

Abstract
Survivin is overexpressed by 70–80% of pancreatic cancers, and is associated with resistance to chemotherapy and a poor prognosis. Gemcitabine has been a standard treatment for patients with advanced pancreatic cancer for a decade. Recent reports have demonstrated that gemcitabine treatment attenuates the tumor-suppressive environment by eliminating CD11b+/Gr-1+ myeloid-derived suppressor cells (MDSCs). We hypothesize that a cancer vaccine targeting survivin can achieve enhanced efficacy when combined with gemcitabine. In this study, we tested this hypothesis using modified vaccinia Ankara (MVA) expressing full-length murine survivin. The poorly immunogenic mouse pancreas adenocarcinoma cell line, Pan02, which expresses murine survivin and is syngeneic to C57BL/6, was used for this study. Immunization with MVA-survivin resulted in a modest therapeutic antitumor effect on established Pan02 tumors. When administered with gemcitabine, MVA-survivin immunization resulted in significant tumor regression and prolonged survival. The enhanced vaccine efficacy was associated with decreased CD11b+/Gr-1+ MDSCs. To analyze the survivin-specific immune response to MVA-survivin immunization, we utilized a peptide library of 15mers with 11 residues overlapping from full-length murine survivin. Splenocytes from mice immunized with MVA-survivin produced intracellular γ-interferon in response to in vitro stimulation with the overlapping peptide library. Increased survivin-specific CD8+ T cells that specifically recognized the Pan02 tumor line were seen in mice treated with MVA-survivin and gemcitabine. These data suggest that vaccination with MVA-survivin in combination with gemcitabine represents an attractive strategy to overcome tumor-induced peripheral immune tolerance, and this effect has potential for clinical benefit in pancreatic cancer.
Keywords: Modified vaccinia Ankara (MVA), Survivin, Gemcitabine, Pancreatic cancer, Cancer vaccine
Introduction
Pancreatic cancer, with an overall 5-year survival rate of 5% [1], remains one of the most deadly cancers, and success in its treatment has progressed little in past decades. Surgical resection improves the outlook, although fewer than 20% of patients with pancreatic cancer are eligible for resection [2, 3]. Most resected patients fail treatment due to local recurrence, hepatic metastases, or both within 1–2 years after surgery [4, 5]. The effect of traditional cytotoxic chemotherapy on pancreatic cancer is very limited. Gemcitabine is a chemotherapeutic drug (cytidine analog) that is currently the most effective treatment for advanced pancreatic cancer patients, resulting in a modest increase in survival [6]. However, recent trials have failed to show an improvement in survival when gemcitabine is combined with other chemotherapeutic drugs [7]. Erlotinib, an epidermal growth factor receptor inhibitor, was recently reported to produce a statistically significant improvement in survival, when combined with gemcitabine. However, the increase in survival was modest [8]. Clearly, novel treatments for pancreatic cancer are desperately needed. One approach which might have significant therapeutic potential is to combine gemcitabine with immunotherapy. Recent studies have revealed substantial cell-mediated immune responses in pancreatic cancer patients, suggesting that immunity may be relevant [9]. Other studies indicated that gemcitabine treatment is not immunosuppressive and may enhance responses to specific immunotherapy by driving effector immunity to cancer cells [10, 11].
Gemcitabine not only exerts direct antitumor activity through induction of death pathways leading to apoptosis [12], but also mediates immunological effects relevant to tumor immunotherapy [11, 13]. Enhanced cross-presentation of tumor antigens by DCs after gemcitabine treatment leads to enhanced tumor recognition by cytotoxic T lymphocyte (CTLs) in vivo [14]. Recent reports demonstrate that gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid-derived suppressor cell (MDSC) in tumor-bearing animals thereby enhancing antitumor immune activity [15, 16]. Pancreatic carcinoma cells can be recognized by tumor-specific T cells [17, 18] and tumor-reactive T cells capable of tumor rejection can also be isolated from blood of pancreatic cancer patients [19]. Tumor infiltration with CTLs and T helper cells is associated with a favorable prognosis in pancreatic cancer patients [20]. Therefore, combination of immunotherapy and gemcitabine might be a promising strategy for treatment of pancreatic cancer patient.
Survivin, a member of the inhibitor of apoptosis (IAP) family, regulates important pathways implicated in cell cycle progression, cell proliferation and cell death [21]. This intracellular protein is broadly expressed during embryonic development [22], mostly absent in differentiated cells, but then strongly up-regulated in human cancer cells of various origins [23, 24]. Survivin expression is associated with enhanced tumor cell viability and resistance to cancer therapies [25]. Accordingly, knock-down of survivin expression in tumor cells has highly detrimental effects on cancer cell viability and tumor progression [26]. Widespread expression in several types of human cancers, general absence in normal adult tissues and a requirement for tumor cell survival essentially identifies survivin as an almost ideal “universal” tumor-associated antigen (TAA). Spontaneous cellular, as well as humoral immune responses against survivin have been detected in patients with several types of cancer [27, 28], further validating survivin as a TAA that can be exploited for therapeutic purposes. Importantly, CD8+ and CD4+ T cell responses restricted to different human and mouse MHC molecules have been characterized and several relevant survivin epitopes were identified. All together, these features make survivin a highly attractive target for T cell-based immune strategies against cancer. Dendritic cell- and gene-based vaccines targeting survivin have been tested in preclinical settings where induction of T cell responses and tumor protection were observed [29, 30]. Because antigen-escape mutants would not be able to survive, survivin makes an excellent target for antigen-specific cell-mediated immunotherapy. Survivin is overexpressed in 70–82% of pancreatic cancers, and is associated with resistance to chemotherapy and radiotherapy, and a poor prognosis [31, 32]. Survivin has also been found in more than 56% of intraductal papillary-mucinous tumors, a precancerous neoplasm in the pancreatic duct, suggesting that it plays a role in oncogenesis [33].
An optimal immunotherapy approach to survivin would involve an immunization strategy which generates vigorous T cell responses, without the requirement for a haplotype (HLA-matched)- or patient-specific vaccine. The highly attenuated strain of vaccinia, modified vaccinia Ankara (MVA) has been employed as a vaccine vector to deliver TAA [34–36]. MVA vaccines have demonstrated safety, immunogenicity, and evidence of clinical benefit in patients with cancer [37]. In this study, we generated MVA expressing mouse survivin and evaluated vaccine efficacy. We evaluated the antitumor effect of MVA-survivin against a poorly immunogenic pancreatic carcinoma in a preclinical murine model. We evaluated the effect of gemcitabine in combination with the MVA-survivin vaccine. We have demonstrated that the combination of MVA-survivin and gemcitabine generates enhanced antitumor immunity in an established pancreatic carcinoma model.
Materials and methods
Mice and cell lines
Six-week-old female C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and maintained in a specific pathogen-free animal facility. All studies were approved by the Research Animal Care Committee of the City of Hope National Medical Center and performed under the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC) guidelines. The poorly immunogenic mouse pancreas adenocarcinoma cell line, Pan02 (also known as Panc02) [38] was kindly provided by Dr. DC. Linehan, Washington University School of Medicine [39]. Pan02 was cultured in RPMI-1640 medium (Cellgro, Herndon, VA) supplemented with 10% fetal bovine serum (ISC Bioexpress, Kaysville, UT), 1% penicillin/streptomycin (GIBCO, Carlsbad, CA), 1% l-glutamine (GIBCO, Carlsbad, CA), and 1% HEPES buffer (Irvine Scientific, Santa Ana, CA). Baby hamster kidney cells (BHK-21) [40] were purchased from American Type Culture Collection (Manassas, VA) and grown in MEM (Cellgro, Herndon, VA) supplemented with non-essential amino acids, l-glutamine, and 10% FCS.
Antibodies
Fluorescein isothiocyanate (FITC)-conjugated CD4 monoclonal antibodies (mAb), FITC-conjugated CD11b mAb, phycoerythrin (PE)-conjugated CD8 mAb, PE-conjugated Gr-1 mAb, or allophycocyanin (APC)-conjugated gamma interferon (IFN-γ) mAbs were purchased from BD Pharmingen (San Diego, CA). Anti-CD4 (GK1.5) [41] was purchased from American Type Culture Collection (Manassas, VA). Anti-CD8 (H35) [42] was kind gift from James P. Allison (Memorial Sloan-Kettering Cancer Center, New York, NY). Antibodies were produced using a CELLine Device (BD Biosciences, Bedford, MA). IgG antibodies were purified by absorbance over protein G-Sepharose (Amersham Biosciences, Uppsala, Sweden) followed by elution with 0.1 M glycine–HCl (pH 2.7). The product was then dialyzed against PBS and concentrated using a Centriplus centrifugal filter device (Millipore, Bedford, MA). Anti-asialo GM1 was purchased from Wako Chemicals (Richmond, VA).
Generation of recombinant MVA
Construction of recombinant (r) MVA was performed according to published protocols [34–36, 43]. In brief, BHK cells were infected with wildtype MVA and then transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) with a recombination shuttle plasmid containing the mouse survivin expression cassette to promote homologous recombination. The recombinant virus was screened by isolation of Venus™-positive BHK cells under the fluorescence microscope. PCR to detect wildtype MVA was run with the following conditions: 95°C for 5 min and then 40 cycles of 95°C for 1 min, 55°C for 1 min, and 72°C for 1 min, followed by 72°C for 5 min. Primers used in PCR and specific for the flanking IGR3 regions were IGR3F 5′-TCCGCTCCCATTCAATTC-3′ and IGR3R 5′-AACACCCGCACAAGTCTG-3′ [44]. PCR products were visualized by gel electrophoresis on a 1.2% agarose/ethidium bromide gel under UV light. The survivin expression cassette is ~1.6 kb in length, while the wildtype IGR3 product is ~300 bp in length. Viruses were screened for expression levels, candidates selected, and purified by sucrose density ultracentrifugation. High-titer stocks were stored at −80°C. The protein expression levels of mouse survivin from recombinant MVA-infected BHK-21 cells, normal mouse pancreas tissue, and Pan02 cells were evaluated by Western blot analysis. Membranes were incubated first with the rabbit anti-mouse survivin antibody (Ab) (AbD Serotec, Oxford, UK), followed with a peroxidase-labeled goat anti-rabbit Ab (Sigma-Aldrich, St. Louis, MO).
In vivo evaluation of gemcitabine on Gr-1+/CD11b+ myeloid-derived suppressor cells
C57BL/6 mice received s.c. injections of 2.5 × 105 Pan02 cells. Tumor volumes were calculated from caliper measurements of length, width, and height of the tumor mass. When tumors reached a minimal volume of 500 mm3, mice were injected i.p. with a single dose of 60 mg/kg of gemcitabine (Eli Lilly, Indianapolis, IN). Forty-eight hours after treatment, spleen cells were harvested and the single cell suspensions were subjected to flow cytometry [15].
In vivo tumor challenge experiments
To develop an established pancreatic carcinoma model, mice were challenged with 2.5 × 105 Pan02 cells by subcutaneous injection into the right lower flank. When the tumors were palpable and reached 3–4 mm in diameter, mice were immunized twice at a 1-week interval with 5 × 107 plaque-forming units (pfu) of MVA-survivin or control MVA (recombinant MVA carrying human carcinoembryonic antigen (CEA) [45]). For the gemcitabine combination experiment, 60 mg/kg of gemcitabine was administrated i.p. 48 h prior to prime immunization. For the in vivo mAb depletions, MVA-survivin with gemcitabine-treated mice were injected i.p. with 200 μg of anti-CD8 mAb (H35), anti-CD4 mAb (GK1.5), or anti-asialo GM1 (dilution 1/20) with a maintenance dose every 3 days until killing of the animals [34, 35]. Tumors were measured twice weekly in three dimensions with calipers. Growth curves, representing mean tumor size, were truncated when the first mouse in the respective group died.
Cellular cytotoxicity assay
Cytotoxicity against gemcitabine treated or non-treated Pan02 cells was determined with a standard 51Cr release assay at E:T ratios of 100, 20 and 4 [34]. Effectors used in the assay were derived from spleens of Pan02-bearing C57BL6 mice (n = 3) immunized with a single dose of MVA-survivin at day 3 post-tumor challenge. Briefly, Pan02-bearing mice were killed 1 week post-immunization and splenocytes were co-incubated with irradiated Pan02 cells (20,000 rads) for 1 week. Effectors were then co-incubated for 4 h with 5,000 Pan02 target cells in 96-well plates (in triplicate) that had been incubated overnight in DMEM, without or with gemcitabine at a final concentration of 10 nM, and then labeled with 51Cr. Radioactivity released into the supernatant was measured using a Cobra Quantum gamma counter (PerkinElmer). Percent-specific lysis was calculated using the formula: (experimental release − spontaneous release)/(maximum release − spontaneous release) × 100.
Overlapping peptide library
A 15-mer overlapping peptide library derived from full-length mouse survivin was composed of 34 peptides, which covered all 140 amino acids of the survivin protein with an 11-amino acid overlap between peptides (Table 1). Peptides were synthesized in our laboratory with a Symphony Quartet peptide synthesizer (Protein Technologies Inc., Tucson, AZ) using standard FMOC protocols and purified to >95% purity by HPLC (Agilent 1200) using an Atlantis™ preparative HPLC column (Waters, Watertown, MA). The identity of the peptides was confirmed by MALDI TOF mass spectrometric analysis using a Kompact Probe mass spectrometer (Kratos Analytical, Shimadzu, Kyoto, Japan).
Table 1.
Mouse survivin overlapping peptide library
| No. | Sequence |
|---|---|
| 1 | MGAPALPQIWQLYLK |
| 2 | ALPQIWQLYLKNYRI |
| 3 | IWQLYLKNYRIATFK |
| 4 | YLKNYRIATFKNWPF |
| 5 | YRIATFKNWPFLE |
| 6 | IATFKNWPFLEDCA |
| 7 | FKNWPFLEDCACTPE |
| 8 | PFLEDCACTPERMAE |
| 9 | DCACTPERMAEAGFI |
| 10 | TPERMAEAGFIHCPTE |
| 11 | MAEAGFIHCPTENE |
| 12 | AGFIHCPTENEPDLA |
| 13 | HCPTENEPDLAQCFF |
| 14 | ENEPDLAQCFFCFKE |
| 15 | DLAQCFFCFKELEGW |
| 16 | CFFCFKELEGWEPDD |
| 17 | FKELEGWEPDDNPIE |
| 18 | EGWEPDDNPIEEHRK |
| 19 | PDDNPIEEHRKHSPG |
| 20 | NPIEEHRKHSPGCAF |
| 21 | EHRKHSPGCAFLTVK |
| 22 | HSPGCAFLTVKKQME |
| 23 | CAFLTVKKQMEELTV |
| 24 | TVKKQMEELTVSEFL |
| 25 | KQMEELTVSEFLKLD |
| 26 | ELTVSEFLKLDRQRA |
| 27 | SEFLKLDRQRAKNKI |
| 28 | KLDRQRAKNKIAKET |
| 29 | RQRAKNKIAKETNNKQK |
| 30 | NKIAKETNNKQKEFE |
| 31 | KETNNKQKEFEETAK |
| 32 | NKQKEFEETAKTTRQS |
| 33 | EFEETAKTTRQSIE |
| 34 | ETAKTTRQSIEQLAA |
A 15-mer overlapping peptide library derived from full-length mouse survivin was composed of 34 peptides, which covered all 140 amino acids of the survivin protein with an 11-amino acid overlap between peptides
Intracellular cytokine assays
A prime immunization was conducted by i.p. injection and consisted of either 5 × 107 pfu of recombinant MVA-survivin or control MVA on day 0. A boost immunization was administered on day 14. One week after the boost immunization, mice were killed and splenocytes were incubated in the presence of irradiated and peptide library (10 μg/ml each peptide) pulsed LPS blasts, as described [36]. After a 7-day in vitro stimulation, the splenocytes were assayed for intracellular IFN-γ production. The cells were exposed to 10 μg/ml of peptide library for 1 h after which GolgiPlug (BD Pharmingen, San Diego, CA) was added. Following an overnight incubation, the cells were washed and labeled with FITC-conjugated Ab to mouse CD4 and PE-conjugated Ab to mouse CD8. The cells were then permeabilized and labeled with APC-conjugated Ab to IFN-γ for 30 min at 4°C. The cells were washed and analyzed on a FACSCanto flow cytometer (Becton-Dickinson, San Jose, CA).
Statistical methods
Comparisons between two groups were evaluated by the Student’s t test using GraphPad Prizm 5 software. Comparisons with more than two groups were done by ANOVA. Values of the results were expressed as mean and SEs. Differences were considered to be statistically significant when P < 0.05. The survival time was evaluated using Kaplan–Meier plots and log-rank (Mantel–Cox) tests.
Results
Construction of recombinant MVA-survivin
Recombinant MVA carrying the mouse survivin gene (MVA-survivin) inserted in the IGR3 site [44] was generated by infection of BHK cells with wildtype MVA followed by transfection of a transfer vector containing the mouse survivin expression cassette (Fig. 1a). Following transfection/infection, MVA-survivin isolates were selected using the Venus fluorescent marker, and Venus-positive BHK cells were presumed to house the recombinant virus. Following large-scale expansion and ultrapurification of isolate C7 MVA-survivin, DNA was extracted from uninfected BHK cells (negative control), BHK cells infected with wildtype MVA or MVA-survivin (with or without ultrapurification), and used in PCR analysis to detect for mouse survivin and wildtype MVA for prevention of contamination. As shown in Fig. 1b, bands were observed at 1.6 kb, the length of the mouse survivin expression cassette, but not at 300 bp in the lanes carrying PCR product from viral DNA of C7 or ultrapurified C7 isolates. These results suggest the absence of wildtype MVA in the ultrapurified stocks. To verify that the mouse survivin gene was still present in the ultrapurified C7 stock, Western blot analysis for mouse survivin expression was conducted. Lysates from BHK cells infected with ultrapurified isolate C7 were shown to contain mouse survivin, as indicated by the band present at ~17 kDa (Fig. 1c). For the tumor challenge experiment, Pan02 cells were tested for expression of survivin by Western blot analysis. Survivin protein was clearly detected as a protein of ~17 kDa in Pan02 cells (Fig. 1d).
Fig. 1.
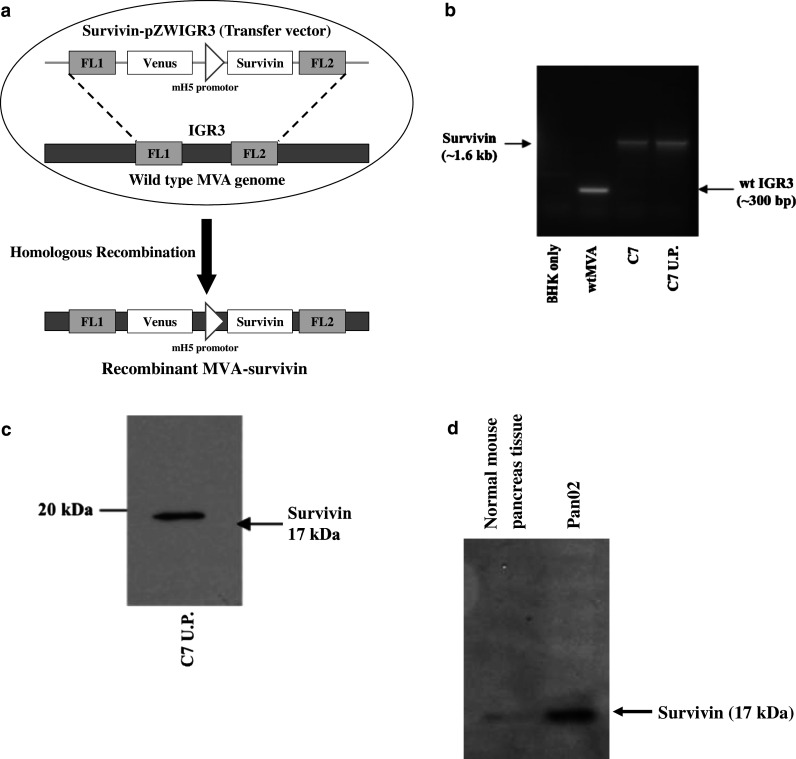
Construction of recombinant MVA-survivin. a BHK cells were infected with MVA and transfected with mouse survivin transfer vector. The mouse survivin expression cassette was inserted into the wildtype IGR3 site by homologous recombination. b DNA was extracted from ultrapurified isolate C7 and then used in PCR to detect for the presence of wildtype MVA. Isolates from uninfected and wtMVA-infected BHK cells were used as negative and positive controls, respectively. The mouse survivin expression cassette and wildtype IGR3 site have lengths of 1.6 and 300 bp, respectively. c Ultrapurified C7 lysate was tested for expression of survivin by Western blot analysis. Survivin is a protein of ~17 kDa. d Pan02 tumor cell line and normal pancreas were tested for expression of survivin by Western blot analysis
MVA-survivin immunization results in modest antitumor effects in an established mouse pancreatic cancer model
We examined the potential of MVA-survivin to suppress tumor growth in a clinically relevant established tumor model. C57BL/6 mice were inoculated with 2.5 × 105 cells of Pan02 by s.c. injection into right lower flank. When the palpable tumors reached 3–4 mm in diameter, mice were immunized i.p. with 5 × 107 pfu of MVA-survivin or MVA-control. Booster immunization was performed 7 days after the prime immunization. Despite multiple immunization, MVA-survivin alone resulted in a modest therapeutic antitumor effect on established pancreatic tumors (Fig. 2).
Fig. 2.

Antitumor effect of MVA-survivin vaccine. C57BL/6 mice received s.c. injections of 2.5 × 105 Pan02 cells on day 0. When the tumors were palpable and reached 3–4 mm in diameter, mice were immunized i.p. with 5 × 107 pfu of MVA-survivin. Booster immunization was performed 7 days after the prime immunization. Tumors were measured twice weekly in three dimensions with calipers. Bars represent SE
Gemcitabine reduces tumor-associated MDSC in mice inoculated with Pan02 pancreatic cancer cells
As an initial evaluation of gemcitabine, we tested whether gemcitabine could effect splenic MDSC in Pan02 tumor-bearing mice. Tumor-bearing mice with large, >500 mm3 Pan02 tumors, were treated with a single dose of 60 mg/kg of gemcitabine. Forty-eight hours after treatment, spleen cells were evaluated. Using two-color flow cytometry, physiological CD11b+/Gr-1+ MDSC constituted 4–6% of the splenocytes in naïve C57BL/6 mice (data not shown). In contrast, mice-bearing large Pan02 tumors showed marked increases in splenic MDSC. As shown in Fig. 3, CD11b+/Gr-1+ MDSC made up to 23.58% of the splenocytes in an animal bearing a large Pan02 tumor (500 mm3 in size). The average percentage of CD11b+/Gr-1+ MDSC in mice-bearing tumors increased significantly. Gemcitabine administration resulted in a reduction in MDSC in mice-bearing tumors. Two days after treatment with a single dose of gemcitabine (60 mg/kg), the average percentage of CD11b+/Gr-1+ MDSC decreased significantly (P < 0.05).
Fig. 3.

Gemcitabine attenuates splenic MDSCs in Pan02 tumor-bearing mice. C57BL/6 mice received s.c. injections of 2.5 × 105 Pan02 cell. Single dose of gemcitabine (60 mg/kg) or PBS was administered i.p. when Pan02 tumors reached a minimal volume of 500 mm3. MDSCs from the spleen of large Pan02 tumor-bearing mice were harvested 48 h after gemcitabine or PBS treatment. The average percentage of double-positive CD11b+/Gr-1+ MDSCs is shown. *P < 0.05 by unpaired t test. Bars SE
MVA-survivin combined with gemcitabine elicits potent antitumor effects in an established mouse pancreatic cancer model
Because the MVA-survivin vaccine alone was not sufficient to induce a strong antitumor response in a therapeutic model, we tested whether the addition of gemcitabine would result in enhanced priming of survivin-specific antitumor responses. C57BL/6 mice were first injected with 2.5 × 105 Pan02 cells. Once palpable tumors developed, gemcitabine was injected i.p. on days 3. Two days after gemcitabine administration, mice were immunized i.p. with 5 × 107 pfu of MVA-survivin, MVA-control, or PBS. A booster immunization, or control was administered i.p. 7 days after the prime immunization. As shown in Fig. 4a, treatment with gemcitabine in combination with MVA-survivin immunization resulted in potent antitumor effects relative to immunizations with MVA-control with gemcitabine or MVA-survivin without gemcitabine. MVA-survivin combined with gemcitabine also showed improved survival versus gemcitabine treatment groups without MVA-survivin immunization (Fig. 4b). Survival results are displayed using a Kaplan–Meier plot which shows that 50% of the mice in the MVA-survivin with gemcitabine group are alive at 50 days, whereas all other animals expired by 38 days post-tumor inoculation. There was a significant difference in survival in the MVA-survivin with gemcitabine group versus the gemcitabine treatment group without MVA-survivin (P < 0.05 by log-rank test).
Fig. 4.

Therapeutic antitumor effect of MVA-mouse. Survivin administered with gemcitabine in an established Pan02 tumor model. a C57BL/6 mice received s.c. injections of 2.5 × 105 Pan02 cells. Gemcitabine (60 mg/kg) was injected i.p. on day 3. Mice were immunized twice i.p. with 5 × 107 pfu of MVA-survivin or MVA-control (human CEA) or PBS control on day 5 and 12. *P < 0.05 comparing MVA-survivin with gemcitabine to MVA-survivin alone groups by one-way ANOVA. b Kaplan–Meier graph representing cumulative survival of mice in the indicated treatment groups. *P < 0.05 comparing MVA-survivin with gemcitabine to PBS, **P < 0.05 comparing MVA-survivin with gemcitabine to gemcitabine alone, ***P < 0.05 comparing MVA-survivin with gemcitabine to MVA-control with gemcitabine groups by log-rank (Mantel–Cox) test
Gemcitabine augments induction of survivin-specific T cell responses in MVA-survivin immunization
To evaluate immunogenicity against mouse survivin, we immunized 8-week-old C57BL/6 mice with MVA-survivin (5 × 107 pfu). The initial i.p. vaccination was followed 2 weeks later by an additional boost vaccination. Splenocytes from immunized mice were restimulated in vitro with the mouse survivin peptide library for 1 week prior to conducting an ICC assay to detect IFN-γ release. As shown in Fig. 5, survivin-specific IFN-γ responses in both CD8+ and CD4+ T cells were induced in mice immunized with MVA-survivin. Furthermore, when MVA-survivin immunization was combined with gemcitabine treatment, there was a trend toward enhanced survivin-specific IFN-γ release in CD8+ T cells in excess of that seen with MVA-survivin immunization alone. Interestingly, a minimal survivin-specific IFN-γ response was seen in mice immunized with a control MVA with gemcitabine. Gemcitabine treatment may have resulted in de novo in vitro, peptide library-specific CD8+ T cell immunity.
Fig. 5.

Immunogenicity test against mouse survivin. C57BL/6 mice were immunized twice by i.p. injection with MVA-survivin with or without gemcitabine. ICC assays for IFN-γ were performed on splenocytes from immunized mice. Bars represent the percentage of CD8+ and CD4+ T cells expressing IFN-γ in response to the mouse survivin peptide library. Black bar CD8+ T cell response to mouse survivin. Gray bar CD4+ T cell response to mouse survivin
Role of immune subsets and gemcitabine in control of Pan02 tumor growth
To determine the relative contribution of T cell and NK cell subsets on enhanced antitumor effect of Pan02 tumors following MVA-survivin immunization and gemcitabine administration, mice were treated with i.p. injections of depleting doses of CD4+, CD8+, or NK cell-specific antibodies. As shown in Fig. 6a, mice depleted of CD8+ cells developed rapidly lethal tumors. In contrast, CD4+ T cell and NK cell depletion resulted in only partial abrogation of the response to the vaccine. We then assessed whether gemcitabine could alter the sensitivity of Pan02 tumor cells to CTL killing. To do this, we carried out chromium release assays using Pan02 targets untreated or treated overnight with gemcitabine. As shown in Fig. 6b, treatment of Pan02 targets with gemcitabine significantly increased susceptibility to killing (P < 0.05 by Student’s t test) by CTL obtained from the spleen of Pan02-bearing mice vaccinated with MVA-survivin.
Fig. 6.

Role of immune subsets and gemcitabine in control of Pan02 growth. a Effect of depletion of the CD8+ T cells, CD4+ T cells, or NK cells in mice immunized with MVA-survivin and gemcitabine. C57BL/6 mice received s.c. injections of 2.5 × 105 Pan02 cells. Gemcitabine (60 mg/kg) was injected i.p. on day 3. Mice were immunized twice i.p. with 5 × 107 pfu of MVA-survivin. Mice treated with MVA-survivin with gemcitabine were i.p. injected with 200 μg of anti-CD8 mAb (H35) or anti-CD4 mAb (GK1.5), or anti-asialo GM1 (dilution 1/20) with a maintenance dose every 3 days until killing of the animals. b Effect of gemcitabine on susceptibility of Pan02 to cytotoxic T lymphocytes using chromium release assay. To generate effectors, Pan02-bearing C57BL6 mice (n = 3) were first immunized with MVA-survivin 3 days after tumor inoculation. Splenocytes were then harvested 1 week post-immunization and incubated for 1 week with irradiated Pan02 cells. These effectors were then incubated in 96-well plates (in triplicate) for 4 h with 51Cr-labeled Pan02 cells that had been treated overnight either in the presence or absence of 10 nM gemcitabine. *P < 0.05, Student’s t test
Discussion
Vaccine immunotherapy of tumors aims to recruit and activate T cells that recognize tumor-specific antigens to eliminate tumors. Vaccine immunotherapy is an attractive adjunct to chemotherapy for the treatment of pancreatic cancer, because immunotherapy acts through a mechanism that is distinct from chemotherapy, and represents a non-cross-resistant treatment with an entirely different and minimal spectrum of toxicity. New insights into the mechanisms by which T cells are successfully activated and by which tumors evade immune recognition are driving the development of new combinatorial immunotherapeutic approaches. In addition, recent advances in gene-expression analysis have allowed for the identification of new pancreatic cancer targets, including candidate tumor antigens that might serve as T cell targets. These advances now make it possible to exploit the immune system in the fight against pancreatic cancer.
Survivin is an attractive T cell target for pancreatic cancer. The feasibility and efficacy of targeting survivin as a cancer vaccine have been investigated in several cancer models [29, 30]. MVA engineered with recombinant genes (rMVA) have shown promise as a vaccine in rodents and macaques for infectious disease and cancer. MVA infection leads to effective antigen presentation by human antigen presenting cells, and the generation of TAA-specific CTL [34]. Previously, we generated and extensively characterized an attenuated poxvirus referred to as MVA expressing either human or murine p53 [35–37]. These MVA vaccines resulted in enhanced tumor-specific immunity and tumor rejection of immunogenic tumor lines. In this study, however, MVA-survivin immunization alone did not result in significant antitumor activity for the poorly immunogenic Pan02 mouse pancreatic tumor despite its overexpression of survivin.
Gemcitabine can increase the antitumor activity of CD8+ T cells and activated NK cells by selectively reducing the CD11b+/Gr-1+ MDSCs, known to be significantly increased in the spleen and tumor of tumor-bearing mice [15, 46]. MDSCs in mice are heterogeneous myeloid cells primarily comprised of CD11b+/Gr-1+ cells [47]. Tumor-associated MDSCs and their suppressive function against tumor-specific T cells have been well described previously [48, 49]. Recently, many drugs including Cox-2 inhibitors and PDE-5 inhibitors have been evaluated as MDSC inhibitors in preclinical tumor models, and they have been used to limit the MDSC-mediated suppressive environment [50, 51]. Sunitinib has also been reported to be an effective suppressor of MDSCs [52]. Gemcitabine is a standard chemotherapeutic drug with activity in pancreatic cancer, and its combination with MVA-survivin is a reasonable strategy. Treatment of an established tumor with gemcitabine could induce apoptosis of tumor cells and, hence, prime antitumor immunity by increasing antigen cross-presentation [11]. Based on these findings, we attempted to combine gemcitabine with MVA-survivin immunotherapy. Pan02 cells were used to model a combination strategy of MVA-survivin and gemcitabine in a preclinical pancreatic cancer model. We found that gemcitabine treatment works in synergy with immunotherapy to delay tumor growth. To minimize the adverse effect of the chemotherapeutic agent on activated tumor-specific CTL, gemcitabine was administered at a low dose 48 h before MVA-survivin immunization [15]. Splenic MDSCs in mice-bearing Pan02 tumor were selectively attenuated by the low dose gemcitabine treatment. Together, gemcitabine treatment and immunotherapy significantly augmented therapeutic antitumor immunity against established pancreatic tumors. It is likely that the attenuation of MDSCs resulting from gemcitabine treatment augmented the effect of immunotherapy. The enhanced antitumor effect was probably related to augmented immunogenicity against survivin when MVA-survivin immunization was combined with gemcitabine treatment.
In our study, gemcitabine treatment enhanced survivin-specific CD8+ IFN-γ immune responses in MVA-survivin immunized mice. This is further supported by the in vivo mAb depletion experiments, which demonstrated that the antitumor effect of the combination was mediated primarily by CD8+ T cells and to a lesser degree by NK cells. We also show that gemcitabine treatment of Pan02 cells significantly increases susceptibility to killing by CTL when compared to untreated Pan02 cells. In summary, the synergistic effect of this combined chemoimmunotherapy using gemcitabine and subsequent immunotherapy is dependent primarily on CD8+ T cells. Gemcitabine succeeded in breaking tolerance to survivin. Gemcitabine treatment resulted in diminished MDSCs. Taken together, these data suggest that a two-pronged approach, the generation of effector cells against self-tumor antigen and the attenuation of the tumor-suppressive environment, can act in synergy for the induction of antitumor immunity in pancreatic cancer.
Acknowledgments
The authors thank the staff of the Animal Resource Center at City of Hope for their expert animal handling and assistance in husbandry. Grant support for these studies was from NCI (CA077544 and CA030206) to Don J. Diamond, a minority supplement award to E. Manuel (CA030206S2) and a RAID supplement (CA030206S3) to DJD. Grants from the Riley Foundation and FAMRI also provided partial support for the project to Joshua D. I. Ellenhorn. The COH Cancer Center is supported by CA033572. D. Diamond dedicates this report to the memory of his dear friend, David VP Marks.
Conflict of interest
The authors declare that there are no conflicts of interest in regard to this work.
Footnotes
H. Ishizaki and E. R. Manuel contributed equally to this work.
References
- 1.Jemal A, Siegel R, Ward E, et al. Cancer statics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Sohn TA, Yeo CJ, Cameron JL, et al. Resected adenocarcinoma of the pancreas-616 patients: results, outcomes, and prognostic indicators. J Gastrointest Surg. 2000;4:567–579. doi: 10.1016/S1091-255X(00)80105-5. [DOI] [PubMed] [Google Scholar]
- 3.Li D, Xie K, Wolff R, et al. Pancreatic cancer. Lancet. 2004;363:1049–1057. doi: 10.1016/S0140-6736(04)15841-8. [DOI] [PubMed] [Google Scholar]
- 4.Griffin JF, Smalley SR, Jewell W, et al. Patterns of failure after curative resection of pancreatic carcinoma. Cancer. 1990;66:56–61. doi: 10.1002/1097-0142(19900701)66:1<56::AID-CNCR2820660112>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 5.Sperti C, Pasquali C, Piccoli A, et al. Recurrence after resection for ductal adenocarcinoma of the pancreas. World J Surg. 1997;21:195–200. doi: 10.1007/s002689900215. [DOI] [PubMed] [Google Scholar]
- 6.Burris HA, Moore MJ, Andersen J, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15:2403–2413. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 7.Louvet C, Labianca R, Hammel P, et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: results of a GERCOR and GISCAD phase III trial. J Clin Oncol. 2005;23:3509–3516. doi: 10.1200/JCO.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 8.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–1966. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 9.Plate JM, Shott S, Harris JE. Immunoregulation in pancreatic cancer patients. Cancer Immunol Immunother. 1999;48:270–279. doi: 10.1007/s002620050575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daikeler T, Maas K, Hartmann JT, et al. Weekly short infusions of gemcitabine are not associated with suppression of lymphatic activity in patients with solid tumors. Anticancer Drugs. 1997;8:643–644. doi: 10.1097/00001813-199707000-00014. [DOI] [PubMed] [Google Scholar]
- 11.Plate JM, Plate AE, Shott S, et al. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol Immunother. 2005;54:915–925. doi: 10.1007/s00262-004-0638-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casneuf VF, Demetter P, Boterberg T, et al. Antiangiogenic versus cytotoxic therapeutic approaches in a mouse model of pancreatic cancer: An experimental study with multitarget tyrosine kinase inhibitor (sunitinib), gemcitabine and radiotherapy. Oncol Rep. 2009;22:105–113. doi: 10.3892/or_00000412. [DOI] [PubMed] [Google Scholar]
- 13.Nowak AK, Robinson BW, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. 2003;63:4490–4496. [PubMed] [Google Scholar]
- 14.Nowak AK, Lake RA, Marzo AL, et al. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J Immunol. 2003;170:4905–4913. doi: 10.4049/jimmunol.170.10.4905. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki E, Kapoor V, Jassar AS, et al. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res. 2005;11:6713–6721. doi: 10.1158/1078-0432.CCR-05-0883. [DOI] [PubMed] [Google Scholar]
- 16.Ko HJ, Kim YJ, Kim YS, et al. A combination of chemoimmunotherapies can efficiently break self-tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer Res. 2007;67:7477–7486. doi: 10.1158/0008-5472.CAN-06-4639. [DOI] [PubMed] [Google Scholar]
- 17.Ito M, Shichijo S, Tsuda N, et al. Molecular basis of T cell-mediated recognition of pancreatic cancer cells. Cancer Res. 2001;61:2038–2046. [PubMed] [Google Scholar]
- 18.Schnurr M, Galambos P, Scholz C, et al. Tumor cell lysate-pulsed human dendritic cells induce a T-cell response against pancreatic carcinoma cells: an in vitro model for the assessment of tumor vaccines. Cancer Res. 2001;61:6445–6450. [PubMed] [Google Scholar]
- 19.Schmitz-Winnenthal FH, Volk C, Z’graggen K, et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005;65:10079–10087. doi: 10.1158/0008-5472.CAN-05-1098. [DOI] [PubMed] [Google Scholar]
- 20.Fukunaga A, Miyamoto M, Cho Y, et al. CD8+ tumor-infiltrating lymphocytes together with CD4+ tumor-infiltrating lymphocytes and dendritic cells improve the prognosis of patients with pancreatic adenocarcinoma. Pancreas. 2004;28:e26–e31. doi: 10.1097/00006676-200401000-00023. [DOI] [PubMed] [Google Scholar]
- 21.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 22.Adida C, Crotty PL, McGrath J, et al. Developmentally regulated expression of the novel cancer anti-apoptosis gene survivin in human and mouse differentiation. Am J Pathol. 1998;152:43–49. [PMC free article] [PubMed] [Google Scholar]
- 23.Ambrosini G, Adida C, Altieri DC. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med. 1997;3:917–921. doi: 10.1038/nm0897-917. [DOI] [PubMed] [Google Scholar]
- 24.Velculescu VE, Madden SL, Zhang L, et al. Analysis of human transcriptomes. Nat Genet. 1999;23:387–388. doi: 10.1038/70487. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto H, Ngan CY, Monden M. Cancer cells survive with survivin. Cancer Sci. 2008;99:1709–1714. doi: 10.1111/j.1349-7006.2008.00870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altieri DC. Targeted therapy by disabling crossroad signaling networks: the survivin paradigm. Mol Cancer Ther. 2006;5:478–482. doi: 10.1158/1535-7163.MCT-05-0436. [DOI] [PubMed] [Google Scholar]
- 27.Andersen MH, Pedersen LO, Capeller B, et al. Spontaneous cytotoxic T-cell responses against survivin-derived MHC class I-restricted T-cell epitopes in situ as well as ex vivo in cancer patients. Cancer Res. 2001;61:5964–5968. [PubMed] [Google Scholar]
- 28.Rohayem J, Diestelkoetter P, Weigle B, et al. Antibody response to the tumor-associated inhibitor of apoptosis protein survivin in cancer patients. Cancer Res. 2000;60:1815–1817. [PubMed] [Google Scholar]
- 29.Xiang R, Mizutani N, Luo Y, et al. A DNA vaccine targeting survivin combines apoptosis with suppression of angiogenesis in lung tumor eradication. Cancer Res. 2005;65:553–561. [PubMed] [Google Scholar]
- 30.Zeis M, Siegel S, Wagner A, et al. Generation of cytotoxic responses in mice and human individuals against hematological malignancies using survivin-RNA-transfected dendritic cells. J Immunol. 2003;170:5391–5397. doi: 10.4049/jimmunol.170.11.5391. [DOI] [PubMed] [Google Scholar]
- 31.Sarela AI, Verbeke CS, Ramsdale J, et al. Expression of survivin, a novel inhibitor of apoptosis and cell cycle regulatory protein, in pancreatic adenocarcinoma. Br J Cancer. 2002;86:886–892. doi: 10.1038/sj.bjc.6600133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kami K, Doi R, Koizumi M, et al. Downregulation of survivin by siRNA diminishes radioresistance of pancreatic cancer cells. Surgery. 2005;138:299–305. doi: 10.1016/j.surg.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 33.Satoh K, Kaneko K, Hirota M, et al. Expression of survivin is correlated with cancer cell apoptosis and is involved in the development of human pancreatic duct cell tumors. Cancer. 2001;92:271–278. doi: 10.1002/1097-0142(20010715)92:2<271::AID-CNCR1319>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 34.Espenschied J, Lamont J, Longmate J, et al. CTLA-4 blockade enhances the therapeutic effect of an attenuated poxvirus vaccine targeting p53 in an established murine tumor model. J Immunol. 2003;170:3401–3407. doi: 10.4049/jimmunol.170.6.3401. [DOI] [PubMed] [Google Scholar]
- 35.Daftarian P, Song GY, Ali S, et al. Two distinct pathways of immuno-modulation improve potency of p53 immunization in rejecting established tumors. Cancer Res. 2004;64:5407–5414. doi: 10.1158/0008-5472.CAN-04-0169. [DOI] [PubMed] [Google Scholar]
- 36.Song GY, Gibson G, Haq W, et al. An MVA vaccine overcomes tolerance to human p53 in mice and humans. Cancer Immunol Immunother. 2007;56:1193–1205. doi: 10.1007/s00262-006-0270-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrop R, Connolly N, Redchenko I, et al. Vaccination of colorectal cancer patients with modified vaccinia Ankara delivering the tumor antigen 5T4 (TroVax) induces immune responses which correlate with disease control: a phase I/II trial. Clin Cancer Res. 2006;12:3416–3424. doi: 10.1158/1078-0432.CCR-05-2732. [DOI] [PubMed] [Google Scholar]
- 38.Corbett TH, Roberts BJ, Leopold WR, et al. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984;44:717–726. [PubMed] [Google Scholar]
- 39.Tan MC, Goedegebuure PS, Belt BA, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182:1746–1755. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macpherson I, Stoker M. Polyoma transformation of hamster cell clones—an investigation of genetic factors affecting cell competence. Virology. 1962;16:147–151. doi: 10.1016/0042-6822(62)90290-8. [DOI] [PubMed] [Google Scholar]
- 41.Dialynas DP, Wilde DB, Marrack P, et al. Characterization of the murine antigenic determinant, designated L3T4a, recognized by monoclonal antibody GK1.5: expression of L3T4a by functional T cell clones appears to correlate primarily with class II MHC antigen-reactivity. Immunol Rev. 1983;74:29–56. doi: 10.1111/j.1600-065X.1983.tb01083.x. [DOI] [PubMed] [Google Scholar]
- 42.Miconnet I, Coste I, Beermann F, et al. Cancer vaccine design: a novel bacterial adjuvant for peptide-specific CTL induction. J Immunol. 2001;166:4612–4619. doi: 10.4049/jimmunol.166.7.4612. [DOI] [PubMed] [Google Scholar]
- 43.Wang Z, La Rosa C, Mekhoubad S, et al. Attenuated poxviruses generate clinically relevant frequencies of CMV-specific T cells. Blood. 2004;104:847–856. doi: 10.1182/blood-2003-10-3469. [DOI] [PubMed] [Google Scholar]
- 44.Manuel ER, Wang Z, Li Z, et al. Intergenic region 3 of modified vaccinia Ankara is a functional site for insert gene expression and allows for potent antigen-specific immune responses. Virology. 2010;403:155–162. doi: 10.1016/j.virol.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ishizaki H, Song GY, Srivastava T, et al. Heterologous prime/boost with p53-based vaccines combined with toll-like receptor stimulation enhances tumor regression. J Immunother. 2010;33:609–617. doi: 10.1097/CJI.0b013e3181e032c6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gallina G, Dolcetti L, Serafini P, et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest. 2006;116:2777–2790. doi: 10.1172/JCI28828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabrilovich DI, Velders MP, Sotomayor EM, et al. Mechanism of immune dysfunction in cancer mediated by immature Gr-1+ myeloid cells. J Immunol. 2001;166:5398–5406. doi: 10.4049/jimmunol.166.9.5398. [DOI] [PubMed] [Google Scholar]
- 49.Serafini P, Borrello I, Bronte V. Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Semin Cancer Biol. 2006;16:53–65. doi: 10.1016/j.semcancer.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 50.Kusmartsev S, Cheng F, Yu B, et al. All-trans-retinoic acid eliminates immature myeloid cells from tumor-bearing mice and improves the effect of vaccination. Cancer Res. 2003;63:4441–4449. [PubMed] [Google Scholar]
- 51.Serafini P, Meckel K, Kelso M, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med. 2006;203:2691–2702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514–2522. doi: 10.1158/0008-5472.CAN-08-4709. [DOI] [PMC free article] [PubMed] [Google Scholar]