

Abstract
ErbB2 and ErbB3 receptor tyrosine kinases are key regulators of proliferation, migration, differentiation and cell survival; however, their roles in gastrointestinal biology remain poorly defined. We hypothesized that ErbB2 and ErbB3 promote colon epithelial cell survival in the context of the wound healing response following colitis. In this study, mice bearing intestinal epithelial-specific deletion of ErbB2 or ErbB3 were treated with dextran sulfate sodium (DSS). Colon sections were examined for injury, cytokine expression, epithelial cell proliferation and apoptosis. Deletion of epithelial ErbB2 did not affect the extent of intestinal injury in response to DSS, while deletion of ErbB3 slightly increased injury. However, the roles of both receptors were more apparent during recovery from DSS colitis in which ErbB2 or ErbB3 epithelial deletion resulted in greater inflammation and crypt damage during the early reparative period. Moreover, loss of ErbB3 prevented normal epithelial regeneration in the long-term with damage persisting for at least 6 weeks following a single round of DSS. Delayed recovery in mice with epithelial deletion of ErbB2 or ErbB3 was associated with increased colonic expression of TNF-α and increased epithelial apoptosis. Furthermore, epithelial ErbB3 deletion increased apoptosis at baseline and during DSS injury. Additionally, epithelial cell hyperproliferation during recovery was exacerbated by deletion of either ErbB2 or ErbB3. These results suggest that ErbB2 and ErbB3 play important cytoprotective and reparative roles in the colonic epithelium following injury by promoting colon epithelial cell survival.
Keywords: apoptosis, colitis, ErbB2, ErbB3, proliferation, TNF-α
Growth factors play significant roles in maintaining epithelial integrity through the regulation of intestinal epithelial cell migration, proliferation and cell survival1, 2. Disruption of intestinal epithelial integrity contributes to the pathogenesis of several gastrointestinal disorders, including inflammatory bowel disease. The ErbB family of receptor tyrosine kinases, consisting of ErbB1 (EGFR), ErbB2 (Her2), ErbB3 (Her3), and ErbB4 (Her4), can be activated by direct interaction with ligands or by forming heterodimers with other ErbBs, leading to increased kinase activity and downstream signal transduction. The ErbB family members are potential therapeutic targets as evidenced by the protective effects of EGF in experimental colitis and in clinical trials with ulcerative colitis3, 4. However, the direct influence of individual ErbBs on intestinal injury and repair in vivo is relatively not well defined.
ErbB2 has been extensively studied in many types of cancers5; however, determination of its role in intestinal development and epithelial response to injury and inflammation has been hampered by the fact that ErbB2 deletion is embryonic lethal due to both cardiac and neural defects6. Although no ligands have been identified for ErbB2, its activation can be induced by heterodimerization with ligand-occupied EGFR, ErbB3 or ErbB47. ErbB2 has been implicated in mediating myoblast cell survival and in inhibiting cancer cell apoptosis8, 9. In addition, our group has shown that ErbB2 is transactivated by tumor necrosis factor alpha (TNF-α), which in turn protects intestinal epithelial cells from TNF-α-induced apoptosis10.
Interestingly, increased ErbB3 expression has been found in many tumors that overexpress ErbB211, 12. In contrast to ErbB2, ErbB3 binds numerous ligands including neuregulin; however, ErbB3 lacks intrinsic kinase activity and downstream signal transduction relies on heterodimerization with other ErbBs. For example, the ErbB2-ErbB3 dimer is crucial for ErbB2-mediated proliferation in ErbB2-overexpressing tumors13. ErbB3 signaling likely plays an important role in cell survival given that there are six putative PI3 kinase binding sites within its C-terminal domain14. Indeed, ErbB3 silencing by siRNA promotes apoptosis in lung adenocarcinoma cells15. However, delineation of the physiological role of ErbB3 has been difficult since, like ErbB2, ErbB3 deletion also causes embryonic lethal cardiac and neural defects16. ErbB3 deletion in the intestinal epithelium sensitizes the colonic epithelium to dextran sulfate sodium (DSS)-induced colitis17; however, the mechanism by which ErbB3 protects from injury and the effect of ErbB3 on intestinal epithelial recovery following injury are not known.
Numerous studies have implicated ErbB ligands and EGFR in the protection against DSS-induced colitis18–20. Since EGFR and ErbB2 promote intestinal epithelial cell survival in vitro and in vivo in the presence of TNF-α, we hypothesized that ErbB2 and ErbB3 protect the intestinal epithelium during injury and inflammation in vivo. DSS causes a chemically induced colitis21, 22, with direct cytotoxic insult to the colon epithelium. This results in a well-characterized pattern of increased apoptosis, subsequent disruption of crypt structure, followed by severe inflammation22, 23 and extensive proliferation to restore the epithelial barrier23. Using this model, we studied the role of ErbB2 and ErbB3 in injury/repair responses using mice with intestinal epithelial cell-specific ErbB2 or ErbB3 deletion. Our data show that intestinal epithelium-specific ErbB3 knockout mice develop worse colitis than controls and that ErbB2 and ErbB3 both play important roles in epithelial recovery following injury. Deletion of ErbB2 or ErbB3 increases colonic tumor necrosis factor alpha (TNF-α) production, epithelial apoptosis, and worsens colitis. In addition, deletion of ErbB3 resulted in histological evidence of sustained colitis for at least six weeks following a single DSS insult. These findings provide important insights into the specific roles of ErbB2 and ErbB3 in vivo in the regulation of crypt survival and epithelial recovery following colonic injury.
Materials and Methods
Mice and DSS Treatment
All animal procedures were approved by the Vanderbilt Institutional Animal Care and Use Committee. ErbB2flox/flox (FVB background) or ErbB3flox/flox (129 background) mice were crossed with villin-Cre24 (C57BL/6) mice to generate constitutive intestinal epithelium-specific ErbB2 knockout (ErbB2 KOIEC) or ErbB3 knockout mice (ErbB3 KOIEC)8, 17. Littermate ErbB2flox/flox (wild-type) or ErbB3flox/flox (wild-type) mice were used as control mice. ErbB3flox/flox mice were also crossed with villin-Cre ERT224 (C57BL/6) mice to generate tamoxifen-inducible ErbB3flox/flox/Cre ERT2 mice. Intestinal epithelium-specific ErbB3 knockout mice (ErbB3 KOIEI) were obtained by giving 6–8 week old ErbB3flox/flox/Cre ERT2 mice tamoxifen (1 mg/mouse, i.p., Sigma Chemical Co, St Louis, MO) for five days, as reported24. ErbB3flox/flox/Cre ERT2 mice receiving vehicle only (sunflower oil, Sigma) were used as controls (wild-type) for inducible ErbB3 deletion. Experiments began four weeks following tamoxifen to allow for efficient Cre-mediated recombination of the floxed ErbB3 allele.
To induce colitis, mice received DSS (36–50 kDa; MP Biomedicals, Solon, OH) dissolved in water (3 % w/v) for 4–8 days, as indicated. Control groups received normal drinking water without DSS. Mice were either sacrificed immediately after DSS treatment or allowed to recover for the indicated times on normal drinking water. All groups contained balanced numbers of age-matched male and female mice.
Epithelium Isolation and Western Blot Analysis
Epithelial cell isolation and western blot analysis was performed as previously described25. Colons were cut into 4–5 mm pieces and incubated in Cell Recovery Solution (BD Biosciences, San Jose, CA) at 4 °C overnight followed by manual shaking to release the epithelial fraction; the stromal fraction consisted of the remaining tissue. Western blotting against E-cadherin was used to determine the purity of epithelial and stromal fractions. Tissue lysates were made with RIPA buffer26 (1% Triton X-100, 1mM EDTA, 10mM Tris, pH 7.4, 150 mM NaCl, 0.1% SDS, 0.2% sodium deoxycholate) with protease and phosphatase inhibitors (Sigma Chemical Co, St. Louis, MO). Cell lysates were subjected to SDS-PAGE and transferred onto nitrocellulose membranes. Membranes were blocked with milk, incubated with primary antibodies at 4°C overnight, and then incubated with horseradish peroxidase-conjugated secondary antibodies. Membranes were developed using Western Lighting (Perkin Elmer, Waltham, MA).
Histological Scoring
The extent of injury and inflammation was scored by a gastrointestinal pathologist blinded to genotype and treatment. After the mice were sacrificed, colons were removed, opened longitudinally, rolled from the distal to proximal end and fixed in 10% neutral-buffered formalin; paraffin-embedded sections (5 μm) were then prepared and H&E stained as previously reported25. Histological scores were evaluated using a protocol described by Dieleman et al27 that quantifies colitis according to the following five categories in which each category has a score range from 0–3: the amount of inflammation, depth of inflammation, percentage of the crypts involved by inflammation, crypt damage, and the percentage of crypts involved by crypt damage. The total injury and inflammation score is the sum of the individual scores assigned to each category according to severity and extent of colitis as we have previously reported25.
Real-Time PCR analysis
Total RNA was isolated from whole colon tissue using a Qiagen RNA isolation kit (Qiagen, Valencia, CA) following the manufacturer’s instructions, including DNase digestion. One microgram of RNA sample was then reverse transcribed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystem, Foster City, CA). Quantitative real-time PCR was performed on an iCycler iQ5™ system (Bio-Rad, Hercules, CA, USA) using SYBR green PCR Master Mix (Bio-Rad, Hercules, CA, USA) for TNF-α, or on an ABI7000 system (Applied Biosystems, Foster City, CA) using a primer and probe set (Assay ID: Mm99999071_m1; Applied Biosystems) for IFN-γ. Actin expression was used as internal reference in all PCR experiments. The PCR primer sequences used were as follows: TNF-α, 5′-CTGTGAAGGGAATGGCTGTT-3′ and 5′-GGTCACTGTCCCAGCATCTT-3′; actin, 5′-GAAGCATTTGCGGTGGACGAT-3′ and 5′-CCAGGTCATCACCATTGGCAA-3′.
Apoptosis
Apoptotic cells were detected within tissue samples using terminal deoxynucleotidyl transferase-mediated dNTP nick-end labeling (TUNEL) as previously reported28. Briefly, 5 μm paraffin-embedded colon sections were deparaffinized, rehydrated and then pretreated with proteinase K (20 μg/ml) for 15 minutes at room temperature, followed by incubation with 3% H2O2 for 5 minutes to quench endogenous peroxidase. Apoptotic cells were labeled using a TUNEL assay kit according to the manufacturer’s instructions (ApopTag, #S7100; Millipore) and sections were counterstained with hematoxylin. TUNEL positive cells were counted in a blinded fashion and expressed as the number of apoptotic cells per 100 colonic crypts as we have previously reported29.
Immunohistochemistry
Slides were stained using previously described approaches25. Paraffin-embedded colon sections were deparaffinized, rehydrated and subjected to antigen retrieval by boiling in either citrate buffer (Vector Labs) or target solution (DAKO). Sections were then blocked with 10% goat serum (Zymed, Carlsbad, CA) and stained with rabbit anti ErbB2 (Abcam, Cambridge, MA) or rat anti Ki67 antibody (DAKO, for determining epithelial cell proliferation) as previously reported.25 Slides were incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-rat antibodies and developed with DAB substrate (Sigma Chemical Co, St. Louis, MO). Ki67 positive-staining epithelial cells in 30 crypts from distal colon for each mouse were counted in a blinded fashion and expressed as the number of proliferating cells per crypt.
For colonic expression of ErbB3, 5 μm frozen colon sections were fixed in methanol (5 minutes at −20°C) and blocked in 1% BSA/0.2% nonfat dry milk in PBS for 20 minutes at room temperature. Sections were stained with antibody against ErbB3 (Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C overnight and then with rhodamine-conjugated anti-rabbit antibody at room temperature for 30 minutes. Slides were mounted with aqueous solution containing DAPI to counterstain nuclei. ErbB3 expression was visualized with standard fluorescence microscopy.
Statistics
Statistical significance for injury scores, mRNA transcript expression, and proliferative and apoptotic indices were determined using the nonparametric Mann-Whitney test. Body weights were analyzed using unpaired Student’s t tests. Two-sided P values are reported for all tests and P values less than 0.05 were considered significant. Statistical analyses were performed using GraphPad Prism software (GraphPad Software Inc., San Diego, CA).
Results
Deletion of colonic epithelial ErbB2 delays colon epithelial recovery following DSS-induced colitis
ErbB2 mediates cell survival in addition to its more established roles in regulating cell migration, proliferation and differentiation8, 10, 15. Since global deletion of ErbB2 leads to embryonic lethality6, 16, we used intestinal epithelium-specific ErbB2 (ErbB2 KOIEC) knockout mice to test the hypothesis that ErbB2 protects the intestinal epithelium from injury and promotes epithelial recovery following injury in experimental colitis. ErbB2 KOIEC mice were generated by crossing ErbB2flox/flox (wild-type) mice with mice expressing a constitutively-active Cre recombinase driven by the villin promoter. Intestinal epithelium-specific deletion of ErbB2 was confirmed by western blot analysis of isolated epithelial cells and by immunohistochemistry (Figure 1A); ErbB2 protein was efficiently deleted in the colonic epithelium, but not in the stroma of ErbB2 KOIEC mice. There were no changes in the expression of EGFR or ErbB3 in the ErbB2 KOIEC colon epithelium or stroma (data not shown).
Figure 1. Deletion of colon epithelial ErbB2 or ErbB3 expression in ErbB2 KOIEC and ErbB3 KOIEI mice.

Epithelial and stromal fractions were isolated from colons of the indicated mice. (A and B) Western blot and immunohistochemical analysis with antibodies against ErbB2 (A) or ErbB3 (B). Omission of primary antibody was used as a control for immunohistochemistry. Actin expression was used as loading control. Brown indicates ErbB2 staining. Red indicates ErbB3 staining, blue indicates DAPI staining. Scale bars, 50 μm. These results are representative of independent analyses of at least three different mice in each group.
In contrast to global ErbB2 knockout mice, ErbB2 KOIEC mice survived normally and exhibited no obvious phenotypic changes. Furthermore, these mice showed normal architecture of the epithelium in the small intestine (data not shown) and colon (Figure 2A). ErbB2 KOIEC mice also displayed no basal defects in epithelial cell proliferation or migration (data not shown).
Figure 2. Epithelial ErbB2 is required for short-term recovery from DSS-induced colitis.

ErbB2 KOIEC and wild-type controls were treated with 3% DSS for 4 or 7 days (A, C, E), or were treated with 3% DSS for 4 days followed by a recovery period of 3 days or 6 weeks on normal water (B, D, F); n=6–12. (A, B) Representative H&E sections of colons from the indicated mice. Scale bars, 100 μm. (C, D) Colonic injury scores were quantified by a pathologist blinded to the treatment and group. Bars indicate mean value. (E) Body weight loss during DSS administration; values indicate mean±SD. (F) Body weight loss during the 3 day recovery period following 4 days of DSS administration; values indicate mean±SD.
Administration of DSS induces superficial mucosal erosions mostly through direct cytotoxic damage to the colonic epithelium leading to mucosal inflammation21, 22, 27, and has been widely used to delineate the requirement of specific gene products and/or signaling pathways in response to tissue damage and inflammation25, 30. Therefore, we used this model to dissect the role of epithelial ErbB2 in intestinal injury and repair following injury. ErbB2 KOIEC mice received either water alone or 3% DSS for 4 d to induce colitis. After DSS treatment, wild-type and ErbB2 KOIEC mice showed a similar degree of colonic injury and inflammation (Figure 2A and Figure 2C). During this 4 d DSS treatment, neither ErbB2-deleted mice nor their wild-type controls exhibited colitis-induced weight loss.
To assess whether loss of ErbB2 alters the response to a more severe injury, we then extended the length of DSS treatment. ErbB2 KOIEC mice treated with DSS for 7 d showed about 12 % body weight loss (Figure 2E), after which treatment was discontinued to prevent extensive morbidity; in contrast, during this period, wild-type controls did not lose any body weight. After 7 d of DSS, wild-type and ErbB2-deleted mice each demonstrated extensive crypt damage and inflammation (Figure 2A and 2C); although there was an apparent increase in the ErbB2 KOIEC injury scores, this difference did not reach statistical significance. However, since the ErbB2 knockout mice lost more body weight than wild-type controls during prolonged DSS treatment, this indicates that epithelial ErbB2 may contribute to protection from colitis that is not reflected in our injury score data.
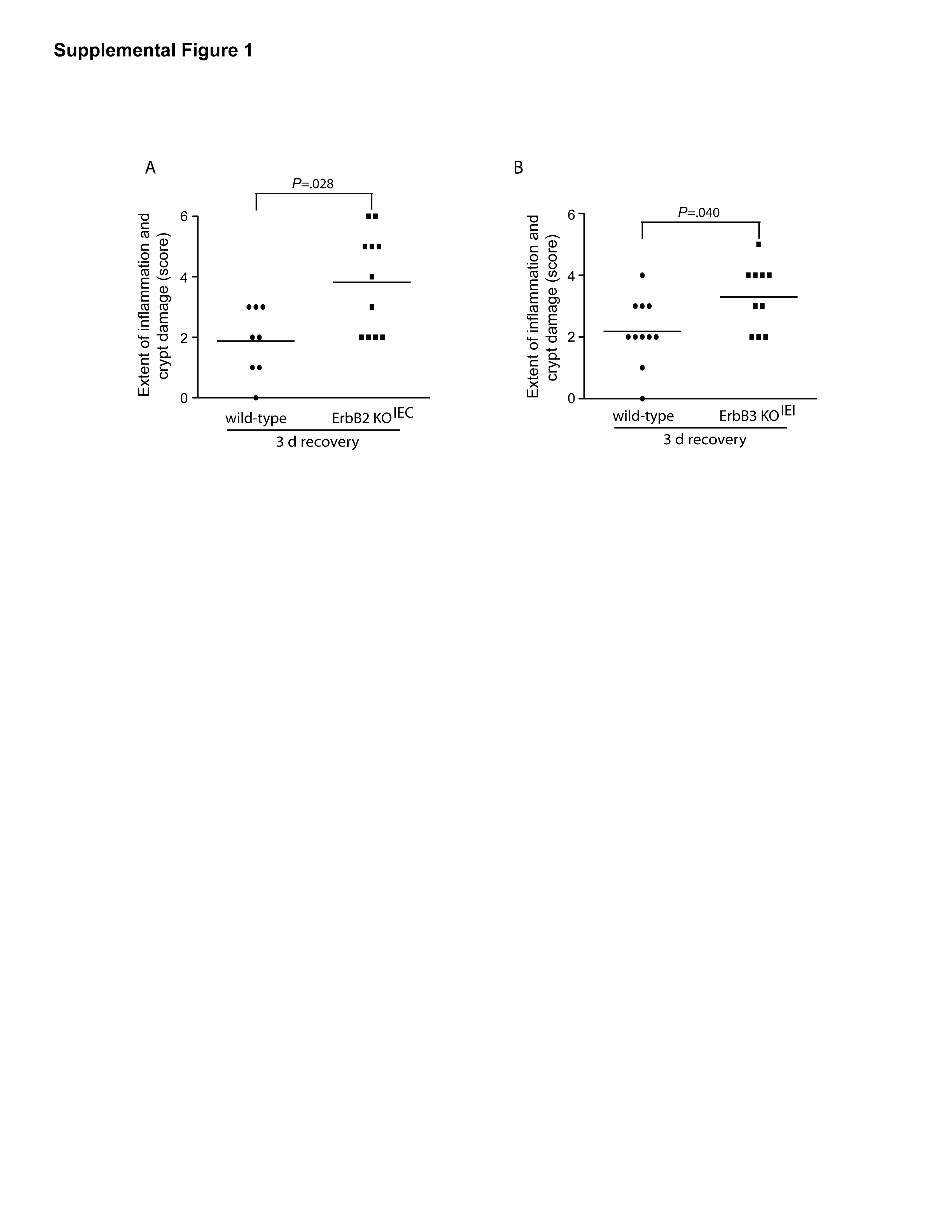
To determine if epithelial ErbB2 plays a role in epithelial reestablishment following injury, ErbB2 KOIEC mice were allowed to recover on normal drinking water for a 3 d period following 4 d of DSS treatment. Although no histological difference was observed between wild-type and ErbB2 KOIEC mice in 4 d DSS-induced injury (Figure 2A and 2C), in this short-term recovery period, ErbB2 KOIEC mice exhibited exacerbated colitis with significantly increased injury and inflammation scores compared to wild-type controls (Figure 2B and 2D). Indeed, the extent of injury during this recovery period in ErbB2 KOIEC mice was greater and involved most of the mid- and distal colon; in fact, some specimens displayed a complete denudation of the epithelium with full-thickness mucosal inflammation throughout the majority of the length of the colon. In contrast, although wild-type mice during this recovery period also displayed epithelial injury and mucosal inflammation, this occurred with a patchy distribution, sparing much of the mid colon. The percentage of the colon involved with crypt damage and inflammation was significantly higher in recovering ErbB2 KOIEC mice compared to control mice (Supplemental Figure 1). Interestingly, the body weight of ErbB2 knockout mice decreased after DSS withdrawal and was significantly lower during the second and third day of recovery than that of wild-type mice (Figure 2F). These results suggest that epithelial ErbB2 is crucial in the regenerative response of the epithelium following injury, with its absence leading to heightened inflammatory responses during recovery.
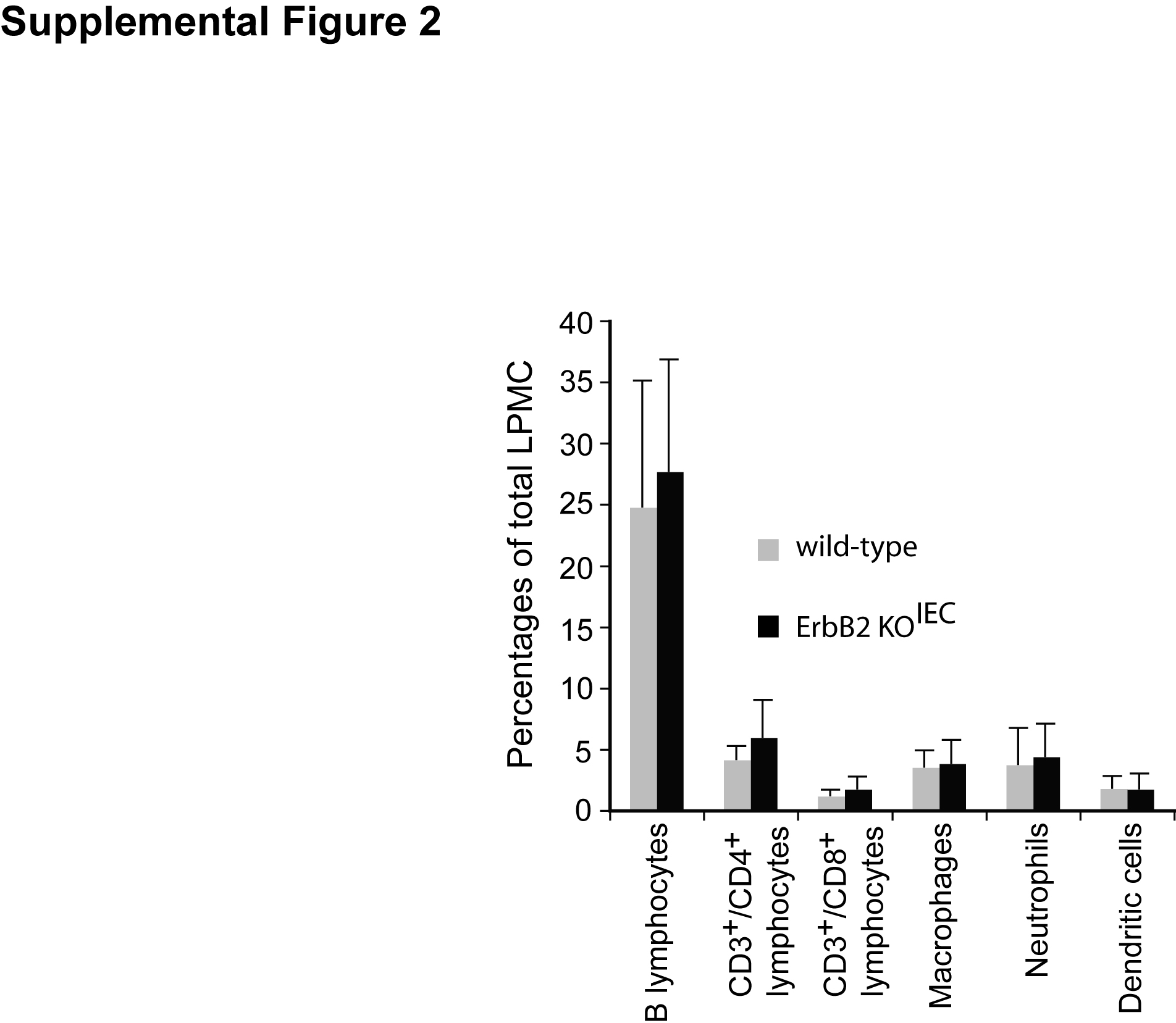
Since DSS treatment induced a significant inflammatory response in recovering ErbB2 knockout mice, we determined whether epithelial ErbB2 deletion altered the phenotype of the immune response. However, flow cytometry analysis failed to detect any alterations in immune cell populations in the lamina propria of ErbB2 KOIEC mice when compared to wild-type controls (Supplemental Figure 2).
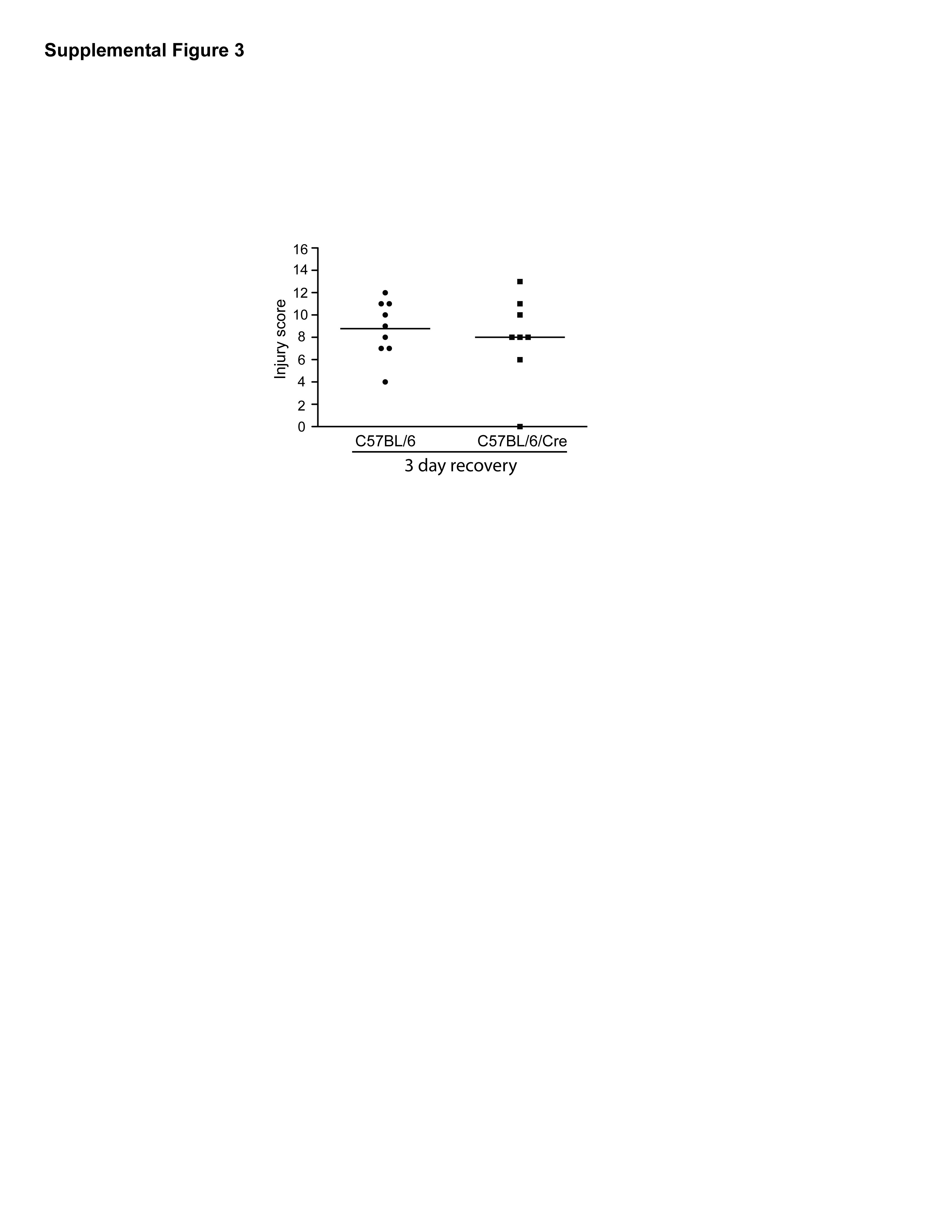
Furthermore, to rule out the possibility that Cre recombinase expression itself resulted in increased sensitivity to DSS, we treated mice carrying the villin-Cre recombinase alone with 3% DSS for four days and then allowed the mice to recover for three days. No differences were observed in these mice compared to non-Cre expressing control mice during recovery phase as shown in Supplemental Figure 3. These data suggest that ErbB2 expression contributes to epithelial recovery in response to DSS-induced acute injury.
Given this exacerbated injury and inflammation following an initial DSS insult, we determined if this abnormal restitution in ErbB2 epithelial-null mice would lead to chronic changes. Thus, we extended the DSS recovery period to 6 wk in ErbB2 KOIEC mice and wild-type controls following a single insult of 4 d of DSS. Both wild-type and ErbB2 KOIEC mice showed complete weight recovery after DSS withdrawal for six weeks (not shown). Histological analysis indicated that the colons of ErbB2 KOIEC mice and controls had very mild colonic injury, with no differences observed between these two genotypes (Figure 2B and 2D). Thus, although the data suggest that epithelial ErbB2 was critical for short-term recovery from DSS injury, it appears to be dispensable during a longer recovery period.
Deletion of ErbB3 in colonic epithelium exacerbates DSS-induced injury and impairs recovery
Since global deletion of ErbB3 also leads to embryonic lethality6, 16, we used inducible intestinal epithelium-specific ErbB3 (ErbB3 KOIEI) knockout mice to determine the role that ErbB3 plays in DSS-induced colitis. ErbB3 KOIEI mice were generated by crossing ErbB3flox/flox (wild-type) mice with mice expressing a tamoxifen-inducible Cre recombinase under control of the villin promoter. Tamoxifen-induced intestinal epithelium-specific deletion of ErbB3 was confirmed as shown in Figure 1B; there were no changes in the expression of EGFR or ErbB2 (not shown). Deletion of epithelial ErbB3 did not cause defects in architecture of the epithelium in the small intestine (data not shown) or colon (Figure 3A), as well as basal epithelial cell proliferation or migration (data not shown).
Figure 3. Epithelial ErbB3 is required for short-term and long-term recovery from DSS-induced colitis.

ErbB3 KOIEI (A, B, C, Di, E, F) or ErbB3 KOIEC (B, Dii) mice and their respective wild-type controls were treated with 3% DSS for 4 or 8 days (A, C, E), or were treated with 3% DSS for 4 days followed by a recovery period of 3 days or 6 weeks on normal water (B, D, F), as indicated; n=7–13. (A, B) Representative H&E sections of colons from the indicated mice. Scale bars, 100 μm. (C, D) Colonic injury scores were quantified by a pathologist blinded to the treatment and group. Bars indicate mean value. (E) Body weight loss during DSS administration; values indicate mean±SD. (F) Body weight loss during the 3 day recovery period following 4 days of DSS administration; values indicate mean±SD.
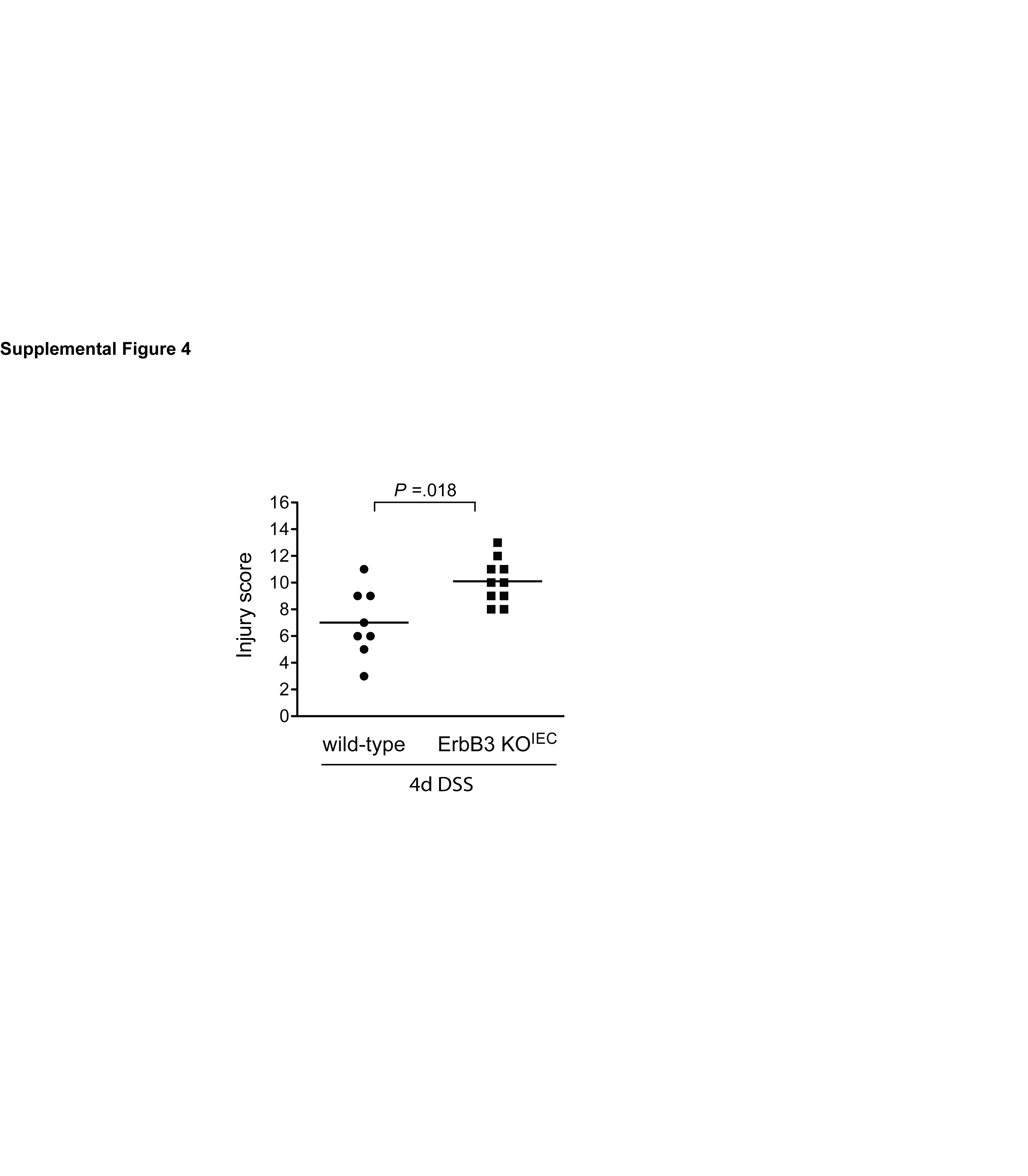
For DSS-induced colitis, ErbB3 KOIEI mice received either water alone or 3 % DSS for 4 d to induce injury, 4 wk following tamoxifen-induced ErbB3 deletion. After this DSS treatment, ErbB3 KOIEI mice showed slightly more histological damage compared to controls, although this did not reach statistical significance using conservative nonparametric methods (Figure 3A and 3C). However, ErbB3 KOIEI mice showed increased inflammatory infiltrate, greater depth of inflammation, and a greater distribution of inflammation throughout the colon following acute injury versus wild type controls. Neither genotype lost body weight during 4 d DSS treatment (Figure 3E). To confirm that these results were specific to the role of epithelial ErbB3 in acute DSS injury in mice, we also studied constitutive deletion of epithelial ErbB3 (ErbB3 KOIEC). In agreement with results published by Lee et al.17, ErbB3 KOIEC mice displayed greater histological injury following 4 d DSS treatment than wild-type controls (Supplemental Figure 4).
As with the ErbB2 KOIEC mice, we also tested the response of ErbB3 KOIEI mice to extended DSS treatment. ErbB3 KOIEI mice and wild-type controls were treated with DSS for 8 d; during this period ErbB3 KOIEI mice lost 10 % of their starting body weight, while wild-type controls maintained normal body weights (Figure 3E). Although both genotypes displayed extensive colonic injury, there was no difference in their injury scores following prolonged DSS treatment (Figure 3A and 3C).
We then allowed ErbB3 KOIEI mice to recover on normal drinking water for a 3 d period following 4 d of DSS treatment to determine the role of ErbB3 in epithelial recovery after injury. In this short-term recovery period, significantly more severe damage was seen in ErbB3 KOIEI mice when compared to wild-type controls (Figure 3B and 3Di). As with the ErbB2 KOIEC mice, the extent of injury during this recovery period in the ErbB3 KOIEI mice was greater and involved most of the colon with epithelial denudation and full-thickness mucosal inflammation. In addition, ErbB3 KOIEI mice lost significantly more body weight than wild-type mice following DSS withdrawal (Figure 3F). These data provide evidence that epithelial ErbB3 is an important regulator of colonic epithelial repair following injury.
Given this exacerbated injury and inflammation in ErbB3 KOIEI mice during initial repair from a DSS insult, we tested whether ErbB3 regulates long-term recovery. Thus, we allowed wild-type and ErbB3 knockout mice to recover for 6 wk after a single round of 4 d of DSS treatment. To study the long-term effect of ErbB3 deletion on recovery from DSS-induced colitis, we used ErbB3 KOIEC mice. After 6 wk recovery, ErbB3 KOIEC mice had significantly higher injury and inflammation scores when compared to wild-type littermate controls (Figure 3B and 3Dii). Initially, we had also used the inducible ErbB3 knockout (ErbB3 KOIEI) for these studies. Although the tamoxifen-induced ErbB3 KOIEI mice initially exhibited significantly slower weight recovery following DSS withdrawal (Figure 3F), after six weeks, ErbB3 KOIEI mice only had a moderate, but similar degree of colitis to that of wild-type mice (Supplemental Figure 5). Given that inducible loss of ErbB3 caused such an extensive loss of the epithelium during the 3 d recovery period, this raised the possibility that there might be a strong selective pressure for rare ErbB3-expressing epithelial cells (i.e. not recombined during prior tamoxifen treatment) during this 6 wk recovery period. Indeed, Western blot analysis indicated that epithelial ErbB3 expression was partially restored 6 wk following DSS treatment in ErbB3 KOIEI mice (Supplemental Figure 5), whereas water-treated ErbB3 KOIEI mice did not show restoration of epithelial ErbB3 during the same time period. This suggests that selection for epithelial ErbB3-expressing cells may account for normal regeneration in these mice. Taken together, these data show that ErbB3 plays a critical role in regeneration following DSS-induced injury.
Increased cytokine expression ErbB2 KOIEC and ErbB3 KOIEI during recovery from DSS injury
DSS-induced colitis involves the release of proinflammatory cytokines such as TNF-α and IFNγ by infiltrating immune cells in the lamina propria, which in turn further worsen subsequent injury31. Therefore, we measured inflammatory cytokines in the colon during recovery in ErbB2 KOIEC and ErbB3 KOIEI mice. While mRNA levels of IL-1β and IL-6 were not changed (data not shown), there were significantly higher mRNA levels of TNF-α and IFN-γ in ErbB2 KOIEC mice following 3 d recovery (Figure 4A and 4B). Furthermore, in ErbB3 KOIEI mice, mRNA levels of TNF-α were also increased compared to controls following this 3 d recovery period (Figure 4C). Both ErbB2 KOIEC and ErbB3 KOIEI mice showed redistribution of the tight junctional protein, ZO-1 in colonic sections during the recovery period from DSS colitis (Supplemental Figure 6). Taken together, these data are consistent with increased histological inflammation seen in recovering ErbB2 KOIEC and ErbB3 KOIEI mice.
Figure 4. Colonic TNF-α expression is increased in ErbB2 KOIEC and ErbB3 KOIEI mice during recovery from DSS-induced colitis.

TNF-α (A, C) and IFN-γ (B, D) mRNA transcript levels were measured by real-time RT-PCR from whole colon RNA of the indicated genotypes and treatment groups. Bars indicate mean expression of transcripts normalized for actin; n=4–7.
Colonic epithelium of ErbB2 KOIEC and ErbB3 KOIEI mice retain proliferative capacity during recovery from DSS injury
During recovery from acute injury, colon epithelial cells undergo extensive proliferation to reestablish the damaged epithelium23. We examined crypt proliferation to determine whether epithelial-specific ErbB2- and ErbB3-null colons retained the capacity to repopulate the epithelium during the recovery phase following DSS-induced injury using immunohistochemistry for the proliferative marker Ki67. Deletion of ErbB2 or ErbB3 did not affect basal levels of proliferation in unchallenged mice (Figure 5). In contrast, following 4 d DSS treatment, there was a significant decrease in the level of proliferation in ErbB3 KOIEI compared to wild-type controls (Figure 5D). Despite this fact, this effect was relatively small and the ErbB3-null epithelium retained approximately 75 % the proliferative capacity of wild-type controls. The effect on proliferation was most dramatic during the recovery period, in which loss of either ErbB2 or ErbB3 significantly increased rather than decreased the level of epithelial proliferation (144 % and 145 % increase compared to wild-type controls, respectively). It is likely that this increased proliferation is an effect secondary to increased injury in these mice during recovery. Therefore, although the ErbB2- and ErbB3-null epithelium experiences more extensive damage during recovery from DSS-induced injury, these findings suggest that this is not likely due to an impairment in epithelial proliferation, per se, and moreover raises the possibility that ErbB2 and ErbB3 regulate epithelial regeneration by promoting cell survival.
Figure 5. Loss of ErbB2 or ErbB3 exacerbates epithelial hyperproliferative response during recovery from DSS-induced colitis.

(A, B) Immunostaining for the proliferative marker Ki67 (brown nuclei) in colonic sections of ErbB2 KOIEC (A), and ErbB3 KOIEI mice (B) and their respective wild-type controls treated with water, DSS for 4 days or DSS followed by a 3 day recovery period, as indicated. Images are representative of 8–12 mice per group. Scale bars, 100 μm. (C, D) The number of Ki67-positive cells per crypt were quantified in a blinded manner. Bars indicate mean values.
ErbB2 and ErbB3 are differentially required to prevent colon epithelial cell apoptosis
Since apoptosis is a pathological feature of DSS colitis22, 23, we hypothesized that the increased severity of crypt damage and inflammation in ErbB2- and ErbB3-null mice may result from decreased epithelial cell survival. We therefore measured basal and DSS-induced apoptosis in the remaining intact crypts in ErbB2- and ErbB3-null mice during the injury and recovery period using TUNEL staining. In unchallenged mice, epithelial deletion of ErbB2 had no effect on the level of epithelial apoptosis (Figure 6A and 6C), while ErbB3 deletion increased basal epithelial apoptosis (Figure 6B and 6D) even without DSS injury. Following 4 d DSS injury, all genotypes (ErbB2 KOIEC, ErbB3 KOIEI and their respective control mice) showed an increased rate of apoptosis compared to water-treated mice. However, during this injury period, only ErbB3 expression had an effect on apoptosis with a significantly increased apoptotic rate in the ErbB3-null epithelium compared to wild-type mice controls (327 % increase compared to wild-type controls; Figure 6B and 6D). This appears to be consistent with the more significant effect of ErbB3 deletion on short-term DSS injury compared to that of ErbB2 deletion (Figure 3 and Supplemental Figure 4). Interestingly, apoptotic rates were significantly increased by either ErbB2 or ErbB3 deletion during the recovery period (267 % and 258 % increase compared to controls, respectively; Figure 6A–D). Again, these data are also consistent with histopathologic findings that ErbB2- and ErbB3-null mice had extensive injury specifically during this recovery period (Figures 2 and 3). These data demonstrate that ErbB2 and ErbB3 protect against colitis through promoting epithelial cell survival, and that ErbB3 regulates epithelial apoptosis basally, during injury and during recovery, while ErbB2 is important only during recovery from injury.
Figure 6. Loss of ErbB2 or ErbB3 increases colonic epithelial apoptosis.

(A, B) Apoptotic cells (arrows) in colon sections were detected by TUNEL staining from wild-type or knockout mice treated with water, DSS for 4 days or DSS followed by a 3 day recovery period, as indicated. Images are representative of 8–12 mice per group. Scale bars, 50 μm. (C, D) The number TUNEL-positive cells per 100 crypts were quantified in a blinded manner. Bars indicate mean values.
Discussion
In this study, using mice with intestinal epithelium-specific deletion of ErbB2 or ErbB3, we uncovered a direct reparative role for colon epithelial ErbB2 and ErbB3 following DSS-induced injury and a lesser role for ErbB3 in regulating the extent of the injury itself. We show that ErbB2 KOIEC and ErbB3 KOIEI mice had markedly reduced recovery following DSS-induced colitis, most likely due to increased epithelial cell apoptosis and increased TNF-α production. Interestingly, a single round of DSS treatment in ErbB3 KOIEC mice resulted in long-term histological alterations, with a failure to resolve inflammation and injury by six weeks following DSS administration. This study is the first to uncover the roles of ErbB2 and ErbB3 as important reparative factors following colonic injury.
Immediately following DSS administration, neither ErbB2 KOIEC nor ErbB3 KOIEI mice exhibited increased injury; however, both genotypes presented with a greater loss of body weight than controls (Figures 2 and 3). However, we did observe a small protective role for epithelial ErbB3 during this injury period since ErbB3 KOIEC mice (constitutive deletion of ErbB3) had greater injury scores (Supplemental Figure 4), which confirms previous findings from Lee et al17. Moreover, deletion of ErbB3 in ErbB3 KOIEI mice (inducible deletion of ErbB3 in adult mice) did result in increased epithelial cell apoptosis and decreased cell proliferation during DSS injury despite there being no differences in injury scores. The reasons for this discrepancy between the two different lines of epithelial ErbB3 knockout may involve strain-specific differences or compensatory changes in other factors resulting from embryonic deletion of ErbB3 in ErbB3 KOIEC mice. Nevertheless, we conclude that epithelial ErbB3, but not epithelial ErbB2, plays a small protective role from colonic injury during DSS administration, likely by promoting epithelial cell survival and the maintenance of cell proliferation.
Importantly, we found that epithelial ErbB2 and ErbB3 are important reparative factors during recovery from injury. For these studies, we made a distinction between short-term (i.e. 3 days) and long-term (i.e. 6 weeks) recovery periods following a single round of DSS administration. Epithelial ErbB2 was required for normal short-term recovery from colitis, and ErbB2 KOIEC mice showed elevated injury scores, cytokine expression, hyperproliferation and epithelial apoptosis during this period. However, the effect of epithelial ErbB2 knockout was confined to the acute period; following 6 weeks of recovery, we found no histological differences between wild type and ErbB2 KOIEC mice. Thus, epithelial ErbB2 appears to be important for optimal early recovery of the epithelium from injury, but may be dispensable for long-term regeneration. However, it is important to note that the model used in this study involved only a single inflammatory insult; upon repeated injury or chronic injury, such as that which occurs in inflammatory bowel disease patients, the role of ErbB2 in ongoing epithelial repair could be more important.
While epithelial ErbB3 appeared to play a small protective role in determining the extent of injury during DSS administration, we found that it was required for optimal regeneration both in the short-term (3 days) and in the long-term (6 weeks). Mice with inducible ErbB3 deletion (ErbB3 KOIEI) had increased colonic injury, TNF-α expression, hyperproliferation and epithelial apoptosis during short-term recovery from DSS. Moreover, the extent of this injury was much greater than wild-type controls and involved much of the colon with areas of complete epithelial ulceration. Although epithelial ErbB2 knockouts showed a similar phenotype during this period, loss of ErbB2 increased TNF-α and IFN-γ transcripts; in contrast ErbB3 deletion only affected TNF-α expression. Interestingly, in the long-term recovery period, we found that expression of epithelial ErbB3 in the tamoxifen-inducible strain (ErbB3 KOIEI) was restored, which we conclude contributed to the observed normal regeneration in these mice (Supplemental Figure 5). Indeed, this type of strong selective pressure for the restoration of wild-type intestinal epithelial cells following inducible deletion has been reported by others32. In contrast, the permanent and constitutive deletion of epithelial ErbB3 (ErbB3 KOIEC) produced substantial long-term changes in epithelial recovery, with injury and inflammation that persisted throughout this 6-week period. These differences emphasize the importance of ErbB3 in long-term regeneration, since it appeared that a strong selective pressure was involved in the reestablishment of ErbB3 expression in the inducible strain.
ErbB2 and ErbB3 are implicated in cell proliferation, particularly in breast tumor cells13. During recovery from DSS colitis, epithelial cells undergo extensive hyperproliferation to regenerate damaged crypts and repopulate the denuded epithelium23. Although we observed a significant decrease in proliferation in ErbB3 KOIEI mice relative to wild-type controls after 4 days DSS administration, this decrease may be related to the greater degree of crypt damage and/or a greater degree of epithelial apoptosis in these mice during injury (Figure 6), indirectly leading to a decrease in epithelial progenitors during this injury phase. For instance, it has been reported that DSS preferentially affects stem cells and progenitor cells in the base of the crypt28; thus this reduction in proliferation may be due to increased apoptosis of progenitor cells caused by DSS in an ErbB3-null epithelium.
Elevated apoptosis is important in the etiology of colon disorders and is seen in both ulcerative colitis patients and in the mouse DSS colitis model23, 28, 33. Indeed, epithelial apoptosis appears to be one of the mechanisms through which DSS induces colitis. Increased apoptosis leads to a disruption of the intestinal barrier and increased exposure of lamina propria immune cells to luminal antigens; in turn, subsequent colonic inflammation further increases epithelial injury34–36. Apoptotic rates were comparable between ErbB2 KOIEC and wild-type mice in the unchallenged state and following DSS injury. However, during epithelial recovery, ErbB2 KOIEC mice had higher apoptotic rates than those of littermate controls. In addition, there was a dramatic reduction in the number of surviving crypts that could support proliferation in the recovery phase in ErbB2 KOIEC and ErbB3 KOIEI mice compared to their wild-type controls. Thus, during this early recovery period, increased cell death in epithelial cells leads to more severe colitis and early recovery failure in ErbB2 KOIEC mice. Our previous report demonstrated that transactivation of ErbB2 by TNF-α is required for colonic epithelial survival following exposure to TNF-α10. Indeed, TNF-α levels are elevated in both DSS-induced colitis and in inflammatory bowel disease patients; furthermore, anti-TNF-α antibodies reduce colitis progression37. In our study, we confirmed that TNF-α transcripts were increased in recovering ErbB2 and ErbB3 knockout mice. Thus, one potential mechanism for the delayed short-term recovery following DSS withdrawal in ErbB2 KOIEC mice is due to increased TNF-α-induced apoptosis in the absence of ErbB2 transactivation.
Surprisingly, inducible deletion of ErbB3 resulted in increased epithelial cell death in unchallenged mice. In a recent study, Lee et al. showed that constitutive deletion of ErbB3 in the colon epithelium did not alter basal intestinal epithelial cell survival17. Our studies involved inducible deletion of ErbB3 in adult mice with a mature gastrointestinal system. In contrast, constitutive villin-Cre constructs used in their study delete ErbB3 starting from embryonic day 924. One possibility for this discrepancy is that early embryonic deletion of ErbB3 may lead to developmental compensation by other ErbB family members, which may explain the differences in our respective studies.
Although loss of ErbB3 increased injury during DSS administration, the protective role of epithelial ErbB3 was even more apparent during recovery in which ErbB3 KOIEI colons displayed greatly exaggerated injury and apoptosis. Furthermore, these changes failed to resolve in ErbB3 KOIEC mice over a 6-week period. These data suggest that the ErbB3-deleted intestinal epithelium shows different patterns of DSS-induced toxicity than that with ErbB2 deletion. Although these studies were performed in mice with different genetic backgrounds, when compared to their respective wild-type controls, epithelial ErbB3 appeared to play a greater role in regulating levels of apoptosis basally, and during injury and short- and long-term recovery. A mechanistic reason may be that ErbB3 is a potent activator of PI3 kinase since it possesses multiple PI3 kinase binding sites; since this is known to be an important regulator of cell survival5, 14, this may explain why ErbB3 is more potent than ErbB2 to prevent apoptosis. Another mechanism through which ErbB3 deletion resulted in severe crypt damage and inflammation might be that the ErbB3-deleted intestinal epithelium is more sensitive to TNF-α-induced toxicity compared to that containing wild type ErbB3. Our group has shown that TNF-α can activate EGFR, ErbB2 as well as ErbB4 to promote epithelial cell survival and knock down of any of these ErbBs significantly potentiates TNF-α-induced apoptosis10, 38. Activation of ErbB3 depends on heterodimerization with other ErbBs; thus, it is also possible that increased TNF-α-induced transactivation may also promote ErbB3-regulated signaling pathways. Indeed, the ErbB2-ErbB3 heterodimer is the most potent signaling pair amongst the ErbB heterodimers5, and ErbB3 is the favored heterodimerization partner for ErbB239. Cell death following the loss of ErbB3 may therefore represent a higher sensitivity of the epithelium to TNF-α-induced apoptosis. Therefore, our data suggest that ErbB2 and ErbB3 each contribute complementary yet non-redundant roles in regulating intestinal injury and repair.
In summary, the results of our study demonstrate that ErbB2 and ErbB3 are important regulators of colonic epithelial cell responses playing a critical role in optimizing intestinal recovery from injury. These findings expand the known range of activities of EGFR family receptor tyrosine kinases, suggesting that strategies designed to activate ErbB2 and ErbB3 may be of therapeutic value in promoting recovery following colon injury.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Grant Support: Supported by NIH grant R01DK056008 (to D.B.P), a Research Fellowship Award from the Crohn’s and Colitis Foundation of America (to Y.Q.Z) a Fellowship from the Canadian Institutes of Health Research (to P.E.D.), and the Vanderbilt Digestive Diseases Research Center (VDDRC; NIH Award P30DK058404), including the VDDRC Histology core laboratory.
The authors would like to thank William Muller for providing the ErbB2 floxed mice, David Threadgill for providing the ErbB3 floxed mice, Sylvie Robine for providing the villin-Cre and villin-Cre ERT2 mice, and Frank Revetta for providing technical assistance in immunohistochemical staining.
Abbreviations
- DSS
dextran sulfate sodium
- IFN-γ
interferon gamma
- KOIEC
constitutive intestinal epithelium-specific knockout
- KOIEI
inducible intestinal epithelium-specific knockout
- TNF-α
tumor necrosis factor alpha
- TUNEL
terminal deoxynucleotidyl transferase-mediated dNTP nick-end labeling
Footnotes
Disclosure of Interest: The authors disclose no conflicts.
References
- 1.Carpenter GCS. EGF: receptor interactions and the stimulation of cell growth. In: Lefkowitz R, editor. Receptors and recognition, Series B. Vol. 13. London: Chapman and Hall; 1981. pp. 43–66. [Google Scholar]
- 2.Polk DB. Epidermal growth factor receptor-stimulated intestinal epithelial cell migration requires phospholipase C activity. Gastroenterology. 1998 Mar;114(3):493–502. doi: 10.1016/s0016-5085(98)70532-3. [DOI] [PubMed] [Google Scholar]
- 3.Procaccino F, Reinshagen M, Hoffmann P, et al. Protective effect of epidermal growth factor in an experimental model of colitis in rats. Gastroenterology. 1994 Jul;107(1):12–7. doi: 10.1016/0016-5085(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 4.Sinha A, Nightingale J, West KP, et al. Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N Engl J Med. 2003 Jul 24;349(4):350–7. doi: 10.1056/NEJMoa013136. [DOI] [PubMed] [Google Scholar]
- 5.Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009 Jul;9(7):463–75. doi: 10.1038/nrc2656. [DOI] [PubMed] [Google Scholar]
- 6.Lee KF, Simon H, Chen H, et al. Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature. 1995 Nov 23;378(6555):394–8. doi: 10.1038/378394a0. [DOI] [PubMed] [Google Scholar]
- 7.Jackson-Fisher AJ, Bellinger G, Ramabhadran R, et al. ErbB2 is required for ductal morphogenesis of the mammary gland. Proc Natl Acad Sci U S A. 2004 Dec 7;101(49):17138–43. doi: 10.1073/pnas.0407057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andrechek ER, Hardy WR, Girgis-Gabardo AA, et al. ErbB2 is required for muscle spindle and myoblast cell survival. Mol Cell Biol. 2002 Jul;22(13):4714–22. doi: 10.1128/MCB.22.13.4714-4722.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lucs AV, Muller WJ, Muthuswamy SK. Shc is required for ErbB2-induced inhibition of apoptosis but is dispensable for cell proliferation and disruption of cell polarity. Oncogene. 2010 Jan 14;29(2):174–87. doi: 10.1038/onc.2009.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaoka T, Yan F, Cao H, et al. Transactivation of EGF receptor and ErbB2 protects intestinal epithelial cells from TNF-induced apoptosis. Proc Natl Acad Sci U S A. 2008 Aug 19;105(33):11772–7. doi: 10.1073/pnas.0801463105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Travis A, Pinder SE, Robertson JF, et al. C-erbB-3 in human breast carcinoma: expression and relation to prognosis and established prognostic indicators. Br J Cancer. 1996 Jul;74(2):229–33. doi: 10.1038/bjc.1996.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bobrow LG, Millis RR, Happerfield LC, et al. c-erbB-3 protein expression in ductal carcinoma in situ of the breast. Eur J Cancer. 1997 Oct;33(11):1846–50. doi: 10.1016/s0959-8049(97)00244-x. [DOI] [PubMed] [Google Scholar]
- 13.Holbro T, Beerli RR, Maurer F, et al. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci U S A. 2003 Jul 22;100(15):8933–8. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soltoff SP, Carraway KL, 3rd, Prigent SA, et al. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994 Jun;14(6):3550–8. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sithanandam G, Fornwald LW, Fields J, et al. Inactivation of ErbB3 by siRNA promotes apoptosis and attenuates growth and invasiveness of human lung adenocarcinoma cell line A549. Oncogene. 2005 Mar 10;24(11):1847–59. doi: 10.1038/sj.onc.1208381. [DOI] [PubMed] [Google Scholar]
- 16.Erickson SL, O’Shea KS, Ghaboosi N, et al. ErbB3 is required for normal cerebellar and cardiac development: a comparison with ErbB2-and heregulin-deficient mice. Development. 1997 Dec;124(24):4999–5011. doi: 10.1242/dev.124.24.4999. [DOI] [PubMed] [Google Scholar]
- 17.Lee D, Yu M, Lee E, et al. Tumor-specific apoptosis caused by deletion of the ERBB3 pseudo-kinase in mouse intestinal epithelium. J Clin Invest. 2009 Sep;119(9):2702–13. doi: 10.1172/JCI36435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Egger B, Procaccino F, Lakshmanan J, et al. Mice lacking transforming growth factor alpha have an increased susceptibility to dextran sulfate-induced colitis. Gastroenterology. 1997 Sep;113(3):825–32. doi: 10.1016/s0016-5085(97)70177-x. [DOI] [PubMed] [Google Scholar]
- 19.Egger B, Buchler MW, Lakshmanan J, et al. Mice harboring a defective epidermal growth factor receptor (waved-2) have an increased susceptibility to acute dextran sulfate-induced colitis. Scand J Gastroenterol. 2000 Nov;35(11):1181–7. doi: 10.1080/003655200750056664. [DOI] [PubMed] [Google Scholar]
- 20.Lee D, Pearsall RS, Das S, et al. Epiregulin is not essential for development of intestinal tumors but is required for protection from intestinal damage. Mol Cell Biol. 2004 Oct;24(20):8907–16. doi: 10.1128/MCB.24.20.8907-8916.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okayasu I, Hatakeyama S, Yamada M, et al. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990 Mar;98(3):694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 22.Cooper HS, Murthy SN, Shah RS, et al. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993 Aug;69(2):238–49. [PubMed] [Google Scholar]
- 23.Fukata M, Chen A, Klepper A, et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology. 2006 Sep;131(3):862–77. doi: 10.1053/j.gastro.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.el Marjou F, Janssen KP, Chang BH, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004 Jul;39(3):186–93. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 25.Edelblum KL, Washington MK, Koyama T, et al. Raf protects against colitis by promoting mouse colon epithelial cell survival through NF-kappaB. Gastroenterology. 2008 Aug;135(2):539–51. doi: 10.1053/j.gastro.2008.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Whitehead RH, VanEeden PE, Noble MD, et al. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci U S A. 1993 Jan 15;90(2):587–91. doi: 10.1073/pnas.90.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dieleman LA, Palmen MJ, Akol H, et al. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol. 1998 Dec;114(3):385–91. doi: 10.1046/j.1365-2249.1998.00728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez JA, Williams CS, Amann JM, et al. Deletion of Mtgr1 sensitizes the colonic epithelium to dextran sodium sulfate-induced colitis. Gastroenterology. 2006 Aug;131(2):579–88. doi: 10.1053/j.gastro.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Yan F, John SK, Wilson G, et al. Kinase suppressor of Ras-1 protects intestinal epithelium from cytokine-mediated apoptosis during inflammation. J Clin Invest. 2004 Nov;114(9):1272–80. doi: 10.1172/JCI21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. 2007 Apr;132(4):1359–74. doi: 10.1053/j.gastro.2007.02.056. [DOI] [PubMed] [Google Scholar]
- 31.Froicu M, Zhu Y, Cantorna MT. Vitamin D receptor is required to control gastrointestinal immunity in IL-10 knockout mice. Immunology. 2006 Mar;117(3):310–8. doi: 10.1111/j.1365-2567.2005.02290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato T, van Es JH, Snippert HJ, et al. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011 Jan 20;469(7330):415–8. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iwamoto M, Koji T, Makiyama K, et al. Apoptosis of crypt epithelial cells in ulcerative colitis. J Pathol. 1996 Oct;180(2):152–9. doi: 10.1002/(SICI)1096-9896(199610)180:2<152::AID-PATH649>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 34.Vetuschi A, Latella G, Sferra R, et al. Increased proliferation and apoptosis of colonic epithelial cells in dextran sulfate sodium-induced colitis in rats. Dig Dis Sci. 2002 Jul;47(7):1447–57. doi: 10.1023/a:1015931128583. [DOI] [PubMed] [Google Scholar]
- 35.Satoh Y, Ishiguro Y, Sakuraba H, et al. Cyclosporine regulates intestinal epithelial apoptosis via TGF-beta-related signaling. Am J Physiol Gastrointest Liver Physiol. 2009 Sep;297(3):G514–9. doi: 10.1152/ajpgi.90608.2008. [DOI] [PubMed] [Google Scholar]
- 36.Lorenz RG, McCracken VJ, Elson CO. Animal models of intestinal inflammation: ineffective communication between coalition members. Springer Semin Immunopathol. 2005 Sep;27(2):233–47. doi: 10.1007/s00281-005-0208-4. [DOI] [PubMed] [Google Scholar]
- 37.Siegmund B, Fantuzzi G, Rieder F, et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol. 2001 Oct;281(4):R1264–73. doi: 10.1152/ajpregu.2001.281.4.R1264. [DOI] [PubMed] [Google Scholar]
- 38.Frey MR, Edelblum KL, Mullane MT, et al. The ErbB4 growth factor receptor is required for colon epithelial cell survival in the presence of TNF. Gastroenterology. 2009 Jan;136(1):217–26. doi: 10.1053/j.gastro.2008.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tzahar E, Waterman H, Chen X, et al. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol Cell Biol. 1996 Oct;16(10):5276–87. doi: 10.1128/mcb.16.10.5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.