

Abstract
Mutation T4825I in the type 1 ryanodine receptor (RYR1T4825I/+) confers human malignant hyperthermia susceptibility (MHS). We report a knock-in mouse line that expresses the isogenetic mutation T4826I. Heterozygous RYR1T4826I/+ (Het) or homozygous RYR1T4826I/T4826I (Hom) mice are fully viable under typical rearing conditions but exhibit genotype- and sex-dependent susceptibility to environmental conditions that trigger MH. Hom mice maintain higher core temperatures than WT in the home cage, have chronically elevated myoplasmic[Ca2+]rest, and present muscle damage in soleus with a strong sex bias. Mice subjected to heat stress in an enclosed 37°C chamber fail to trigger MH regardless of genotype, whereas heat stress at 41°C invariably triggers fulminant MH in Hom, but not Het, mice within 20 min. WT and Het female mice fail to maintain euthermic body temperature when placed atop a bed whose surface is 37°C during halothane anesthesia (1.75%) and have no hyperthermic response, whereas 100% Hom mice of either sex and 17% of the Het males develop fulminant MH. WT mice placed on a 41°C bed maintain body temperature while being administered halothane, and 40% of the Het females and 100% of the Het males develop fulminant MH within 40 min. Myopathic alterations in soleus were apparent by 12 mo, including abnormally distributed and enlarged mitochondria, deeply infolded sarcolemma, and frequent Z-line streaming regions, which were more severe in males. These data demonstrate that an MHS mutation within the S4-S5 cytoplasmic linker of RYR1 confers genotype- and sex-dependent susceptibility to pharmacological and environmental stressors that trigger fulminant MH and promote myopathy.—Yuen, B., Boncompagni, S., Feng, W., Yang, T., Lopez, J. R., Matthaei, K. I., Goth, S. R., Protasi, F., Franzini-Armstrong, C., Allen, P. D., Pessah, I. N. Mice expressing T4826I-RYR1 are viable but exhibit sex- and genotype-dependent susceptibility to malignant hyperthermia and muscle damage.
Keywords: Ca2+ regulation, ryanodine receptor mutation, anesthesia, heat stress
Human malignant hyperthermia (MH)-susceptible patients remain subclinical until challenged with one or more pharmacologic triggering agents, including halogenated volatile anesthetics and depolarizing neuromuscular blockers, and/or heat stress (1–3). A fulminant MH episode presents with a very rapid rise in body temperature and metabolic acidosis, progressive skeletal muscle rigidity, increased heart rate, and impaired rhythm during the perioperative period, and is often lethal if not promptly treated with dantrolene (4). The prevalence of MH susceptibility (MHS) is estimated to range from as low as 1 in 250,000 to as high as 1 in 200 for anesthetics, depending on the age and geographic location of the patient population, though accurate measures of MHS prevalence remains difficult due to variable penetrance and the poor epidemiological data (5–7). When a familial history of MH is suspected, MHS is typically diagnosed with an in vitro contracture test that measures contractile responses to halothane and/or caffeine of vastus lateralis or vastus medialis muscle biopsied from patients suspected of being MH positive (1, 8). Over 200 mutations in the gene ryanodine receptor type 1 (RYR1), primarily with autosomal dominant inheritance, have been linked to MHS and collectively account for >50% of known cases (9). RYR1 encodes the calcium release channel protein RYR1 of skeletal muscle sarcoplasmic reticulum (SR) that participates in bidirectional signaling with the dihydropyridine receptor (DHPR), which is essential for excitation-contraction (E-C) coupling (10–12).
In addition to mutations in RYR1, a small number of rare mutations in CACNAS1, which encodes the α1 subunit of the slow voltage-gated L-type Ca2+ channel, DHPR, mediate RYR1 dysregulation and have been linked to MHS (13). This is not unexpected, since DHPR and RYR1 are constituents of tightly regulated calcium release units (CRUs) that span the T-tubule-SR junctions (14, 15), and mutations in either protein could alter the bidirectional signaling between them in a manner that confers MHS and susceptibility to a rare myopathy termed central core disease (CCD).
To date, 2 knock-in mouse models that express human MHS mutations within the N-terminal domain of RYR1, R163C (16), and Y522S (17) have been described. Mice heterozygous for either mutation exhibit anesthetic- and heat stress-triggered fulminant MH episodes, whereas homozygous mice do not survive after birth. Skeletal muscle preparations isolated from these mice have revealed that these mutations not only modify important aspects of RYR1 function (18–20), but also alter several properties of L-type Ca2+ current mediated by DHPR, including causing a leftward shift in voltage dependence and altered inactivation kinetics (21, 22).
Heterozygous Y522S mice display structural abnormalities in their skeletal muscle, as they age with evidence of core-like defects in older animals (23), whereas mice with the R163C mutation, which is also associated with CCD in humans, appear to be much less affected, even in old age (unpublished results). Both MHS mouse lines exhibit evidence of mitochondrial dysfunction and oxidative stress in their skeletal muscles, although the pattern and progression of mitochondrial adaptations and RYR1 dysfunction may differ between R163C and Y522S lines (20, 24).
Much less is known about how MHS mutations within the C-terminal region of RYR1 influences RYR1 and E-C coupling function. I4898T is one of the more common mutations responsible for CCD in humans, and mice heterozygous for this mutation exhibit significant muscle weakness that stems from significantly reduced Ca2+ permeation through the affected RYR1 channels during E-C coupling (25, 26). To advance our understanding of how a MHS mutation located within the C-terminal region of RYR1 alters in vivo and in vitro aspects of muscle function, we used gene targeting to introduce the T4826I mutation that resides within the putative cytoplasmic linker between transmembrane segments S4 and S5 of RYR1 (27). T4626I confers MHS but has not been associated with a clinical diagnosis of CCD in humans (28, 29). Here, we report that, unlike N-terminal mutations studied thus far, both heterozygous RYR1T4826I/+ (Het) and homozygous RYR1T4826I/T4826I (Hom) mice are fully viable under typical rearing conditions and differ in their susceptibility to halothane and heat stress in a manner dependent on gene dose and sex. Male Het mice are significantly more sensitive than females to conditions that trigger fulminant MH. In addition to their MH susceptibility, T4826I mice develop late-onset disturbances in muscle morphology most evident in soleus, including cytoplasmic streaming, altered disposition of mitochondria, and mitochondrial swelling that were more severe in males than females. These results indicate that N- and C-terminal MHS mutations not only differ in their phenotypic and sex penetrance, but also in the onset and pattern of disturbances in muscle morphology. Although rarely documented, individuals homozygous for RYR1 mutations known to confer MHS have been identified (30–33); thus, the current findings may have significant impact on our understanding of the clinical variability of this disorder.
MATERIALS AND METHODS
Generation of T4826I-RYR1 knock-in mice
All experiments on animals from creation of MH mice to establishment of their physiological and biochemical phenotypes were conducted using protocols approved by the institutional animal care and use committees at the University of California at Davis, Harvard Medical School, and the Australian National University.
Site-directed mutagenesis (QuickChange Multi-Site-Directed Mutagenesis Kit; Stratagene, La Jolla, CA, USA) was used to mutate the threonine at codon 4826 in RYR1 cDNA to isoleucine (T4826I). A 9.5-kb EcoRI fragment harboring RYR1 exons 3–13 (Fig. 1A) isolated from a 129Sv/J mouse genomic library was used to construct the targeting vector. Specifically, the same unique KpnI site in both RYR1 cDNA and wild-type RYR1 exon 10 were used to ligate the partial cDNA fragment (after the KpnI site) to the wild-type partial exon 10 (before the KpnI site), forming an intact exon 10 followed by the rest of the whole RYR1 cDNA (Fig. 1A, B). A bacterial LoxP (locus of crossover in P1) recombination site flanked neomycin (G418) and cytosine deaminase (neo/CD) cassette were inserted after the poly(A) sequence, together with a thymidine kinase cassette placed downstream from the 3′ arm in the vector backbone as a negative selection marker to select against random integration (Fig. 1C). The vector was linearized and electroporated into W9.5 129Sv ES cells (34) and subjected to positive selection with G418 and negative selection with ganciclovir using standard techniques, as described previously (35). Polymerase chain reaction (PCR) and Southern blot analysis were used to identify homologous recombination at this location (data not shown). A clone identified as carrying the T4826I mutation was injected into C57BL/6 murine blastocysts and implanted into pseudopregnant mice. Male chimeric mice were mated with female C57BL/6 mice, and germ line transmission was confirmed by the presence of agouti coat color, PCR screening, and Southern blot analysis. These mice were then bred to Tnap-Cre (tissue-nonspecific alkaline phosphatase promoter-driven Cre recombinase) transgenic mice (36), to excise the LoxP-flanked neo/CD cassette (Fig. 1D). The resulting progeny with the neomycin cassette excised, which were used in the present study, were backcrossed >10 generations into a C57BL/6 background and shown to be congenic with WT C57BL/6 by SNP analysis.
Figure 1.

Gene targeting strategy to produce T4826I-RYR1 mouse. A) cDNA insert with T4826I mutation and SV40 polyA signal (1) and 9.5-kb genomic targeting sequence with KpnI cDNA insertion site (2). B) Targeting construct prior to addition of lox-flanked neo insertion. C) Final targeting cassette with loxP-flanked positive selection cassette and TK negative selection cassette. D) Homologously targeted genomic DNA after excision of neo cassette. See Materials and Methods for details.
Basal core temperature and responses to heat stress in vivo
Basal rectal (core) temperature was measured in awake mice at the time they were removed from their home cage (environmental range 24–26°C) using a thermistor probe and recorded digitally (TC-324B Automatic Temperature Controller extension; Warner Instruments, Hamden, CT, USA) after the reading stabilized (∼30 s). To investigate heat stress responses, mice were transferred to an enclosed test chamber equilibrated at either 37°C (WT, n=9, 8–18 mo; Het, n=14, 10–16 mo; Hom, n=14, 4–13 mo), or 41°C (WT, n=13, 2–13 mo; Het, n=18, 12–16 mo), and rectal temperature was measured immediately before and at the end of the 20-min heat stress challenge or at the time of the fulminant MH episode, whichever came first. In a subset of mice, tails were lanced to draw blood immediately before and at the end of heat stress testing (i.e., immediately on triggering MH or at the end of 20 min, whichever came first). Blood pH was measured using an AMANI-650 micro-pH electrode (0.65-mm tip diameter; Innovative Instruments, Tampa, FL, USA) calibrated to standard solutions.
In vivo halothane delivery and anesthesia response
Mice were weighed, placed into an anesthetic chamber, and induced with 2.0% halothane in oxygen using a precision vaporizer (Ohmeda Tec-4 halothane vaporizer system; GE Healthcare, Piscataway, NJ, USA) at 1.5 L/min until there was no detectable response to toe pinches (30–60 s). Next, the mice were rapidly placed atop a bed warmed by recirculating warm water to achieve a bed surface temperature of either 37.0 or 41.0°C and were instrumented with a rectal thermistor. Anesthesia was maintained with halothane, 1.75% in oxygen via nose-cone attachment.
Animals were continuously monitored for fluctuations in body temperature and signs of fulminant MH (limb and tail rigidity). Time points were recorded after every 0.1°C change in temperature, along with all data concerning physiological or behavioral activity during sedation. Anesthesia was maintained with halothane for 40 min or until onset of fulminant MH. MH was determined by close visual surveillance for hyperthermic temperatures, hyperacute rigor in extremities (limb flex response), and tachypnea followed by hyperpnea, and the cessation of respiratory function. Animals surviving anesthesia were returned to their cages and monitored closely for 20 min. At the conclusion of anesthesia recovery or MH episode, mice were euthanized. A subset of WT and Het T4826I-RYR1 mice that did not trigger after being exposed to a halothane anesthetic for 40 min were returned to their home cages and allowed a 72-h recovery period before they were reanesthetized 4 consecutive times with halothane, as described above (with a 72-h recovery between each anesthetic challenge).
Statistical analysis of temperature changes
During halothane exposure, temperature was analyzed from pooled (male+female) data and stratified according to sex, as described in the figure legends. Temperature was averaged over 15-s intervals and plotted over time. Initial temperatures were defined as the average temperature within the first 60 s of maintenance anesthesia (i.e., after induction); final temperatures were defined as the average temperature over the last 60 s of anesthesia or after the onset of fulminant MH, whichever came first. Statistical analyses between groups were performed through Prism software (GraphPad, San Diego, CA, USA) using a 2-tailed Student's t test. Values of P < 0.05 were considered significant.
In vivo assessment of myoplasmic resting Ca2+ concentration and membrane potential
Double-barreled Ca2+-selective microelectrodes were prepared and calibrated, as described previously (37, 38). Only those electrodes with a linear relationship between pCa3 and pCa8 (Nernstian response, 28.5 mV/pCa unit at 24°C) were used experimentally. To better mimic the intracellular ionic conditions, all calibration solutions were supplemented with 1 mM Mg2+. All electrodes were then recalibrated after making measurements of [Ca2+]rest, and if the two calibration curves did not agree within 3 mV from pCa 7 to pCa 8, the data from that microelectrode were discarded.
Recordings of membrane potential (Vm) and myoplasmic resting Ca2+ concentration ([Ca2+]rest) were made in vivo in mice anesthetized with nontriggering ketamine/xylazine (100/5 mg/kg) and whose core temperature maintained euthermic with an automated heating system (ATC1000; WPI, Sarasota, FL, USA). Once mice were fully sedated (lack of tail pinch response), they were intubated with a tracheal cannula and connected to a ventilator (Harvard Minivent, M-845; Harvard Instruments, Holliston, MA, USA); set at a stroke volume of 200 μl, 180 stokes/min; and ventilated with medical air. Small incisions were made to expose the vastus lateralis muscle of the left leg, and muscle fibers were impaled with the double-barreled microelectrode. Potentials were recorded via high-impedance amplifier (FD-223; WPI). The potential from the 3M KCl barrel Vm was subtracted electronically from VCaE, to produce a differential Ca2+-specific potential, VCa, that represents the [Ca2+]rest. Vm and VCa were filtered (30–50 kHz) to improve the signal-to-noise ratio and stored in a computer for further analysis. Once the body temperature recovered to 37°C baseline, Vm and VCa values were recorded, and mice were exposed to halothane (2.0%) in air for 40 min or until they triggered with fulminant MH, whichever came first. Vm and VCa values were recorded intermittently every 2 min during halothane exposure.
Assessment of skeletal muscle morphology
Animals were killed by cervical dislocation. Extensor digitorum longus (EDL) and soleus were dissected out, pinned on Sylgard at resting length, and fixed by immersion in 3.5% glutaraldehyde in 0.1 M Na cacodylate buffer (pH 7.2) at room temperature. Fixed muscles were stored at 4°C for days or up to a few months. Small bundles of fibers were then postfixed in 2% OsO4 in the same buffer for 1 h, en block stained in saturated uranyl acetate for 2 h, dehydrated, and embedded in Epon 812. Ultrathin sections (∼50–60 nm) sections were cut in a Leica Ultracut R (Leica Microsystems, Wien, Austria) using a Diatome diamond knife (Diatome, Biel, Switzerland) and stained in lead citrate. Sections were examined with a Philips 410 microscope (Philips Electron Optics, Mahwak, NJ, USA) equipped with a Hamamatsu C4742–95 digital imaging system (Advanced Microscopy Techniques, Chazy, NY, USA). For histology, 1- to 2-μm-thick sections were mounted on glass slides and examined by phase contrast optics.
RESULTS
Het and Hom T4826I-RYR1 mice are viable, but diverge in susceptibility to heat stress
Correct targeting and expression of the T4826I-RYR1 mutation was confirmed by Southern blot analysis (data not shown) and PCR (Supplemental Fig. S1). Extracted RYR1WT/WT (WT) skeletal muscle DNA (WT template) failed to amplify with T4826I PCR primers, and skeletal muscle DNA from Hom mice did not amplify with the WT PCR primers. DNA sequencing from the start of exon 10 was also used to verify correct targeting.
Unlike previously described MHS mouse lines, RYR1WT/R163C (16) and RYR1WT/Y522S (17), both Het and Hom T4826I-RYR1 mice are viable and phenotypically indistinguishable from WT-RYR1 mice when residing in their home cage. Under a standard laboratory environment, Het and Hom exhibited typical life spans (mean life span ≥2 yr), fecundity, and general physical appearance. Measurements of male and female mice in their home cages indicated that Het mice had a small but significant elevation in their basal core body temperatures compared to WT mice (Supplemental Table S1), primarily attributable to an elevated body temperature in males, rather than in the females. In Hom mice, basal temperature was significantly higher than WT mice, regardless of sex (Supplemental Table S1).
Het and Hom T4826I-RYR1 mice had divergent responses to exposure to mild to severe heat stress. Mice placed in an enclosed chamber set at 37°C for heat stress produced no mortality during or after the 20-min test period regardless of gene dose or sex (Supplemental Table S2). Although all 3 genotypes tested exhibited significantly higher rectal temperatures at the end of the 20-min exposure to heat stress when placed in an enclosed chamber at 41°C (P<0.001; Fig. 2B), only Hom mice developed fulminant MH with acidemia and skeletal muscle rigidity within 20 min of exposure (Supplemental Table S2 and Fig. 2B, C). The mean time to heat stress-induced MH for male and female HOM mice was 14.6 ± 1.8 and 17.1 ± 2.0 min, respectively (P>0.05).
Figure 2.
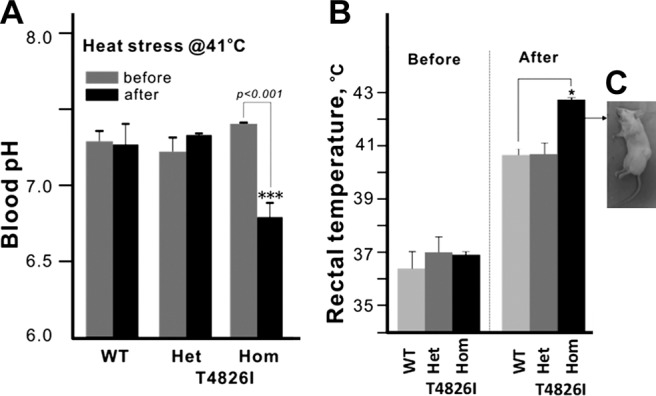
A) Blood pH of WT, HET, and HOM T4826I RYR1 animals before and after 41°C heat stress. Neither WT nor HET displayed changes in blood pH at the end of 20-min heat stress, whereas all Hom animals triggered and attained significantly lower blood pH. B) Core temperatures of HET, HOM T4826I RYR1, and WT animals before and after 41°C heat stress. Animals (n=5/genotype/sex) were placed in an enclosed heating chamber equilibrated to 41°C. T4826I Het and Hom mice had slightly higher starting temperatures compared to WT (also see Supplemental Table S1). All Hom T4826I animals died with short-term heat stress, with significantly higher ending core temperatures compared to Het T4826I and WT animals. Hom T4826I males and females both triggered and showed characteristic signs of malignant hyperthermia onset, including hyperacute rigor in extremities, tachypnea/hyperpnea, the cessation of respiratory function, and hyperthermic temperatures. C) Representative image of a T4826I-RyR1 HOM mouse that underwent MH episode following 41°C heat stress. *P <0.05; ***P <0.001.
Gene dose × sex interaction in susceptibility to halothane-triggered fulminant MH in T4826I-RYR1 mice
Considering the pronounced influence of gene dose on the susceptibility of T4826I-RYR1 mice to heat stress and the more subtle differences in basal body temperature, we investigated whether WT, Het, and Hom mice differed in susceptibility to halothane exposure. Immediately after induction of anesthesia with 2% halothane, mice were placed atop a temperature-controlled 37.0°C bed (temperature at its surface), and anesthesia was maintained at 1.75% halothane by facemask. Core body temperature was continuously monitored for up to 40 min or until the mouse had a fulminant MH episode, whichever came first. Figure 3A shows the mean temperature changes over the 40-min period of anesthesia for WT and Het male and female mice combined. In general, all WT and Het mice experienced an immediate drop in core temperature after induction of anesthesia, which continued to gradually decrease with time during the course of anesthesia when the surface of the bed temperature was set to 37°C. The top inset in Fig. 3A shows a Hom mouse immediately after fulminant MH is triggered, whereas the bottom inset shows a WT mouse that fully recovered from anesthesia (nose cone has been removed). When stratified by sex, WT and Het female mice showed a consistent decline in body temperature over time during halothane exposure (Fig. 3B). However, in WT and Het males, the mean temperature at the start and end of anesthesia did not significantly differ, primarily due to broader individual variability at the end of anesthesia in the case of Het animals (Fig. 3C). One of 6 (17%) Het males had fulminant MH and died, but all WT and the remaining Het mice tested fully recovered (Table 1). At this same bed temperature, 4 Het female mice were rechallenged with up to 4 halothane anesthetics and survived without triggering MH. By comparison, of 5 Het males rechallenged with up to 4 halothane anesthetics, 1 triggered with MH and died on the third anesthetic (data not shown).
Figure 3.
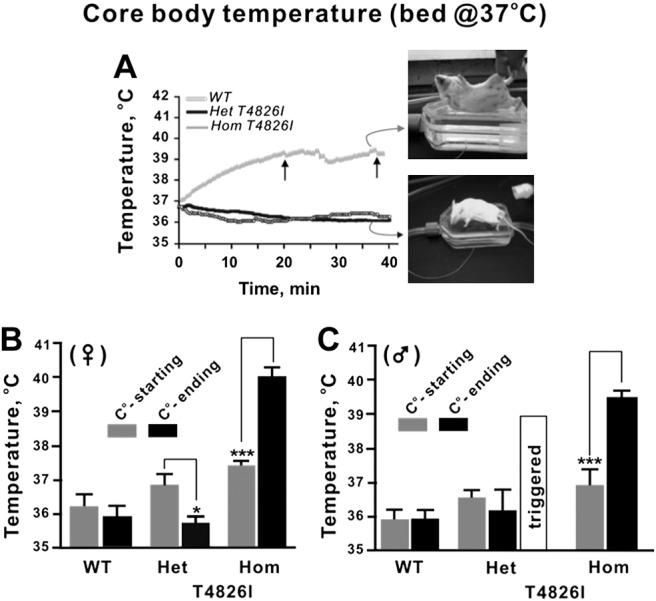
A) Temporal change in core temperature during halothane anesthesia with bed set at 37°C. Animals were induced as described in Materials and Methods and placed on a platform set to 37°C at its surface. Maintenance anesthetic was applied for up to 40 min through a nose cone. Plot shows mean core temperature continuously monitored with WT, HET, and Hom mice (sexes pooled). No WT or Het mice succumbed to MH, whereas 100% of the Hom T4826I animals experienced fulminant MH and died. Arrows indicate mice with the shortest and longest latency to MH. Mice were removed from the moving average of core temperature as they triggered. Inset: representative Hom (top image) and Het (bottom image) mice photographed on the temperature-control bed at the end of the anesthetic procedure (nose cone removed). B, C) Beginning and ending (at the end of 40 min or at the time of MH, whichever came first) core body temperatures, analyzed according to sex and genotype. Female Het T4826I mice (B) displayed reductions in core temperature following halothane exposure (35.7±0.6°C, n=8), whereas T4826I HET males (C) did not (36.5±1.7°C, n=6). Hom T4826I animals (both male and female) displayed marked increases in body temperature following halothane exposure (males, 39.4±0.6°C, n=7; females, 39.9±0.7°C, n=6). *P <0.05; ***P <0.001.
Table 1.
Susceptibility to halothane-triggered fulminant MH by genotype and sex when mice were placed on a 37°C bed (noneuthermic) and 41°C bed that maintained euthermic body temperature
| Body temperature (C°) | WT and T4826I-RYR1 mortality (%) |
|||||
|---|---|---|---|---|---|---|
| Female |
Male |
|||||
| WT | Het | Hom | WT | Het | Hom | |
| 37 | 0 | 0 | 100 | 0 | 17 | 100 |
| 41 | 0 | 40 | – | 0 | 100 | – |
In sharp contrast to Het mice, all Hom mice tested had fulminant MH and died under these test conditions, regardless of sex (Table 1). The mean onset to fulminant MH for Hom mice was 26.3 ± 7.7 min (Fig. 3A; pooled value of n=13 mice; no sex difference, P>0.05), and both sexes exhibited significantly elevated core temperatures during the MH episode compared to corresponding temperatures measured immediately after induction of halothane anesthesia (P< 0.001; Fig. 3B, C).
We further investigated a possible gene dose × sex interaction by equilibrating the bed temperature to 41°C and testing halothane-naive WT and Het T4826I-RYR1 mice of both sexes. In contrast to the influence of this level of heat stress on core temperatures produced by enclosing the mice in a chamber set at 41°C (Fig. 2), male and female WT mice placed on a bed with a surface temperature of 41°C remained euthermic for the duration of the exposure to anesthesia (Fig. 4). Under these conditions, both female and male Het T4826I-RYR1 mice had a progressive rise in their body temperatures (Fig. 4A, B) that culminated in fulminant MH in 100% of the males and 40% of the female Het mice (Table 1). The overall body temperature at the end of anesthesia was higher for males compared to females (Fig. 4C, D). However, females that triggered achieved a mean body temperature that was not significantly different from males that triggered, and the latencies to triggering MH did not differ (data not shown).
Figure 4.

Temporal change in core temperature during halothane anesthesia with bed set at 41°C. Animals were induced as described in Materials and Methods and placed on a platform set to 41°C at its surface, and maintenance anesthetic was applied for up to 40 min through a nose cone. Plot shows mean core temperature continuously monitored with WT and HET T4826I mice (sexes separated). A, B) Both female (A) and male (B) T4826I HET animals exhibited a gradual rise in core temperature and achieved a significantly higher ending core temperature following halothane anesthesia (Het T4826I 40.8±1.2°C vs. WT 37.7±0.6°C). All Het T4826I males triggered with MH and died, compared to 40% of Het T4826I females. C, D) Beginning and ending (at the end of 40 min or at the time of MH, whichever came first) core body temperatures, analyzed according to sex and genotype. C) Female T4826I HET, but not female WT, had significantly elevated body temperatures at the conclusion of halothane anesthesia compared to their initial temperatures. D) Male Het T4826I and WT animals had significantly elevated body temperature at the conclusion of halothane anesthesia compared to their initial temperature. ***P <0.001.
T4826I-RYR1 mice exhibit sex-dependent myoplasmic [Ca2+]rest before and after halothane anesthesia
We proceeded to measure myoplasmic [Ca2+]rest and Vm simultaneously using double-barreled electrodes in the vastus lateralis muscle of mice that were maintained euthermic (core at 37°C) prior to halothane exposure, as described in Materials and Methods. We found no differences in basal Vm among WT, Het, and Hom mice, regardless of sex (range of Vm=81–82±1.7 mV). During exposure to 2% halothane anesthesia, Vm in all genotype groups became less negative (mean +2–4±1.6 mV), indicating no gene-dose or sex influence. In contrast, [Ca2+]rest was sharply dependent on both gene dose and sex. As shown in Table 2, in WT animals, [Ca2+]rest (<120 nM) remained near baseline over the 40-min exposure to halothane, regardless of sex. Both male and female Het mice had basal [Ca2+]rest that was significantly higher than WT (males: [Ca2+]rest=292±14 nM; females: [Ca2+]rest=258 ± 9 nM), and [Ca2+]rest in males was significantly higher than in females (P<0.05). A clear sex difference in Het mice was also observed with respect to the rise in [Ca2+]rest during the course of halothane exposure. Males invariably triggered with fulminant MH after 22 ± 2 min, reaching a peak [Ca2+]rest of 1821 ± 66 nM (n=10 measurements in 3 mice), and all died after <25 min of exposure to halothane. However, in Het females, [Ca2+]rest did not rise to the same extent (1125±253 nM at 24 min of halothane anesthesia; n=10 measures on 2 mice), and they did not succumb to MH after 40 min of halothane exposure despite having a peak [Ca2+]rest of 1636 ± 63 nM (n = 10 measures on 2 mice). Hom mice of both sexes had nearly 3-fold higher basal myoplasmic[Ca2+]rest than WT before anesthesia, which increased rapidly, causing death in all animals within 25 min after 2% halothane exposure (Table 2).
Table 2.
Myoplasmic [Ca2+]rest (nM) for WT, Het, and Hom mice
| Time point and sex | WT | Het | Hom |
|---|---|---|---|
| Before halothane | |||
| Male | 117 ± 5 (9) | 292 ± 14 (20)*** | 345 ± 19 (15)*** |
| Female | 115 ± 3 (11) | 258 ± 9 (14)*** | 324 ± 16 (10)*** |
| After halothane (20–25 min) | |||
| Male | 117 ± 3 (5) | 1821 ± 66 (10)*** | 2276 ± 228 (10)*** |
| Female | 118 ± 4 (5) | 1125 ± 253 (10)*** | 2067 ± 132 (10)*** |
| After halothane (35–40 min) | |||
| Male | 116 ± 4 (5) | DNS | DNS |
| Female | 117 ± 4 (5) | 1636 ± 63 (10)*** | DNS |
Values are means ± sd; numbers in parentheses indicate number of blocks analyzed. DNS, did not survive.
P < 0.001 vs. WT.
Hom T4826I-RYR1 mice show noticeable structural abnormalities in male but not in female soleus muscle, nor in EDL
How the T4826I-RYR1 mutations influenced muscle structure was analyzed by electron microscopy (EM) in EDL and soleus, predominantly fast- and slow-twitch muscles, respectively. Analysis was done at 2 time points: 1 yr and 18–20 mo of age, both in Hom T4826I-RYR1 and WT-RYR1 mice. Qualitative examination allowed quick establishment of significant differences between samples: major alterations were not seen in mutant EDL, which was not further analyzed; at 1 yr, Hom T4826I-RYR1 soleus muscle presented significant structural abnormalities, but only in samples from male animals, not in females; and increasing age caused a mild worsening of the soleus structural phenotype in T4826I-RYR1 males and the appearance of some structural abnormalities in samples from female. On the basis of these initial observations, we decided to limit detailed quantitative analysis to 1 yr of age in soleus of Hom T4826I-RYR1 and WT mice (see Tables 3 and 4). This age was selected because the effects of the T4826I mutation at this stage are easily detectable and will not superimpose on structural changes that may result from normal aging (39, 40).
Table 3.
Subsarcolemmal accumulation (sSA) of mitochondira and/or amorphous material in 1-yr-old mouse soleus fibers
| Genotype and sex | Longitudinal section |
Cross section |
||
|---|---|---|---|---|
| Fiber segments occupied by sSA (%) | Fiber length segments occupied by sSA (%) | Fiber occupied by sSA (%) | sSA area (μm2) | |
| WT | ||||
| Male | 34 | 2.7 ± 4.4 | 60 | 22.9 ± 19.0 |
| Hom | ||||
| Male | 56 | 12.4 ± 15.2 | 83 | 46.3 ± 53.8 |
| Female | 38 | 3.14 ± 4.1 | 67 | 20.8 ± 17.2 |
Length and area values are means ± sd. Student's t test: WT vs. Hom male, P = 5.5 × 10−6; WT vs. Hom female, P = 0.8; Hom male vs. Hom-female P > 7.2 × 10−6. Longitudinal section: WT: 3 mice, 3 muscles, 50 fiber segments, total 10,100 μm; Hom male: 2 mice, 3 muscles, 51 fiber segments, total 13,700 μm; Hom female: 2 mice, 2 muscles, 39 fiber segments, total 10,500 μm. Cross section: WT: 2 mice, 2 muscles, 97 fiber segments; Hom male: 2 mice, 2 muscles, 106 fiber segments; Hom female: 2 mice, 2 muscles, 99 fiber segments.
Table 4.
Frequency and size of Z-line streaming areas in 1-yr-old mouse soleus fibers
| Genotype and sex | Streaming sites (n/100 μm fiber) | Streaming area size (% of total) |
||
|---|---|---|---|---|
| Small | Medium | Large | ||
| WT | ||||
| Male | 0.30 ± 0.57 | 92 | 7 | 1 |
| Hom | ||||
| Male | 0.60 ± 0.37 | 44 | 43 | 12 |
| Female | 0.25 ± 0.30 | 85 | 15 | 0 |
See text for criteria used to classify approximate size streaming areas. WT data were previously published in Boncompagni et al. (41). Hom male: 2 mice, 3 muscles, 51 fiber segments. Hom female: 2 mice, 2 muscles, 39 fiber segments. Note that even though the data for male and female Hom came from only 2 mice/group, we explored several areas of each muscle and found that the data from the 2 representative samples did not differ from each other.
Structural abnormalities most frequently observed in soleus fibers from Hom T4826I-RYR1 male mice are described in the following sections.
Redistribution of mitochondria from intermyofibrillar spaces to abnormal subsarcolemmal (or internal) regions containing amorphous material
In soleus from WT-RYR1 mice, mitochondria are distributed in 3 main populations. Intermyofibrillar mitochondria are disposed transversally at the I-band level, so that in longitudinal sections they appear as small paired profiles on either side of the Z line (Fig. 5A, small arrows and inset); these mitochondria are closely apposed to the triads. Intermyofibrillar longitudinal mitochondria (Fig. 5A, larger arrows) are often interconnected to I-band mitochondria (40, 41). Subsarcolemmal mitochondria are usually clustered in the proximity of capillaries and are predominantly rounded in appearance (Fig. 5A, asterisk).
Figure 5.

Subsarcolemmal accumulation of mitochondria and amorphous electron-dense material in soleus from Hom T4826I-RYR1 males. A) Sectioned profiles of mitochondria in WT fibers are evenly distributed on either side of the Z line (small arrows and inset) and occasionally as single longitudinally oriented elements (larger arrows). Large island of subsarcolemmal mitochondria is also seen (star). Arrowheads indicate the plasmalemma. B) In mutant fibers, peripheral accumulations of mitochondria are larger and also contain abundant amorphous electron-dense material and are in proximity of fiber regions that lack intermyofibrillar mitochondria (dashed box). C) Unusually large peripheral regions devoid of myofibrils (stars) in this light microscope image correspond to accumulations of mitochondria.
One of most notable ultrastructural alterations in soleus fibers from male Hom T4826I-RYR1 mice is a striking change in the disposition of mitochondria (Figs 5B and 6). Many of these organelles lose proper I-band positioning and migrate to subsarcolemmal areas, were they are often surrounded by amorphous electron-dense material (Figs. 5B, star; and 6, asterisks). Proliferation of T-tubules in the form of focal honeycomb networks of variable size is also frequently encountered within these regions (Fig. 6B, dotted semicircle and inset). The mitochondria in peripheral clusters are not only redistributed but also altered in size. The surface area of sectioned mitochondrial profiles in peripheral clusters is slightly smaller in Hom T4826I-RYR1 than in WT soleus muscle (0.206±0.163 and 0.233±0.151 μm, respectively, from 2 and 3 muscles; 454 and 833 mitochondrial profiles; Student's t test: P<0.001). This is confirmed by the shift in size distributions (Supplemental Fig. S2). In soleus fibers from male Hom T4826I-RYR1 mice, extensive subsarcolemmal accumulations of amorphous material and mitochondria (Fig. 5B, star) appear, in longitudinal histological sections, as large areas lacking cross striations that run for long stretches (from 10 to 200 μm in length) along the fiber edge (Fig. 5C, stars). The average frequency and size of subsarcolemmal accumulations, which affect relatively large segments of the fibers, are shown in Table 3; both parameters are greater in Hom T4826I-RYR1 males than in either Hom T4826I-RYR1 females or male WT mice. In the proximity of extended peripheral mitochondrial clusters, intermyofibrillar mitochondria may be completely absent (Figs. 5B and 6, dashed boxes). This correspondence suggests that the peripheral mitochondrial accumulations are likely the result of migration of mitochondria from the fiber interior to the periphery. Some abnormal mitochondrial clusters are also present between the myofibrils (Fig. 6A, C; arrows).
Figure 6.

Redistribution of mitochondria, and T-tubule proliferation in soleus of Hom T4826I-RYR1 males. A, B) Unusual accretions of mitochondria between the myofibrils (arrows) and under the sarcolemma (asterisks) are likely caused by migration from intermyofibrillar regions. A) Dashed box marks a region devoid of mitochondria. B) Proliferation of T-tubules in the form of focal honeycomb networks of variable size is also frequently encountered within these regions (dashed semicircle and inset). C) In cross section, a mitochondria-free region (dashed box) in proximity of subsarcolemmal (asterisk) and internal (arrows) aggregates.
Sarcolemma of soleus fibers is infolded in Hom T4826I-RYR1 male mice
A second frequent alteration found exclusively in soleus muscles from Hom T4826I-RYR1 male mice is the presence of extensive infolding of the plasma membrane that penetrates to variable depths into the fiber (Fig. 7A, arrowheads). There is a clear correspondence between the presence of infoldings and the mitochondrial alterations described above: 80% of the fiber segments showing infoldings (from 2 mice, 51 fiber segments) also present large peripheral clusters (Fig. 7, asterisks). Infoldings do not affect the entire fiber length. The frequency of segments with infoldings varies between muscles from different mice. In 2 mice, 26 and 58% of the total fibers examined (n=51) presented one or more areas with scalloped edges (Fig. 7B). The average fiber cross-sectional areas, normalized vs. sarcomere lengths, did not show any evidence of atrophy, thus excluding this as a factor in the formation of foldings.
Figure 7.

Extensive sarcolemmal infolding in soleus of Hom T4826I-RYR1 males. Deep plasma membrane infoldings (A, arrowheads) and scalloped fibers (B) are only found in Hom male soleus fibers; 80% of the fiber segments showing infoldings also present large peripheral clusters (asterisks).
Z-line streaming is frequent in Hom T4826I-RYR1 soleus fibers
Soleus muscle fibers from male Hom T4826I-RYR1 mice show also high frequency of Z-line streaming, i.e., a structural disarrangement of Z lines in elongated patches. In the extensive areas of Z-line streaming, all elements of the sarcomere are markedly altered, with a loss of thin and thick filaments, sarcomere misalignment, and a very disordered cross striation (Fig. 8A, B). In addition, as previously noted (41), all structural elements (SR/T tubules and mitochondria) are totally missing in these areas (Fig. 8B). As a result, histological sections stained for mitochondrial enzymes and cross-sections for EM at the level of a large area of streaming (Fig. 8C) would appear as cores, by the classic definition of this structural alteration. See also Boncompagni et al. (23).
Figure 8.

Extensive and frequent Z-line streaming regions in soleus of Hom T4826I-RYR1 males. A, B) In areas of Z-line streaming, i.e., a structural disarrangement of Z lines in elongated patches, all elements of the sarcomere are markedly altered, with loss of thin and thick filament, sarcomere misalignment, and a very disordered cross striation (A) and enlargement (B). In addition, all structural elements (SR/T-tubules and mitochondria) are totally missing in these areas. C) In cross sections, these alterations appear as cores, by the classic definition of this structural alteration.
For quantitative purposes, we classified Z-line streaming into 3 categories: small, affecting a limited portion of the Z line and one myofibril; medium, affecting few sarcomeres of several adjacent myofibrils; and large, affecting extended areas of the fiber with severe disruption of the whole sarcomere (see also ref. 41). In soleus of male WT RYR1Z mice, Z-line streaming sites are fairly frequent, but all of the small size. In soleus of male Hom T4826I-RYR1 mice, the overall frequency is significantly higher (Student's t test P<0.005) than in WT, with a higher percentage of medium and large disrupted areas (Table 4). Z-line streaming is associated with large subsarcolemmal accumulations and intermyofibrillar clusters of mitochondria in ∼80% of fibers (Fig. 8).
In Hom T4826I-RYR1 females at 1 yr of age, areas of medium and large streaming are much less frequent than in aged-matched Hom T4826I-RYR1 male mice. No significant difference in frequency of small streaming between Hom females and male WT (P=0.32) was detected. However, Hom T4826I-RYR1 females in 18- to 20-mo-old mice showed some large areas of streaming, although not at the frequency and severity of those in 1-yr-old males and not in association with accumulation of mitochondria.
DISCUSSION
Here, we produce, validate, and describe the phenotype of the first knock-in mouse line that expresses RyR1-T4826I, a mutation first associated with MHS in a large New Zealand Maori pedigree (28). Mice expressing this mutation contribute in several significant ways to our understanding of MHS. First and foremost is the unexpected observation that Hom mice are not only viable but survive into old age under normal rearing conditions, even in the presence of littermates that can increase social stress. In marked contrast, MHS mice expressing 2 other mutated RYR1 alleles within the N-terminal region of the protein (e.g., R163C and Y522S) do not survive past birth (16, 17). In this regard, individuals homozygous for RYR1 mutations C35R (30) and R614C (31) have been described in families with a history of MHS, and muscle biopsies indicate that homozygous individuals have increased sensitivity to halothane and caffeine compared to heterozygous individuals (31). Hom T4826I-RYR1 mice survive without overt clinical pathology, also consistent with the human condition when homozygosis has been identified. More recently a nonsynonymous substitution R3772Q-RYR1 was detected in patients of Indian and Asian descent in both the heterozygous and homozygous states (32). Second, there is a clear gene-dose contribution to responses of T4826I-RyR1 mice to pharmacologic and environmental stress that is known to trigger the fulminant MH syndrome, with Homs being significantly more sensitive than Hets to both acute heat stress and exposure to volatile anesthetic. Third, an important new finding is the existence of a delicate gene dose × sex × environmental temperature interaction that influences the probability of an individual animal progressing to a fulminant MH episode when exposed to volatile anesthetic agents. This interaction is most dramatically manifested when comparing Het males and females, which diverge significantly in their [Ca2+]rest and susceptibility to halothane anesthesia when they are kept in an environment that allows WT mice to maintain euthermic temperatures while under anesthesia. It should be emphasized that subjecting Het animals to a low or high degree of acute heat stress alone is not sufficient to trigger MH in either sex. Fourth, there are subtle through statistically significant differences in basal core body temperature in RYR1-T4826I mice of both sexes that are especially divergent in Hom males compared to WT. This is not surprising, since Het and Hom mice have chronically elevated resting cytoplasmic Ca2+ levels compared WT, which could promote mitochondrial Ca2+ overload and partial uncoupling of oxidative phosphorylation, as already demonstrated for RyR1163/+ and RyR1Y522S/+ muscle cells (18, 20, 24). In support of this possibility, maximum respiratory capacity and mitochondrial mass were found to be reduced in myotubes isolated from Het and Hom T4826I mice compared to WT (unpublished results). Finally, structural alterations are strikingly muscle and sex dependent.
The cause of the divergent penetrance exhibited by T4826I mice compared to R163C and Y522S mice is unclear. One possibility is the location of these mutations within the RYR1 sequence. Mutations that reside within or near NH2-terminal residues 590–609 (termed the DP1 region; ref. 42) are proposed to destabilize domain-domain interactions with a central region of RyR1 (residues 2350–2450), thereby destabilizing the closed state of the channel (4, 43–45). It has been suggested that dantrolene, the clinical intervention for MH, directly stabilizes these interdomain interactions (46); however, more recent findings using cryo-EM place the dantrolene-binding sequence near the FK506-binding protein-binding site but distant to the central domain (47), and the locations of the N-terminal mutations were applied to the recently obtained 2-Å crystal structure of this region (48), showing that while these mutations may interrupt interactions within this domain, any further action must be through allosteric mechanisms. Our results with the T4826I mutation indicate that the domain-domain model cannot explain how mutations outside the N-terminal region cause MH in vivo (present study and ref. 49) and inherent RYR1 channel dysregulation in vitro (18), further supporting long-range allosterism in the pathogenesis of MH. T4826I is located within the C-terminal region of RYR1, most likely near the cytoplasmic loop linking putative transmembrane segments 4 and 5 (S4-S5 linker; refs. 27, 50, 51). It has been recently suggested that T4826I contributes to an α-helical structure near the N-terminal S4-S5 linker known to occur in ion channels such as KV1.2 that are important for regulating gating of the pore (52, 53). Analysis of gating properties of single channels isolated from Het and Hom MHS mice has revealed that the T4826I mutation concomitantly destabilizes the closed state and stabilizes the open state of the channel (54), supporting this hypothesis.
Basal metabolic disturbances have been reported in skeletal muscle of both R163C and Y522Y knock-in mice (20, 24). However, the age-dependent myopathy in Y522S mice does not mimic human CCD pathology, and, surprisingly, few if any myopathic changes are seen in muscles from R163C mice (unpublished observation).
Although clinical muscle impairments or CCD have not been described in human patients heterozygous for the T4826I mutation, significant morphological abnormalities are detected in the mouse model. It is interesting that mitochondria are affected both in the T4826I mutation and in the Y522S, but in different ways. In the latter, mitochondria show clear damage and only a minor tendency toward loosening their adhesion to CRUs, while in the former, there is a clear loss of the positional relationship between mitochondria and CRU. This must imply a specific loss of the tethering links that normally hold the two organelles together (40). The observed changes in mitochondrial size distribution within the peripheral aggregates would indicate that the mitochondria either bud out or fragment.
It is important to note that no areas of contractures were found in T4826I muscles. This indicates that unless an episode is triggered, cytoplasmic calcium levels do not rise above the threshold for contractures, in keeping with the normal behavior and viability of the mice. On the other hand, the increased frequency of Z-line streaming areas relative to the wild type indicates that minor mechanical stress, of the type characteristic of eccentric contractions and/or muscle reloading (55, 56), must occasionally occur within the muscle fibers carrying the mutation. Limited occurrence of these alterations suggests that the usage pattern of the muscle may strongly influence the occurrence of such microdamage. These muscle abnormalities were more prevalent and severe in Hom mice, a finding consistent with the human condition, where homozygous individuals exhibit both MHS and myopathic alterations, whereas heterozygous individuals only present the MH phenotype (32, 57). Therefore, the T4826I mice provide a valuable model to study how gene-dose and sex influence the susceptibility to clinical and environmental triggers of MH, as well as factors that promote the onset of myopathic changes in skeletal muscle. In summary, our findings with the T4826I-RYR1 mice underscore the importance of gene × environment interactions in expression of clinical and subclinical phenotype, and suggest that individuals with RyR1 mutations may represent particularly vulnerable populations to environmental stressors.
Supplementary Material
Acknowledgments
The work was supported by U.S. National Institutes of Health grants 1P01 AR52354-05 (to C.F.A., I.N.P., and P.D.A.), 3R01 AR043140-15 (to P.D.A. and I.N.P.), and 1R01 ES014901 (to I.N.P.), and an unrestricted gift grant from the J. B. Johnson Foundation (to I.N.P.).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- CCD
- central core disease
- CRU
- calcium release unit
- DHPR
- dihydropyridine receptor
- E-C
- excitation-contraction
- EDL
- extensor digitorum longus
- Het
- heterozygous RYR1T4826I/+
- Hom
- homozygous RYR1T4826I/T4826I
- MH
- malignant hyperthermia
- MHS
- malignant hyperthermia susceptibility
- RYR1
- ryanodine receptor type 1
- WT
- wild type
- SR
- sarcoplasmic reticulum.
REFERENCES
- 1. Zhou J., Allen P. D., Pessah I. N., Naguib M. (2010) Neuromuscular disorders and malignant hyperthermia. In Miller's Anesthesia (Miller R. D., ed) pp. 1171–1196, Churchill Livingstone, Philadelphia [Google Scholar]
- 2. Jurkat-Rott K., Lerche H., Lehmann-Horn F. (2002) Skeletal muscle channelopathies. J. Neurol. 249, 1493–1502 [DOI] [PubMed] [Google Scholar]
- 3. Groom L., Muldoon S. M., Tang Z. Z., Brandom B. W., Bayarsaikhan M., Bina S., Lee H. S., Qiu X., Sambuughin N., Dirksen R. T. (2011) Identical de novo mutation in the type 1 ryanodine receptor gene associated with fatal, stress-induced malignant hyperthermia in two unrelated families. Anesthesiology 115, 938–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Parness J., Bandschapp O., Girard T. (2009) The myotonias and susceptibility to malignant hyperthermia. Anesth. Analg. 109, 1054–1064 [DOI] [PubMed] [Google Scholar]
- 5. Litman R. S., Rosenberg H. (2005) Malignant hyperthermia: update on susceptibility testing. JAMA 293, 2918–2924 [DOI] [PubMed] [Google Scholar]
- 6. Robinson R., Carpenter D., Shaw M. A., Halsall J., Hopkins P. (2006) Mutations in RYR1 in malignant hyperthermia and central core disease. Hum. Mutat. 27, 977–989 [DOI] [PubMed] [Google Scholar]
- 7. Brady J. E., Sun L. S., Rosenberg H., Li G. (2009) Prevalence of malignant hyperthermia due to anesthesia in New York State, 2001–2005. Anesth. Analg. 109, 1162–1166 [DOI] [PubMed] [Google Scholar]
- 8. Denborough M. (1998) Malignant hyperthermia. Lancet 352, 1131–1136 [DOI] [PubMed] [Google Scholar]
- 9. Carpenter D., Robinson R. L., Quinnell R. J., Ringrose C., Hogg M., Casson F., Booms P., Iles D. E., Halsall P. J., Steele D. S., Shaw M. A., Hopkins P. M. (2009) Genetic variation in RYR1 and malignant hyperthermia phenotypes. Br. J. Anaesth. 103, 538–548 [DOI] [PubMed] [Google Scholar]
- 10. Takeshima H., Iino M., Takekura H., Nishi M., Kuno J., Minowa O., Takano H., Noda T. (1994) Excitation-contraction uncoupling and muscular degeneration in mice lacking functional skeletal muscle ryanodine-receptor gene. Nature 369, 556–559 [DOI] [PubMed] [Google Scholar]
- 11. Nakai J., Dirksen R. T., Nguyen H. T., Pessah I. N., Beam K. G., Allen P. D. (1996) Enhanced dihydropyridine receptor channel activity in the presence of ryanodine receptor. Nature 380, 72–75 [DOI] [PubMed] [Google Scholar]
- 12. Buck E. D., Nguyen H. T., Pessah I. N., Allen P. D. (1997) Dyspedic mouse skeletal muscle expresses major elements of the triadic junction but lacks detectable ryanodine receptor protein and function. J. Biol. Chem. 272, 7360–7367 [DOI] [PubMed] [Google Scholar]
- 13. Carpenter D., Ringrose C., Leo V., Morris A., Robinson R. L., Halsall P. J., Hopkins P. M., Shaw M. A. (2009) The role of CACNA1S in predisposition to malignant hyperthermia. BMC Med. Genet. 10, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Protasi F., Franzini-Armstrong C., Allen P. D. (1998) Role of ryanodine receptors in the assembly of calcium release units in skeletal muscle. J. Cell Biol. 140, 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Franzini-Armstrong C., Protasi F., Tijskens P. (2005) The assembly of calcium release units in cardiac muscle. Ann. N. Y. Acad. Sci. 1047, 76–85 [DOI] [PubMed] [Google Scholar]
- 16. Yang T., Riehl J., Esteve E., Matthaei K. I., Goth S., Allen P. D., Pessah I. N., Lopez J. R. (2006) Pharmacologic and functional characterization of malignant hyperthermia in the R163C RyR1 knock-in mouse. Anesthesiology 105, 1164–1175 [DOI] [PubMed] [Google Scholar]
- 17. Chelu M. G., Goonasekera S. A., Durham W. J., Tang W., Lueck J. D., Riehl J., Pessah I. N., Zhang P., Bhattacharjee M. B., Dirksen R. T., Hamilton S. L. (2006) Heat- and anesthesia-induced malignant hyperthermia in an RyR1 knock-in mouse. FASEB J. 20, 329–330 [DOI] [PubMed] [Google Scholar]
- 18. Feng W., Barrientos G. C., Cherednichenko G., Yang T., Padilla I. T., Truong K., Allen P. D., Lopez J. R., Pessah I. N. (2011) Functional and biochemical properties of ryanodine receptor type 1 channels from heterozygous R163C malignant hyperthermia-susceptible mice. Mol. Pharmacol. 79, 420–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cherednichenko G., Ward C. W., Feng W., Cabrales E., Michaelson L., Samso M., Lopez J. R., Allen P. D., Pessah I. N. (2008) Enhanced excitation-coupled calcium entry in myotubes expressing malignant hyperthermia mutation R163C is attenuated by dantrolene. Mol. Pharmacol. 73, 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Durham W. J., Aracena-Parks P., Long C., Rossi A. E., Goonasekera S. A., Boncompagni S., Galvan D. L., Gilman C. P., Baker M. R., Shirokova N., Protasi F., Dirksen R., Hamilton S. L. (2008) RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 133, 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andronache Z., Hamilton S. L., Dirksen R. T., Melzer W. (2009) A retrograde signal from RyR1 alters DHP receptor inactivation and limits window Ca2+ release in muscle fibers of Y522S RyR1 knock-in mice. Proc. Natl. Acad. Sci. U. S. A. 106, 4531–4536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bannister R. A., Esteve E., Eltit J. M., Pessah I. N., Allen P. D., Lopez J. R., Beam K. G. (2010) A malignant hyperthermia-inducing mutation in RYR1 (R163C): consequent alterations in the functional properties of DHPR channels. J. Gen. Physiol. 135, 629–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boncompagni S., Rossi A. E., Micaroni M., Hamilton S. L., Dirksen R. T., Franzini-Armstrong C., Protasi F. (2009) Characterization and temporal development of cores in a mouse model of malignant hyperthermia. Proc. Natl. Acad. Sci. U. S. A. 106, 21996–22001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Giulivi C., Ross-Inta C., Omanska-Klusek A., Napoli E., Sakaguchi D., Barrientos G., Allen P. D., Pessah I. N. (2011) Basal bioenergetic abnormalities in skeletal muscle from ryanodine receptor malignant hyperthermia-susceptible R163C knock-in mice. J. Biol. Chem. 286, 99–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Loy R. E., Orynbayev M., Xu L., Andronache Z., Apostol S., Zvaritch E., MacLennan D. H., Meissner G., Melzer W., Dirksen R. T. (2011) Muscle weakness in Ryr1I4895T/WT knock-in mice as a result of reduced ryanodine receptor Ca2+ ion permeation and release from the sarcoplasmic reticulum. J. Gen. Physiol. 137, 43–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zvaritch E., Kraeva N., Bombardier E., McCloy R. A., Depreux F., Holmyard D., Kraev A., Seidman C. E., Seidman J. G., Tupling A. R., MacLennan D. H. (2009) Ca2+ dysregulation in Ryr1(I4895T/wt) mice causes congenital myopathy with progressive formation of minicores, cores, and nemaline rods. Proc. Natl. Acad. Sci. U. S. A. 106, 21813–21818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Du G. G., Sandhu B., Khanna V. K., Guo X. H., MacLennan D. H. (2002) Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1). Proc. Natl. Acad. Sci. U. S. A. 99, 16725–16730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brown R. L., Pollock A. N., Couchman K. G., Hodges M., Hutchinson D. O., Waaka R., Lynch P., McCarthy T. V., Stowell K. M. (2000) A novel ryanodine receptor mutation and genotype-phenotype correlation in a large malignant hyperthermia New Zealand Maori pedigree. Hum. Mol. Genet. 9, 1515–1524 [DOI] [PubMed] [Google Scholar]
- 29. Sato K., Pollock N., Stowell K. M. (2010) Functional studies of RYR1 mutations in the skeletal muscle ryanodine receptor using human RYR1 complementary DNA. Anesthesiology 112, 1350–1354 [DOI] [PubMed] [Google Scholar]
- 30. Lynch P. J., Krivosic-Horber R., Reyford H., Monnier N., Quane K., Adnet P., Haudecoeur G., Krivosic I., McCarthy T., Lunardi J. (1997) Identification of heterozygous and homozygous individuals with the novel RYR1 mutation Cys35Arg in a large kindred. Anesthesiology 86, 620–626 [DOI] [PubMed] [Google Scholar]
- 31. Rueffert H., Olthoff D., Deutrich C., Thamm B., Froster U. G. (2001) Homozygous and heterozygous Arg614Cys mutations (1840C→T) in the ryanodine receptor gene co-segregate with malignant hyperthermia susceptibility in a German family. Br. J. Anaesth. 87, 240–245 [DOI] [PubMed] [Google Scholar]
- 32. Carpenter D., Ismail A., Robinson R. L., Ringrose C., Booms P., Iles D. E., Halsall P. J., Steele D., Shaw M. A., Hopkins P. M. (2009) A RYR1 mutation associated with recessive congenital myopathy and dominant malignant hyperthermia in Asian families. Muscle Nerve 40, 633–639 [DOI] [PubMed] [Google Scholar]
- 33. Monnier N., Krivosic-Horber R., Payen J. F., Kozak-Ribbens G., Nivoche Y., Adnet P., Reyford H., Lunardi J. (2002) Presence of two different genetic traits in malignant hyperthermia families: implication for genetic analysis, diagnosis, and incidence of malignant hyperthermia susceptibility. Anesthesiology 97, 1067–1074 [DOI] [PubMed] [Google Scholar]
- 34. Szabo P., Mann J. R. (1994) Expression and methylation of imprinted genes during in vitro differentiation of mouse parthenogenetic and androgenetic embryonic stem cell lines. Development 120, 1651–1660 [DOI] [PubMed] [Google Scholar]
- 35. Campbell H. D., Fountain S., McLennan I. S., Berven L. A., Crouch M. F., Davy D. A., Hooper J. A., Waterford K., Chen K. S., Lupski J. R., Ledermann B., Young I. G., Matthaei K. I. (2002) Fliih, a gelsolin-related cytoskeletal regulator essential for early mammalian embryonic development. Mol. Cell. Biol. 22, 3518–3526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lomeli H., Ramos-Mejia V., Gertsenstein M., Lobe C. G., Nagy A. (2000) Targeted insertion of Cre recombinase into the TNAP gene: excision in primordial germ cells. Genesis 26, 116–117 [PubMed] [Google Scholar]
- 37. Eltit J. M., Li H., Ward C. W., Molinski T., Pessah I. N., Allen P. D., Lopez J. R. (2011) Orthograde dihydropyridine receptor signal regulates ryanodine receptor passive leak. Proc. Natl. Acad. Sci. U. S. A. 108, 7046–7051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eltit J. M., Yang T., Li H., Molinski T. F., Pessah I. N., Allen P. D., Lopez J. R. (2010) RyR1-mediated Ca2+ leak and Ca2+ entry determine resting intracellular Ca2+ in skeletal myotubes. J. Biol. Chem. 285, 13781–13787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boncompagni S., d'Amelio L., Fulle S., Fano G., Protasi F. (2006) Progressive disorganization of the excitation-contraction coupling apparatus in aging human skeletal muscle as revealed by electron microscopy: a possible role in the decline of muscle performance. J. Gerontol. A Biol. Sci. Med. Sci. 61, 995–1008 [DOI] [PubMed] [Google Scholar]
- 40. Boncompagni S., Rossi A. E., Micaroni M., Beznoussenko G. V., Polishchuk R. S., Dirksen R. T., Protasi F. (2009) Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell 20, 1058–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Boncompagni S., Loy R. E., Dirksen R. T., Franzini-Armstrong C. (2010) The I4895T mutation in the type 1 ryanodine receptor induces fiber-type specific alterations in skeletal muscle that mimic premature aging. Aging Cell 9, 958–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yamamoto T., El-Hayek R., Ikemoto N. (2000) Postulated role of interdomain interaction within the ryanodine receptor in Ca2+ channel regulation. J. Biol. Chem. 275, 11618–11625 [DOI] [PubMed] [Google Scholar]
- 43. Ikemoto N., Yamamoto T. (2002) Regulation of calcium release by interdomain interaction within ryanodine receptors. Front. Biosci. 7, d671–d683 [DOI] [PubMed] [Google Scholar]
- 44. Shtifman A., Ward C. W., Yamamoto T., Wang J., Olbinski B., Valdivia H. H., Ikemoto N., Schneider M. F. (2002) Interdomain interactions within ryanodine receptors regulate Ca2+ spark frequency in skeletal muscle. J. Gen. Physiol. 119, 15–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murayama T., Oba T., Kobayashi S., Ikemoto N., Ogawa Y. (2005) Postulated role of interdomain interactions within the type 1 ryanodine receptor in the low gain of Ca2+-induced Ca2+ release activity of mammalian skeletal muscle sarcoplasmic reticulum. Am. J. Physiol. Cell Physiol. 288, C1222–C1230 [DOI] [PubMed] [Google Scholar]
- 46. Kobayashi S., Bannister M. L., Gangopadhyay J. P., Hamada T., Parness J., Ikemoto N. (2005) Dantrolene stabilizes domain interactions within the ryanodine receptor. J. Biol. Chem. 280, 6580–6587 [DOI] [PubMed] [Google Scholar]
- 47. Wang R., Zhong X., Meng X., Koop A., Tian X., Jones P. P., Fruen B. R., Wagenknecht T., Liu Z., Chen S. R. (2011) Localization of the dantrolene-binding sequence near the FK506-binding protein-binding site in the three-dimensional structure of the ryanodine receptor. J. Biol. Chem. 286, 12202–12212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tung C. C., Lobo P. A., Kimlicka L., Van Petegem F. (2010) The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature 468, 585–588 [DOI] [PubMed] [Google Scholar]
- 49. Protasi F., Paolini C., Dainese M. (2009) Calsequestrin-1: a new candidate gene for malignant hyperthermia and exertional/environmental heat stroke. J. Physiol. 587, 3095–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhao M., Li P., Li X., Zhang L., Winkfein R. J., Chen S. R. (1999) Molecular identification of the ryanodine receptor pore-forming segment. J. Biol. Chem. 274, 25971–25974 [DOI] [PubMed] [Google Scholar]
- 51. Samso M., Feng W., Pessah I. N., Allen P. D. (2009) Coordinated movement of cytoplasmic and transmembrane domains of RyR1 upon gating. PLoS Biol. 7, e85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Long S. B., Campbell E. B., Mackinnon R. (2005) Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science 309, 897–903 [DOI] [PubMed] [Google Scholar]
- 53. Long S. B., Campbell E. B., Mackinnon R. (2005) Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science 309, 903–908 [DOI] [PubMed] [Google Scholar]
- 54. Feng W., Yang T., Padilla I. T., Barreintos G. C., Allen P. D., Pessah I. N. (2010) Enhanced RyR1 channel activity by the knock-in mouse that expresses human malignant hyperthermia mutation T4826I. Biophys. J. 98, 511a [Google Scholar]
- 55. Newham D. J., McPhail G., Mills K. R., Edwards R. H. (1983) Ultrastructural changes after concentric and eccentric contractions of human muscle. J. Neurol. Sci. 61, 109–122 [DOI] [PubMed] [Google Scholar]
- 56. Krippendorf B. B., Riley D. A. (1994) Temporal changes in sarcomere lesions of rat adductor longus muscles during hindlimb reloading. Anat. Rec. 238, 304–310 [DOI] [PubMed] [Google Scholar]
- 57. Zhou H., Jungbluth H., Sewry C. A., Feng L., Bertini E., Bushby K., Straub V., Roper H., Rose M. R., Brockington M., Kinali M., Manzur A., Robb S., Appleton R., Messina S., D'Amico A., Quinlivan R., Swash M., Muller C. R., Brown S., Treves S., Muntoni F. (2007) Molecular mechanisms and phenotypic variation in RYR1-related congenital myopathies. Brain 130, 2024–2036 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.