

Abstract
Cryptosporidium parasites are important waterborne pathogens of both humans and animals. The C. parvum and C. hominis genomes indicate that the only route to guanine nucleotides is via inosine 5'-monophosphate dehydrogenase (IMPDH). Thus the inhibition of the parasite IMPDH presents a potential strategy for treating Cryptosporidium infections. A selective benzimidazole-based inhibitor of C. parvum IMPDH (CpIMPDH) was previously identified in a high throughput screen. Here we report a structure-activity relationship study of benzimidazole-based compounds that resulted in potent and selective inhibitors of CpIMPDH. Several compounds display potent antiparasitic activity in vitro.
Cryptosporidiosis is a waterborne diarrheal disease caused by protozoan parasites of the genus Cryptosporidium1, 2. While Cryptosporidium hominis is specific to humans, others such as C. parvum infect humans and a wide range of animals and can be transmitted zoonotically. Cryptosporidiosis is a major cause of malnutrition in the developing world and can be life threatening in immunocompromised patients. Cryptosporium oocysts are resistant to commonly employed methods of water treatment, leading to frequent outbreaks in the developed world. In addition, oocysts are relatively easy to obtain, and therefore pose a credible biowarfare threat. No vaccines exist for Cryptosporidium infections and the approved drugs are not particularly effective. Therefore, the tools currently available to combat a massive outbreak are limited.
Like other apicomplexan parasites, Cryptosporidium is unable to synthesize purine nucleotides de novo. Instead, Cryptosporidium relies on a highly streamlined purine salvage pathway3–5. The parasite obtains adenosine from the host, which is converted sequentially to AMP and IMP. The enzyme inosine 5'-monophosphate dehydrogenase (IMPDH) converts IMP to XMP (Scheme 1). XMP is subsequently converted to GMP. Cryptosporidium does not contain guanine salvage enzymes, so this pathway appears to be the only route to guanine nucleotides.
Scheme 1.

The IMPDH reaction. R = ribose-5'-phosphate
Interestingly, Cryptosporidium acquired its IMPDH gene by lateral gene transfer from an ε-proteobacterium and consequently the enzyme is highly divergent from the host counterpart6. Thus, selective inhibition of Cryptosporidium IMPDH presents a potential strategy for treating cryptosporidiosis with minimal effects on its mammalian host7–9. The benzimidazole analog C was identified in a high throughput screen targeting the highly diverged NAD binding site of C. parvum IMPDH (CpIMPDH; this protein is identical to C. hominis IMPDH) (Figure 1).7 Compound C is a moderately potent but highly selective inhibitor for CpIMPDH (IC50 = 1.2 μM) with no detectable activity against the human IMPDH1 and IMPDH2 (IC50 > 50 μM).
Figure 1.

CpIMPDH selective inhibitor C identified by HTS
The structure of CpIMPDH in complex with IMP and the C derivative C64 was recently solved10. This structure revealed the presence of a cavity next to the aniline ring of C64, suggesting that more potent inhibitors would be obtained if the 4-bromoaniline group was replaced with bulkier groups. This information was used to guide the design of C90 and C9710. As expected, the substitution of 2-naphthyl for the 4-bromoaniline increased potency, with C90 and C97 exhibiting values of IC50 of 7–8 nM (Table 1). Herein, we report a more comprehensive structure-activity relationship (SAR) study for this class of inhibitors.
Table 1.
SAR of the aniline ring.
| Compound | X | Y | R | IC50 (nM) | IC50 (nM) BSA |
|---|---|---|---|---|---|
| C a | NH | CH2 | 4-OMePh | 1200 ± 200 | n.d. |
| C39 | NH | CH2 | 4-SMePh | 120 ±40 | 69 ± 7 |
| C43 | NH | CH2 | 4-i-PrPh | ~ 5000 | n.d. |
| C9 | NH | CH2 | 4-FPh | 900 ± 100 | n.d. |
| C10 b | NH | CH2 | 4-ClPh | 120 ± 40 | 200 ± 20 |
| C11 | NH | CH2 | 4-CF3Ph | 220 ± 40 | 330d |
| C58 | NH | CH2 | 4-CNPh | 370 ± 60 | n.d. |
| C14 b | NH | CH2 | 4-BrPh | 60 ± 30 | n.d. |
| C40 | NH | CH2 | 4-SO2MePh | ~ 5000 | n.d. |
| C45 | NH | CH2 | 4-OCF3Ph | 140 ± 50 | n.d. |
| C20 | NH | CH2 | 2-ClPh | ~ 5000 | n.d. |
| C48 | NH | CH2 | 3-ClPh | 490 ± 40 | 1430d |
| C86 b | NH | CH2 | 3,4-di-ClPh | 30 ± 10 | 90 ± 2 |
| C93 | NH | CH2 | 3-CN, 4-ClPh | 30 ± 10 | 60 ± 20 |
| C90 b | NH | CH2 | 2-Naph | 7 ± 4 | 20 ± 10 |
| C28 | NH | CH2 | 1-(4-Cl)Naph | ~ 5000 | n.d. |
| C79 | NH | CH(CH3) | 4-ClPh | 60 ± 10 | 80 ± 20 |
| C87 | NH | CH(CH3) | 3,4-diClPh | 240 ± 40 | 300 ± 70 |
| C24 | NH | CH(iPr) | 4-ClPh | ~ 5000 | n.d. |
| C18 | CH2NH | CH2 | 4-ClPh | ~ 5000 | n.d. |
The benzimidazole analogs were synthesized following the procedure outlined in Scheme 2. Various acetylamide derivatives 3 were prepared by treating substituted anilines 1 with bromo acetylchlorides, 2, in dichloromethane (DCM) and in the presence of catalytic amounts of 4-N, N-dimethylaminopyridine (DMAP). Various 2-substituted benzimidazoles 6 were prepared by condensing o-phenylene diamine 4 with aromatic aldehydes followed by oxidation in the presence of sodium metabisulfite using a slight modification of published procedures11. Finally, 2-substituted benzimidazoles were coupled with the acetylamides 3 in the presence of potassium carbonate to yield benzimidazoles 7. CpIMPDH inhibiton was measured by monitoring the production of NADH in the presence of varying inhibitor concentrations9. Inhibition was also determined in the presence of 0.05% fatty acid free bovine serum albumin (BSA) in order to evaluate the effects of non-specific binding. Gratifyingly, none of the CpIMPDH inhibitors displayed activity against human IMPDH2 (IC50 > 5 μM). Selected compounds were also evaluated for antiparasitic activity12.
Scheme 2.

General procedure for synthesizing analogs of C.
The first region of the molecule examined was the anilide substituent. Replacing the 4-methoxy of C with a thiomethyl (C39) resulted in a ten-fold increase in activity (Table 1). However, a branched aliphatic group (C43) at the same position resulted in decreased activity. Interestingly, replacing the 4-methoxy with electron withdrawing groups (C9 – C11, C58, C14, C45) resulted in compounds with increased activity, except for sulfone C40. Larger more hydrophobic groups such as chlorine (C10) and bromine (C14) were best. Translocation of the chlorine from the 4-position to either the 3- or 2- positions (C20, C48) was detrimental. Several compounds containing electron withdrawing groups in the 3- and 4-positions (C86 and C93) also demonstrated potent inhibitory activity. Surprisingly, addition of a chlorine to the 2-naphthyl (C28) was not tolerated. Introduction of a methyl onto the methylene between the amide carbonyl and the imidazole resulted in increased potency in one case (C79 vs. C10), but decreased activity in another case (C87 vs. C86). Increasing the steric bulk of the methyl group to i-Pr (C24) was detrimental. Finally, inserting a methylene between the amide NH and the phenyl ring (C18) was not tolerated.
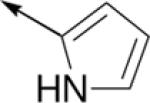
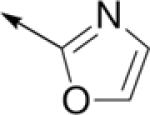
Subsequently, the SAR of the 4-thiazolyl ring was examined (Table 2). As reported previously, changing the connectivity to a 2-thiazolyl increased activity for several analogs (C61 vs. C10, C64 vs. C14, C74 vs. C79) and retained potent activity for another analog (C97 vs. C90)10. The 5-thiazolyl was also comparatively active (C67 vs. C61). In addition, several other heterocycles (C62, C100, C16) also retained potent activity. However, the 2-pyrrolyl (C65) and 2-oxazolyl (C69) derivatives demonstrated reduced potency. Likewise, replacing the thiazole ring by various phenyls (C17, C31, C59) or a methyl (C38) resulted in significant losses in activity.
Table 2.
SAR of thiazole ring.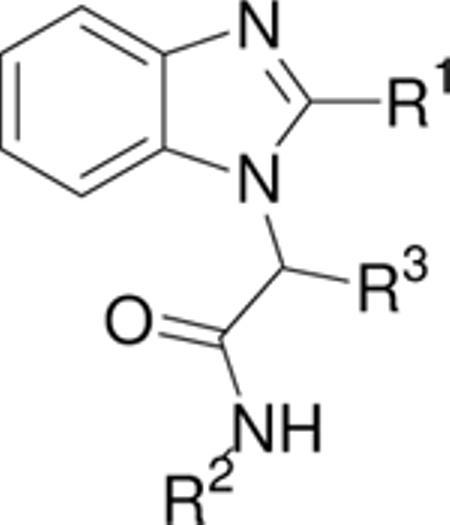
| Compound | R1 | R2 | R3 | IC50 (nM) | IC50 (nM) BSA |
|---|---|---|---|---|---|
| C a |
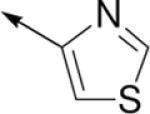
|
4-OMePh | H | 1200 | n.d.c |
| C61 b |

|
4-ClPh | H | 30 ± 10 | 50 ± 10 |
| C64 b | 4-BrPh | H | 28 ± 9 | 27d | |
| C74 | 4-ClPh | Me | 23 ± 4 | 30 ± 6 | |
| C84 b | 3,4-diClPh | H | 18 ± 5 | 50 ± 20 | |
| C97 b | 2-Naph | H | 8 ± 3 | 20 ± 20 | |
| C67 |

|
4-ClPh | H | 35 ±9 | 60d |
| C62 |
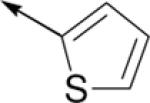
|
4-ClPh | H | 20 ±20 | 60d |
| C100 |

|
4-ClPh | H | 35 ±8 | 42d |
| C16 |
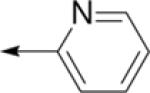
|
4-ClPh | H | 43 ± 9 | 90 ± 30 |
| C85 | 3,4-diClPh | H | 22 ± 5 | 40 ± 7 | |
| C91 | 2-Naph | H | 8 ± 3 | 14 ± 5 | |
| C92 | 3-CN, 4-ClPh | H | 22 ± 10 | 40 ± 10 | |
| C65 |
|
4-ClPh | H | 80 ±10 | 120d |
| C69 |
|
4-ClPh | H | 170 ±10 | 230 ± 10 |
| C17 | Ph | 4-ClPh | H | 210 ±30 | 280 ± 30 |
| C31 | 4-ClPh | 4-ClPh | H | 450 ± 20 | n.d. |
| C59 | 4-FPh | 4-ClPh | H | 870 ± 20 | 1300d |
| C38 | Me | 4-ClPh | H | ~5000 | n.d. |
In order to further analyze the SAR results, select molecules were docked using GLIDE (Schrödinger Inc.) into a CpIMPDH model based on the previously determined co-crystal structure. Free energy perturbation (FEP) calculations were then determined (calculated as ΔΔG relative to inhibitor C) and the results were highly correlated (r2 = 0.93) to the observed IC50 determinations (Table 3, Figure 2)13. Inhibitor potency appears to be driven largely by two major contributions: (1) a hydrogen bond between E329 of CpIMPDH and the amide NH of the inhibitors; (2) an entropic effect of displacing water molecules from the binding cavity by large hydrophobic substituents. The presence of strong electron withdrawing groups on the arylamide increases potency by increasing the strength of the E329-NH H-bond provided no steric clashes are present. Thus the balance between conformational state and electron withdrawing ability appears critical for determining the final potency of the inhibitors. For example, 3,4-dichloro substituted analog C86 is predicted to be more potent than the 3-chloro analog C48. For the 2-chloro analog C20 a steric clash is predicted to change the orientation of the phenyl ring lowering the inductive effect of the chlorine substituent weakening the E329-NH bond. For the 2-naphthyl analog C90, displacement of ordered water molecules from the active site of the protein is entropically favored resulting in ΔΔG relative to C of −5.87 kcal/mol and an IC50 value of 7 nM.
Table 3.
Relative affinity of CpIMPDH inhibitors based on docking experiments.13
| Compound | ΔΔG relative to C (kcal/mol) |
|---|---|
| C | 0 |
| C39 | −2.49 |
| C43 | 4.8 |
| C10 | −2.34 |
| C48 | −1.39 |
| C20 | 3.33 |
| C86 | −4.12 |
| C90 | −5.87 |
| C40 | 3.99 |
| C11 | −1.51 |
| C28 | 5.77 |
Figure 2.

Correlation of calculated relative affinity with experimental values.
Compounds with a value of IC50 less than 30 nM were candidates for testing in a Toxoplasma gondii model of C. parvum infection12. Preference was given to compounds that displayed little non-specific binding as judged by changes in the value of IC50 in the presence of BSA. Wild type T. gondii expresses a eukaryotic IMPDH that is resistant to the CpIMPDH inhibitors. In contrast, in the T. gondii/CpIMPDH model parasite, the endogenous IMPDH gene has been replaced with CpIMPDH. In addition, the gene encoding hypoxanthine-guanine-xanthine phosphoribosyltransferase was knocked out, so this strain relies on the activity of CpIMPDH to obtain guanine nucleotides. Both T. gondii strains express yellow fluorescent protein enabling easy monitoring of parasite proliferation. T. gondii strains were cultured in human foreskin fibroblasts immortalized with hTERT, so this assay also reports on host cell toxicity. Compounds C64, C84, C90, C91 and C97 all displayed sub-micromolar activity against T. gondii/CpIMPDH (Table 4). C64 and C97 displayed selectivity • 30 versus the wild-type strain, strongly indicating that antiparasitic activity results from the inhibition of CpIMPDH.
Table 4.
Antiparasitic activity of selected compounds.
| Compound | T. gondii modela | C. parvum model | ||
|---|---|---|---|---|
| EC50(μM) | Selectivity | |||
| Toxo/WT | Toxo/CpIMPDH | EC50 (μM) | ||
| C64 | >23 | 0.3 ± 0.1 | >73 | 0.7 ± 0.2c |
| C84 | 3 ± 2 | 0.7 ± 0.3 | 5 | 1.7 ± 0.8c |
| C90 | 5 ± 1 | 0.6 ± 0.1 | 9 | 0.9 ± 0.5 |
| C91 | 2.7 ± 0.9c | 0.3 ± 0.2 | 9 | n.d. |
| C97 | 17 ± 9 | 0.5 ± 0.4 | 30 | < 0.8d |
All values are the average of three independent trials unless otherwise stated.
T. gondii model12. Toxo/WT, strain with endogenous IMPDH; Toxo/CpIMPDH, strain that depends on CpIMPDH. Selectivity = EC50(Toxo/WT)/EC50(Toxo/CpIMPDH);
C. parvum in vitro infection model;
two determinations;
Average growth inhibition 80 ± 10 % at 0.8 μM.
Compounds C64, C84, C90 and C97 were also tested in an in vitro model of C. parvum infection12. Importantly, all four compounds are approximately two orders of magnitude more potent than paromomycin, the standard control for anticryptosporidial activity (literature paromomycin EC50 values are 65–130 μM7, 12, 14–16). The potencies of C64, C84, C90 and C97 were similar to that observed in the T. gondii model (Table 4).
In conclusion, a SAR study of benzimidazole-based CpIMPDH inhibitors revealed that variations to the aniline and to the heterocycle attached to the 2-position of the benzimidazole could be altered in order to increase inhibitory activity, while retaining excellent selectivity over human IMPDH2. The benzimidazole-based CpIMPDH inhibitors described herein could serve as useful molecular probes for studying C. parvum and other related organisms in addition to providing lead compounds for the development of effective treatments of cryptosporidiosis.
Supplementary Material
Acknowledgments
This work was supported by funding from the National Institute of Allergy and Infectious Diseases (U01AI075466) to LH. GDC thanks the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NERCE/BEID), and the Harvard NeuroDiscovery Center for financial support. BS is a Georgia Research Alliance Distinguished Inverstigator. IC50 data for these compounds are maintained using ChemAxon, http://www.chemaxon.com/.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Fayer R. Vet Parasitol. 2004;126:37. doi: 10.1016/j.vetpar.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 2.Huang DB, White AC. Gastroenterol Clin North Am. 2006;35:291. doi: 10.1016/j.gtc.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 3.Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, Buck GA, Xu P, Bankier AT, Dear PH, Konfortov BA, Spriggs HF, Iyer L, Anantharaman V, Aravind L, Kapur V. Science. 2004;304:441. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- 4.Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, Puiu D, Manque P, Akiyoshi D, Mackey AJ, Pearson WR, Dear PH, Bankier AT, Peterson DL, Abrahamsen MS, Kapur V, Tzipori S, Buck GA. Nature. 2004;431:1107. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]
- 5.Striepen B, Pruijssers AJ, Huang J, Li C, Gubbels MJ, Umejiego NN, Hedstrom L, Kissinger JC. Proc Natl Acad Sci U S A. 2004;101:3154. doi: 10.1073/pnas.0304686101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Jr., Liu C, Abrahamsen MS. Proc Natl Acad Sci U S A. 2002;99:6304. doi: 10.1073/pnas.092525699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. Chem Biol. 2008;15:70. doi: 10.1016/j.chembiol.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. J Biol Chem. 2004;279:40320. doi: 10.1074/jbc.M407121200. [DOI] [PubMed] [Google Scholar]
- 9.Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. J Med Chem. 2009;52:4623. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacPherson IS, Kirubakaran S, Gorla SK, Riera TV, D'Aquino JA, Zhang M, Cuny GD, Hedstrom L. J Am Chem Soc. 2010;132:1230. doi: 10.1021/ja909947a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang D, Fokas D, Li J, Yu L, Baldino CM. Synthesis. 2005:47. [Google Scholar]
- 12.Sharling L, Liu X, Gollapalli DR, Maurya SK, Hedstrom L, Striepen B. PLoS Negl Trop Dis. 2010;4:e794. doi: 10.1371/journal.pntd.0000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Protein data base (PDB) file 3KHJ was used to generate a model for the protein bearing the missing side chains, loops and hydrogen atoms. The loop positions were refined using PRIME, IMP and C64 were modeled and the overall structure was minimized. GLIDE (Schrödinger Inc.) was used to calculate a grid around C64 with a single positional constraint on the carbon attached to the benzimidazole nitrogen. The grid was restricted to a size similar to C64. The inhibitor structures were converted to 3-D coordinates, pre-processed using ligprep to generate tautomers and ionization states at pH 2.0 – 7.0 and stored as sd files. GLIDE calculations were carried out in standard precision mode with the same single constraint. GLIDE automatically generated a pool of conformation for each inhibitor (~5000 each) and the best pose was chosen based on total number of productive interactions (both Van der Walls and electrostatic) with the protein. No water molecules were included in this calculation. The structural overlay of the docked inhibitors showed < 1.5 Å deviation from C64 indicating that GLIDE was able to reproduce the conformation of the inhibitors in the active site of CpIMPDH. For FEP calculations, DESMOND molecular dynamics engine (D.E Shaw Research and Schrödinger Inc.) with the OPLS2005 force field was used. In the current setup, a 22-window scheme was adopted for both A (WT) and B (B) states to achieve reasonably high accuracy. A simulation production time of 44 ns (2 ns × 22 windows) was used for all the calculations and the jobs were run on an in-house 24 node 3.4 GHz Beowulf cluster. Inhibitor C (IC50 = 1200 nM) was used as the initial structure ( =0) to generate other structure in the series using alchemical FEP mutations.
- 14.Perkins ME, Wu TW, Le Blancq SM. Antimicrob Agents Chemother. 1998;42:843. doi: 10.1128/aac.42.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woods KM, Nesterenko MV, Upton SJ. FEMS Microbiol Lett. 1995;128:89. doi: 10.1111/j.1574-6968.1995.tb07505.x. [DOI] [PubMed] [Google Scholar]
- 16.You X, Arrowood MJ, Lejkowski M, Xie L, Schinazi RF, Mead JR. FEMSMicrobiol Lett. 1996;136:251. doi: 10.1111/j.1574-6968.1996.tb08057.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.