage may cause severe impairment of oxygenation and intensive care is thus required.[10,15,16] The patient often does not seek medical attention until hemoptysis is seen. The patient may be anemic due to chronic pulmonary hemorrhage. In GS, unlike many other conditions that cause similar symptoms, lung hemorrhage most often occurs in smokers and those with additional pulmonary damage from infection or exposure to fumes.[10,15,16] It has been speculated that some environmental stimuli, such as viral infections (especially by influenza) or kidney surgery could trigger the autoimmune phenomenon in GS. Other possible triggers include the presence of an inherited genetic component and exposure to selected chemicals, including hydrocarbon solvents and the weed killer N, N’-dimethyl-4,4’-bipyridinium dichloride(Paraquat), one of the most widely used herbicides in the world.[10,15,16]
As with many autoimmune conditions, the precise cause of GS is not yet known. It is believed to be a type II hypersensitivity reaction to Goodpasture antigens on the basement membrane of the renal glomeruli and pulmonary alveoli, specifically the non-collagenous domain of the alpha3 chain of the Type IV collagen.[10,15,16] In GS, the immune system inappropriately recognizes these motifs as foreign antigens and produces antibodies to them . Over the past century, GS has often been a fatal disease. However, due to advances in diagnosis and treatment, patient morbidity is now significantly decreased.[10,15,16] Death from pulmonary hemorrhage can occur before the diagnosis is made, or in the initial stages of treatment. With treatment, however, the patient can usually recover completely from pulmonary damage. In contradistinction, patient kidneys are less able to repair themselves; patients with renal damage must therefore often rely on long-term dialysis or kidney transplantation.[10,15,16] Even with the best management, there is still significant mortality from renal failure, particularly if the patient is otherwise in poor health. Furthermore, immunosuppressive treatment may increase the clinical risk of serious or fatal secondary infections. GS responds well to treatment with corticosteroids and immunosuppressants.[10,15,16] The concentration of anti-GBM antibodies in the blood may be reduced by apheresis; this procedure involves removing a portion of blood plasma and replacing it with an isotonic salt and protein solution.[10,15,16] A multiple apheresis course of treatment usually lasts between three and six months.
Collagen Type IV in Wound Healing
The value of collagen type IV has also been demonstrated in forensic medicine, by studying this molecule in different wound types. In one study, specifically, an antibody to collagen IV was utilized.[17] The authors analyzed 62 human skin wounds (including surgical wounds, stab wounds, and lacerations after surgical treatment), and noted the localization of collagen IV in the cutaneous BMZ by IHC staining.[18] In 27 of these wounds, the distribution of collagen VII was also analyzed. The authors demonstrated a virtually identical co-distribution of both collagen IV and VII in the wound areas, with no significant time-dependent differences in the appearance of both collagen types.[17,18] Fragments of the epithelial BMZ could be detected in the wound area from as early as four days after wounding. After eight days, the earliest complete restitution of the epithelial BMZ was observed. In all cases with a wound age of more than 21 days, the BMZ was completely reformed over the former lesional area. The period between eight and 21 days after wounding was characterized by a wide variability, ranging from complete restitution to deposition of BMZ fragments, or total lack of epidermal BMZ material in the wound. The authors emphasized the importance of collagen type IV in wound healing.[17,18]
Type IV Collagen and the Skin
Immunohistochemical stains with monoclonal antibodies have been utilized to localize two basement membrane components (laminin and type IV collagen) in the nerves and sensory nerve mechanoreceptors supplying human digital skin.[19] The authors colocalized S-100 protein and epithelial membrane antigen in parallel with markers such as neurofilament antibody.[19] In the dermal nerve trunks, immunostaining for laminin and type IV collagen was found to colocalize in the perineurium and the Schwann cells, with stronger immunoreactivity at the external surface of the cells. In the Meissner corpuscles, immunoreactivity for laminin and type IV collagen was primarily observed under the cell surface of lamellar cells, while the cytoplasms were weakly immunolabeled or unlabeled.[19] Finally, within the Pacinian corpuscles, colocalization of the two basement membrane molecules was encountered in the inner core, intermediate layer, outer core, and the capsule. Laminin and type IV collagen immunoreactivities were also found in blood vessels and sweat glands, apparently labeling BMZ. The authors demonstrated evidence for the presence of basement membrane material in all periaxonic cells forming human cutaneous sensory nerve formations, and suggested that all of these cells were able to synthesize and release some basement membrane components, including laminin and type IV collagen.[19]
Other authors attempted to characterize the distributions of type IV collagen alpha chains in the BMZ of the human skin and its appendages, utilizing DIF and IIF with chain-specific monoclonal antibodies.[20] The BMZ contained [alpha1(IV)](2)alpha2(IV) and [alpha5(IV)](2)alpha6(IV), but no alpha3(IV)alpha4(IV)alpha5(IV); this also held true for the eccrine sweat coils and glandular ducts, sebaceous glands, hair follicles, and arrector pili muscles of the hair follicles.[19] The secretory portion of the eccrine sweat glands was rich in [alpha1(IV)](2) alpha2(IV) and had less [alpha5(IV)](2)alpha6(IV), while [alpha5(IV)](2) alpha6(IV) was abundant in the ductal portion.[19,20] The authors suggested that the alpha5(IV) / alpha6(IV) chain negative spots within the BMZ manifested a discrete relationship with melanocytes, and were sites of interactions between the two.[20] Continuous linear staining of IgG was found along the BMZ and around the hair follicles, sebaceous gland acini, and small capillaries.
Type IV Collagen and Distinguishing Epidermal and Subepidermal Blistering Diseases in the Skin
Two methods can be used to differentiate between epidermolysis bullosa acquisita (EBA) and bullous pemphigoid (BP) patients. The first method uses purified antibodies against type IV collagen and laminin, localized to the BMZ by either immunohistochemistry and/or indirect immunofluorescence, with salt split skin. EBA is characterized by the production of autoantibodies against collagen VII in the sublamina densa anchoring fibrils.[21] The authors have described the usefulness of collagen IV immunostaining on paraffin-embedded skin biopsies as an aid for diagnosing EBA in dogs, but it is also useful in humans.[21] In EBA, collagen IV (which forms the fibrous two-dimensional network of the lamina densa), is detected more commonly above the subepidermal blisters than below them. Collagen IV immunostaining thus offers an inexpensive means to assist in making a diagnosis of EBA distinct from the serological determination of the targeted autoantigen.[21]
Salt split skin is normal skin that has been soaked in NaCl 0.1 M solution.[22,23] Over time, the skin is split subepidermally. Salt split skin can be used to differentiate BP from EBA; in EBA, the IIF deposits of the antibodies are usually found on the blister floor, below the split. In the case of bullous pemphigoid, the IIF deposits of immunoglobulin and the complement are usually on the blister roof, above the split. In contradistinction, collagen IV is found on the blister roof in EBA, and on the blister floor in BP. Furthermore, anti-epiligrin cicatricial pemphigoid (AECP) is an uncommon subtype of cicatricial pemphigoid (CP), primarily affecting the mucous membranes. AECP involves autoantibodies against the α subunit of epiligrin, also known as laminin 5 or laminin 332 (α3β3γ2). Bullous lupus erythematosus (BLE) is a manifestation of systemic lupus erythematosus. Similar to EBA, BLE is associated with autoantibodies to the type VII collagen. As previously noted, IIF salt split skin disease antibody binding is present in the epidermal / blister roof side with BP, and in contrast to this, reactivity on the dermal / blister floor side is associated with the other two disorders (AECP and BLE). Finally, a case of crescentic glomerulonephritis, with subepidermal blisters and autoantibodies to the α5 and α6 chains of type IV collagen has also been documented.[32]
Footnotes
Source of Support: Nil.
Conflict of Interest: None declared.
References
- 1.Kalluri R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422–33. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 2.Yurchenco PD. Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harb Perspect Biol. 2011;3:a004911. doi: 10.1101/cshperspect.a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasegawa H, Naito I, Nakano K, Momota R, Nishida K, Taguchi T, et al. The distributions of type IV collagen alpha chains in basement membranes of human epidermis and skin appendages. Arch Histol Cytol. 2007;70:255–65. doi: 10.1679/aohc.70.255. [DOI] [PubMed] [Google Scholar]
- 4.Konomi H, Hayashi T, Nakayasu K, Arima M. Localization of type V collagen and type IV collagen in human cornea, lung, and skin.Immunohistochemical evidence by anti-collagen antibodies characterized by immunoelectroblotting. Am J Pathol. 1984;116:417–26. [PMC free article] [PubMed] [Google Scholar]
- 5.Bunt S, Hooley C, Hu N, Scahill C, Weavers H, Skaer H. Hemocyte-secreted type IV collagen enhances BMP signaling to guide renal tubule morphogenesis in Drosophila. Dev Cell. 2010;17:296–306. doi: 10.1016/j.devcel.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Harris RE, Bayston LJ, Ashe HL. Type IV collagens regulate BMP signaling in Drosophila. Nature. 2008;455:72–7. doi: 10.1038/nature07214. [DOI] [PubMed] [Google Scholar]
- 7.Petty RE, Hunt DW, Rosenberg AM. Antibodies to type IV collagen in rheumatic diseases. J Rheumatol. 1986;13:246–53. [PubMed] [Google Scholar]
- 8.Mackel AM, DeLustro F, DeLustro B, Fudenberg HH, LeRoy EC. Immune response to connective tissue components of the basement membrane. Connect Tissue Res. 1982;10:333–43. doi: 10.3109/03008208209008058. [DOI] [PubMed] [Google Scholar]
- 9.Pescucci C, Longo I, Bruttini M, Mari F, Renieri A. Type IV collagen related diseases. J Nephrol. 2003;16:314–6. [PubMed] [Google Scholar]
- 10.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG. Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N Engl J Med. 2003;348:2543–56. doi: 10.1056/NEJMra022296. [DOI] [PubMed] [Google Scholar]
- 11.Patey-Mariaud de Serre N, Noël LH. [Collagen alpha5 and alpha2 (IV) chain coexpression: The procedure of choice to diagnose Alport syndrome from skin biopsies] Ann Pathol. 2008;28:182–6. doi: 10.1016/j.annpat.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 12.Pescucci C, Mari F, Longo I, Vogiatzi P, Caselli R, Scala E, et al. Autosomal-dominant Alport syndrome: Natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int. 2004;65:1598–603. doi: 10.1111/j.1523-1755.2004.00560.x. [DOI] [PubMed] [Google Scholar]
- 13.Gregory MC, Terreros DA, Barker DF, Fain PN, Denison JC, Atkin CL. Alport syndrome clinical phenotypes, incidence, and pathology. Contrib Nephrol. 1996;117:1–28. doi: 10.1159/000424804. [DOI] [PubMed] [Google Scholar]
- 14.Lagona E, Tsartsali L, Kostaridou S, Skiathitou A, Georgaki E, Sotsiou F. Skin biopsy for the diagnosis of Alport syndrome. Hippokratia. 2008;12:116–8. [PMC free article] [PubMed] [Google Scholar]
- 15.Goodpasture EW. The significance of certain pulmonary lesions in relation to the etiology of influenza. Am J Med Sci. 2009;338:148–51. doi: 10.1097/MAJ.0b013e31818fff94. [DOI] [PubMed] [Google Scholar]
- 16.Salama AD, Levy JB, Lightstone L, Pusey CD. Goodpasture's disease. Lancet. 2001;358:917–20. doi: 10.1016/S0140-6736(01)06077-9. [DOI] [PubMed] [Google Scholar]
- 17.Betz P, Nerlich A, Wilske J, Tübel J, Wiest I, Penning R, et al. The time-dependent rearrangement of the epithelial basement membrane in human skin wounds--immunohistochemical localization of Collagen IV and VII. Int J Legal Med. 1992;105:93–7. doi: 10.1007/BF02340831. [DOI] [PubMed] [Google Scholar]
- 18.Wick G, Glanville RW, Timpl R. Characterization of antibodies to basement membrane (type IV) collagen in immunohistological studies. Immunobiology. 1980;156:372–81. doi: 10.1016/S0171-2985(80)80071-4. [DOI] [PubMed] [Google Scholar]
- 19.Vega JA, Esteban I, Naves FJ, del Valle ME, Malinovsky L. Immunohistochemical localization of laminin and type IV collagen in human cutaneous sensory nerve formations. Anat Embryol (Berl) 1995;191:33–9. doi: 10.1007/BF00215295. [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa H, Naito I, Nakano K, Momota R, Nishida K, Taguchi T, et al. The distributions of type IV collagen alpha chains in basement membranes of human epidermis and skin appendages. Arch Histol Cytol. 2007;70:255–65. doi: 10.1679/aohc.70.255. [DOI] [PubMed] [Google Scholar]
- 21.Weber L, Krieg T, Müller PK, Kirsch E, Timpl R. Immunofluorescent localization of type IV collagen and laminin in human skin and its application in junctional zone pathology. Br J Dermatol. 1982;106:267–73. doi: 10.1111/j.1365-2133.1982.tb01722.x. [DOI] [PubMed] [Google Scholar]
- 22.Olivry T, Dunston SM. Usefulness of collagen IV immunostaining for diagnosis of canine epidermolysis bullosa acquisita. Vet Pathol. 2010;47:565–8. doi: 10.1177/0300985809359609. [DOI] [PubMed] [Google Scholar]
- 23.Mutasim DF, Diaz LA. The relevance of immunohistochemical techniques in the differentiation of subepidermal bullous diseases. Am J Dermatopathol. 1991;13:77–83. doi: 10.1097/00000372-199102000-00013. [DOI] [PubMed] [Google Scholar]
- 24.Abreu Velez AM, Girard JG, Howard MS. Antigen presenting cells in a patient with hair loss and systemic lupus erythematosus. North Am J Med Sci. 2009;1:205–10. [PMC free article] [PubMed] [Google Scholar]
- 25.Abreu Velez AM, Smith JG, Jr, Howard MS. Neutrophil extracellular traps (NETS), IgD, myeloperoxidase (MPO) and antineutrophil cytoplasmic antibody (ANCA) associated vasculitides. North Am J Med Sci. 2009;1:309–13. [PMC free article] [PubMed] [Google Scholar]
- 26.Abreu Velez AM, Howard MS, Restrepo M, Smoller BR. formalin deposition as artifact in biopsies from patients affected by a new variant of endemic pemphigus foliaceus in El-Bagre, Colombia, South America. J Cutan Pathol. 2010;37:835–42. doi: 10.1111/j.1600-0560.2009.01492.x. [DOI] [PubMed] [Google Scholar]
- 27.Abreu Velez AM, Howard MS, Pinto FJ., Jr Dyshidrotic eczema: Relevance to the immune response in situ. North Am J Med Sci. 2009;1:117–20. [PMC free article] [PubMed] [Google Scholar]
- 28.Abreu Velez AM, Howard MS, Hashimoto T. Palms with a polyclonal autoimmune response in patients affected by a new variant of endemic pemphigus foliaceus in Colombia, South America. Eur J Dermatol. 2010;20:74–81. doi: 10.1684/ejd.2010.0834. [DOI] [PubMed] [Google Scholar]
- 29.Abreu Velez AM, Jackson BL, Howard MS. Deposition of immunoreactants in a cutaneous allergic drug reaction. North Am J Med Sci. 2009;1:180–3. doi: 10.4297/najms.2009.4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Abreu Velez AM, Howard MS, Hashimoto K, Hashimoto T. Autoantibodies to sweat glands detected by different methods in serum and in tissue from patients affected by a new variant of endemic pemphigus foliaceus. Arch Dermatol Res. 2009;301:711–8. doi: 10.1007/s00403-009-0972-4. [DOI] [PubMed] [Google Scholar]
- 31.Abreu Velez AM, Howard MS, Loebl AM. Autoreactivity to sweat and sebaceous glands and skin homing T cells in lupus profundus. Clin Immunol. 2009;132:420–4. doi: 10.1016/j.clim.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 32.Ghohestani RF, Rotunda SL, Hudson B, Gaughan WJ, Farber JL, Webster G, et al. Crescentic glomerulonephritis and subepidermal blisters with autoantibodies to α5 and α6 chains of type IV collagen. Lab Invest. 2003;83:605–11. doi: 10.1097/01.lab.0000067497.86646.4d. [DOI] [PubMed] [Google Scholar]