

Abstract
Of all the commercially-available amino acid derivatives for solid phase peptide synthesis, none has a greater abundance of sidechain protection diversity than cysteine. The high reactivity of the cysteine thiol necessitates its attenuation during peptide construction. Moreover, the propensity of cysteine residues within a peptide or protein sequence to form disulfide connectivity allows the opportunity for the peptide chemist to install these disulfides iteratively as a post-synthetic manipulation through the judicious placement of orthogonal pairs of cysteine S-protection within the peptide's architecture. It is important to continuously discover new vectors of deprotection for these different blocking protocol in order to achieve the highest degree of orthogonality between the removal of one species in the presence of another. We report here a complete investigation of the scope and limitations of the deprotective potential of 2,2′-dithiobis(5-nitropyridine) (DTNP) on a selection of commercially-available Cys S-protecting groups. The gentle conditions of DTNP in a TFA solvent system show a remarkable ability to deprotect some cysteine blocking functionality traditionally removable only by more harsh or forcing conditions. Beyond illustrating the deprotective ability of this reagent cocktail within a cysteine-containing peptide sequence, the utility of this method was further demonstrated through iterative disulfide formation in oxytocin and apamin test peptides. It is shown that this methodology has high potential as a stand-alone cysteine deprotection technique or in further manipulation of disulfide architecture within a more complex cysteine-containing peptide template.
Keywords: Cysteine, Protecting Group, Deprotection, DTNP, Thioanisole, Thiol
INTRODUCTION
Cysteine (Cys) is an essential and ubiquitous amino acid, possessing an elevated level of importance in the general function of peptides and proteins due to its ability to form disulfide connectivity with other cysteine residues within the protein’s primary sequence. Specific disulfide architecture is crucial to the effective functioning of many essential proteins as well as for various peptide hormones and other messenger molecules. In order to gain a greater understanding of disulfide-containing peptide systems, it is frequently necessary to carry out their linear construction through solid phase peptide synthesis (SPPS) with proper disulfide closure being brought about as a post-synthetic manipulation. Although there are many proven methodologies for installing post-synthetic disulfide architecture into peptides [1–3], the effectiveness of each approach can be heavily dependent upon the size and sequence of the peptide chain. When multiple disulfide connectivity must be implemented correctly in a stepwise fashion, the avenue through which this is carried out can be anything but trivial.
For no amino acid sidechain is there a greater abundance of sidechain protecting protocol as there is for cysteine [4,5]. This is due in part to the multiple levels of protecting group orthogonality necessary for the requisite construction of multiple disulfide-containing peptides. In a stepwise approach toward entry into multiple disulfide constructs, it often becomes necessary to employ orthogonally-protected cysteine pairs to effect one disulfide closure at a time. For a peptide target in which it is desirable to install each disulfide bond sequentially, it is important that each pair of cysteine thiol protecting groups are orthogonal to each other so that one motif can be removed under conditions to which the remaining blocking protocol are robust. As such, it is desirable to either expand the number of available cysteine protection protocols or to devise more attractive deprotection conditions for existing protection schemes in order to allow the highest degree of blocking-group diversity.
We previously reported a new, gentle methodology for the removal of acetamidomethyl (Acm) and p-methoxybenzyl (Mob) cysteine protection which did not require forcing removal conditions standard for these protecting groups [6]. In this approach, treatment of Cys(Mob)- or Cys(Acm)-containing peptides with varying concentrations of 2,2’-dithiobis(5-nitropyridine) (DTNP) [7] in a 2% thioanisole/TFA solvent resulted in virtually complete deprotection within one hour. In this work, we carried out a methodical assay of a more complete selection of acid-stable commercially-available cysteine protecting groups to these DTNP deprotection conditions. This selection of Cys S-protectants, listed in Table 1, have traditionally required very harsh and/or toxic conditions to effect their removal [8–17]. We present the results of these efforts to illustrate the utility of these comparatively more gentle conditions of deprotection.
TABLE 1.
| Name | Structure | Traditional Removal Conditions |
|
|---|---|---|---|
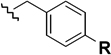
| Acid-Stable Protecting Groups | Bzl R = H |
 |
Na/liq. NH3 (ref. 8) |
| Meb R = CH3 |
50% HF/Anisole (ref. 9) |
||
| Mob R = OCH3 |
AgOTf/TFA/thioanisole (ref. 10) |
||
| Acm | 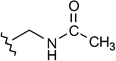 |
Hg(OAc)2 (ref. 11) |
|
| tBu | 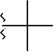 |
Hg(OTf)2 (ref. 12) |
|
| StBu | 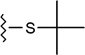 |
P(Ph)3 (ref. 13) |
|
| Acid-Labile Protecting Groups | Trt R = H |
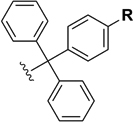 |
95% TFA (ref. 14) |
| Mmt R = OCH3 |
1% TFA/DCM (ref. 15) |
||
| Xan | 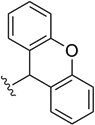 |
1% TFA/DCM (ref. 16) |
|
| Tmob | 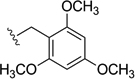 |
7% TFA/DCM (ref. 17) |
|
Bzl = benzyl; Meb = 4-methylbenzyl; Mob = 4-methoxybenzyl; Acm = acetamidomethyl; tBu = tert-butyl; StBu = tert-butylmercapto-; Trt = trityl; Mmt = 4-methoxytrityl; Xan = Xanthenyl; Tmob = 2,4,6-trimethoxybenzyl
In addition to exploring the deprotection profiles of these Cys derivatives on a small test peptide, we investigated the potential for stepwise deprotection/disulfide-closure in model oxytocin systems in which both cysteines were S-protected with identical blocking motifs corresponding to the acid-stable blocking groups in Table 1. In order to strongly illustrate the utility of this method toward stepwise disulfide formation, we utilized a two-disulfide apamin template bearing differentially-protected orthogonal Cys pairings which were iteratively deprotected and cyclized. An understanding of the scope and limitations of these deprotection/cyclization procedures would be an important resource for application to the design of new and more complex disulfide-containing peptides.
MATERIALS AND METHODS
Materials
N,N-dimethylformamide, HPLC-grade acetonitrile, and trifluoroacetic acid were purchased from Fisher Scientific (Pittsburgh, PA). Fmoc-Cys(Acm)-OH, Fmoc-Cys(Mob)-OH, Fmoc-Cys(StBu)-OH, Fmoc-Cys(tBu)-OH, Fmoc-Glu(OtBu)-Thr(ΨMe,Mepro)-OH, and 2-chlorotrityl chloride resin were purchased from Novabiochem (San Diego, CA). All other Fmoc amino acids and O-Benzotriazole-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HBTU) were purchased from RS Synthesis (Louisville, KY). 2-(7-Aza-1H-benzotriazole-1-yl)- N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU) was purchased from Oakwood Products (Jackson Hole, WY). CLEAR-OX resin was purchased from Peptides International (Louisville, KY). 2,2'-dithiobis-5-nitropyridine (DTNP), thioanisole, and all other reagents were purchased from Sigma-Aldrich (Milwaukee, WI). Native apamin was purchased from American Peptide Company (Sunnyvale, CA).
Peptide Syntheses
All peptides were synthesized on a 40 µmole scale using 2-chlorotrityl chloride resin (1.01 mmol/g loading). A Symphony multiple peptide synthesizer (Protein Technologies Inc., Tuscon, AZ) was used to construct the peptide sequences via Fmoc protocol. Double coupling using 1:3 HATU/HBTU activation was employed for peptide elongation. A typical single coupling procedure was as follows: 20% piperidine/DMF (2 × 6 min); DMF washes (6 × 30 s); 5 equiv. Fmoc amino acid and HBTU in 0.4 M NMM/DMF (2 × 30 min); DMF washes (3 × 30 s). Cleavage of peptides from their resins was accomplished through treatment of the resin with 94:2:2:2 TFA/TIPS/H2O/anisole for 2 h. Following filtration of the resin, the cleavage supernatant was evaporated to one tenth its original volume, followed by precipitation of the crude peptide into cold anhydrous diethyl ether.
High Pressure Liquid Chromatography (HPLC)
HPLC analysis was done on a Shimadzu analytical HPLC system with LC-10AD pumps, SPD-10A UV-Vis detector, and SCL-10A controller using a Symmetry® C18 −5 µm column from Waters (4.6 × 150 mm). Aqueous and organic phases were 0.1% TFA in water (Buffer A) and 0.1% TFA in HPLC-grade acetonitrile (Buffer B), respectively. Beginning with 100% Buffer A, a 1.4 ml/min gradient elution increase of 1% Buffer B/min for 50 min was used for all analytical peptide chromatograms. Peptide signals were detected at both 214 and 254 nm. Preparative HPLC purification was carried out on a Shimadzu preparatory HPLC system utilizing LC-8A pumps, an SPD-10A UV-Vis detector, and an SCL-10A controller. A Waters SymmetryPrep C18 preparatory column (7 µm pore size, 1900 × 150 mm) was utilized in these separations. Beginning with 100% Buffer A, a 17 ml/min gradient elution increase of 1% Buffer B/min for 50 min was used for all preparative chromatograms.
Mass Spectrometry
Matrix-Assisted Laser Desorption Ionization / Time-of-Flight (MALDI-TOF) mass spectra were collected on a Voyager DE-Pro instrument under positive ionization and in reflectron mode. All samples were run using a matrix of 10 mg/ml 2,5-dihydroxybenzoic acid (DHB), vacuum-dried from a solution of 1:1 H2O/ACN buffered to 0.1% TFA. Mass spectra of all peptide intermediates and final products are reported in the Supplemental Information for this manuscript.
DTNP deprotection assay conditions for VTGGC(X)A test peptides
1.0 mg (~1.7 µmol) aliquots of VTGGC(X)A test peptides 1–6 were dissolved in 200 µL of either 100% TFA or 2% thioanisole/TFA to a final concentration of ~ 8.5 mM. Each of these solutions was incubated with differing quantities of DTNP corresponding to 1.1 eq. (10 mM), 3.3 eq. (30 mM), 6.7 eq. (60 mM), 11 eq. (100 mM), and 15 eq. (137 mM) with agitation at 25 °C for 1 hr. At the end of this time, cold diethyl ether was added to each reaction and the crude precipitated product isolated by centrifugation. After drying the pellets, the crude isolates were dissolved in 10% ACN/H2O buffered to 0.1% TFA and subjected to analytical HPLC analysis.
DTNP deprotection and disulfide formation in oxytocin test peptides
2.0 mg (~1.7 µmol) aliquots of each di-protected oxytocin peptide 7–10 were dissolved in 200 µL of 2% thioanisole/TFA to a final concentration of ~ 8.5 mM. Each of these solutions was incubated with 20 eq. (183 mM) of DTNP with agitation at varying temperatures and reaction times as listed in Table 2. At the end of the reaction time, cold diethyl ether was added to each reaction and the crude precipitated product isolated by centrifugation. After drying the pellets, the crude isolates were dissolved in 2–3 mL 100 mM NH4HCO3 in 9:1 H2O/ACN [pH: 7.5] to yield a final peptide concentration of 1–2 mM. To a vigorously-stirred solution of each of these intermediates, 1 eq. of DTT dissolved in 500 µL of the same buffer was added in one portion and the solution was allowed to stir for an additional 5 minutes. Following analytical HPLC analysis of the mixture, the entire reaction contents were injected onto the preparative HPLC and the disulfide-containing native oxytocin was isolated by lyophilization of the fractions containing the desired product [m/z: 1010.3 (M+H)].
TABLE 2.
| Protecting Group P |
Peptide Synthetic Yielda |
Deprotection Conditions | ||||
|---|---|---|---|---|---|---|
| Eq. (mM) DTNP | Rxn Time/Temp. | % Deprotection/Cyclization (measured by HPLC)b |
||||
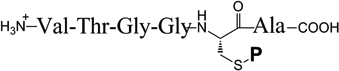 | ||||||
| Bzl (1) | 61% | 15 (137) | 1h / 25°C | 0 | ||
| Meb (2) | 57% | 15 (137) | 1h / 25°C | 16 | ||
| Mob (3) | 58% | 3 (27) | 1h / 25°C | 100 | ||
| Acm (4) | 43% | 15 (137) | 1h / 25°C | 86 | ||
| tBu (5) | 50% | 15 (137) | 1h / 25°C | 92 | ||
| StBu (6) | 45% | 1 (9) | 1h / 25°C | 100 | ||
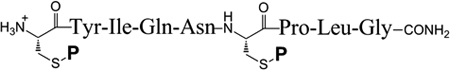 | ||||||
| Mob (7) | 32% | 20 (183) | 3h / 37°C | 100 | 100 | |
| Acm (8) | 33% | 20 (183) | 8h / 50°C | 45 | NA | |
| tBu (9) | 40% | 20 (183) | 3h / 50°C | 100 | 100 | |
| StBu (10) | 34% | 20 (183) | 8h / 50°C | 95 | 100 | |
| Mob (11) | 49% | 32% | 20 (73) | 3h / 37°C | 100 | 100 |
| Acm (12) | 67% | 58% | 20 (73) | 8h / 50°C | 100 | 100 |
| tBu (13) | 76% | 46% | 20 (73) | 8h / 50°C | 100 | 100 |
| (14) | 36% | 20 (73) | 8h / 50°C | 100 | 77 | |
| 100 | 100 | |||||
For apamin constructs 11–13, the first value corresponds to syntnetic yield of uncyclized peptide, and the second value corresponds to isolated yield of CLEAROX-mediated first disulfide closure (illustration shown).
For Apamin constructs 11–14, the first value corresponds to the effectiveness (determined by HPLC) of DTNP deprotection and the second value corresponds to HPLC-derived effectiveness for DTT-cyclization of bis-Npys intermediate.
1st disulfide formation in apamin test peptides 11–13
To install the first disulfide connectivity, 10 mg (~4.5 µmol) of each C1,11-(X), C3,15-(SH) apamin 11–13 were dissolved in 5 mL 100 mM NH4HCO3 in 9:1 H2O/ACN [pH: 7.5] and gently agitated with 0.215 g (10 eq.) CLEAR-OX resin (0.21 mmol/g) [18] for 3 h. The solution containing the peptide was filtered and the resin was washed with 3 × 3 mL 9:1 H2O/ACN. All of the washes were combined with the original filtrate and the solution was frozen to −80 °C and lyophilized. Following lyophilization of the crude isolate, preparative HPLC purification and subsequent lyophilization was carried out to afford the single disulfide-bonded intermediate [11 m/z: 2271.6 (M+H); 12 m/z: 2173.5 (M+H); 13 m/z: 2143.5 (M+H)].
DTNP deprotection and 2nd disulfide formation in apamin test peptides 11–13
5 mg (~2.3 µmol) of each disulfide-bonded intermediate 11–13 was dissolved in 500 µL of 2% thioanisole/TFA to a final concentration of ~ 4.6 mM. Each of these solutions was incubated with differing quantities of DTNP under varying time and temperature conditions dependent upon the identity of the protecting group (see Table 2). At the end of this time, cold diethyl ether was added to each reaction and the crude precipitated product isolated by centrifugation. Following drying of the pellets, the crude isolates were dissolved in 2–3 mL 100 mM NH4HCO3 in 9:1 H2O/ACN [pH: 7.5] to yield a final peptide concentration of 1–2 mM and treated with 1 eq. DTT as previously described to yield the 2-disulfide-containing native apamin [m/z: 2029.5 (M+H)] which, following analytical HPLC analysis, was purified via the injection of the entire reaction contents onto the preparative HPLC. Fractions containing the desired product were frozen to −80 °C and lyophilized. The identity of the synthetic apamin was confirmed by coinjection with an authentic sample (see Supplemental Information).
Stepwise DTNP deprotection and disulfide formation in orthogonally-protected apamin test peptide 14
20 mg (~8.5 µmol) of C1,11 StBu)-C3,13 tBu) apamin peptide 14 was dissolved in 700 µL of TFA to a final concentration of ~ 12.1 mM. This solution was incubated with 54 mg (20 eq.) DTNP under time and temperature conditions as listed in Table 2. At the end of this time, cold diethyl ether was added to each reaction and the crude precipitated product isolated by centrifugation. Following drying of the pellet, the crude isolate was dissolved in 10 mL 100 mM NH4HCO3 in 9:1 H2O/ACN [pH: 7.5] yielding a final peptide concentration of 1–2 mM and treated with 1 eq. DTT as previously described to yield the C3,13-cyclized, C1,11(StBu)-protected intermediate [m/z: 2207.6 (M+H)] which was, following analytical HPLC analysis, purified via the injection of the entire reaction contents onto the preparative HPLC. Fractions containing the desired product were frozen to −80 °C and lyophilized. 7 mg (3.17 µmol) C3,13-cyclized, C1,11 StBu)-protected intermediate was dissolved in 500 µL of 2% thioanisole/TFA, followed by addition of 20 mg (20 eq.) DTNP under time and temperature conditions as listed in Table 2. At the end of this time, Et2O-workup and closure of the remaining disulfide connectivity via treatment with 1 eq. DTT was carried out as previously described to afford the native apamin construct [m/z: 2029.5 (M+H)]. The identity of the synthetic apamin was confirmed by coinjection with an authentic sample (see Supplemental Information).
Results and Discussion
Since it was a goal of this research to examine the deprotection profiles of the common Cys S-protectants shown in Table 1 via this DTNP-mediated approach, we first needed to prioritize the large number of these protecting groups into an assemblage which would illustrate a benefit to their use in SPPS. As such, we immediately disregarded all acid-labile blocking groups due to the TFA-mediated conditions of this protocol. Although the Acm and Mob functionality had been investigated by us previously [6], we chose to reproduce that data along with that from the deprotection assays of the heretofore untested Cys S-protectants in order to directly compare and contrast all of their deprotection profiles.
As illustrated in Figure 1, the general protocol for Cys S-deprotection in the test systems involved the incubation of the protected Cys-containing peptide with DTNP in a 2% thioanisole/TFA (or neat TFA) solvent system under time and temperature constraints specific for the test sequence. During the incubation with DTNP, the Cys S-protectant is removed and substituted by the 2-(5-nitropyridyl) (Npys) group derived from DTNP fragmentation. The reaction was then quenched via precipitation of the entire contents into cold diethyl ether. After isolation of the precipitate via centrifugation, the crude material was either maintained as its Npys conjugate or (in the case of multiple-thiol-containing systems) treated with one equivalent of DTT in order to induce collapse into the desired disulfide.
Figure 1.

General DTNP deprotection overview showing conversion to 2-(5-nitropyridyl) [5-Npys] intermediate derived from DTNP fragmentation. Facile conversion to the cysteine thiol was achieved by treatment of the crude deprotected peptide isolate with excess thiol reductant such as DTT.
The mechanism for this deprotection frequently required thioanisole as an additive, as most of the S-protecting groups studied in this investigation are stable in its absence. As previously reported [6] and summarized in Figure 2a, the assumed reactive intermediate in the deprotection sequence is the trivalent sulfonium thioanisole-Npys conjugate derived from attack of the thioanisole sulfur on the electron-deficient disulfide of DTNP. This intermediate, when in proximity to the protected cysteine, induces attack upon the very reactive sulfur within the conjugate, forming a trivalent sulfonium intermediate at the cysteine residue. This transitory species ultimately collapses into its corresponding Npys conjugate following attack of a scavenger species (Figure 2b).
Figure 2.

Putative mechanism of DTNP-mediated deprotection. (a) Formation of activated DTNP-thioanisole intermediate. (b) Cysteine deprotection brought about by initial attack of cysteine sulfur on activated intermediate to form trivalent sulfonium intermediate, followed by collapse into the 5-Npys intermediate by scavenging of the protecting group fragment.
The basis for determination of the robustness of the acid-stable protecting groups shown in Table 1 was the quantity of DTNP required for removal of each one. In our previous report, these amounts were specified as the number of equivalents of DTNP added to a particular volume of 2% thioanisole/TFA, due to the fact that some species required only stoichiometric amounts of DTNP to effect complete protecting group removal. For the present study we thought it important to measure actual concentration (mM) of DTNP, as it illustrates a more realistic kinetic measure of deprotection effectiveness, especially when studying the deprotection profile of the more robust protecting groups. Further, an absolute concentration value better expresses differences in effective concentration brought about by variations in solution volume when identical stoichiometric amounts of DTNP are used in assays.
Our intention was to study the effectiveness of this deprotection methodology from a number of different standpoints. First, it had become clear since our initial publication that the deprotective potential of this process was heavily dependent upon specific amino acid sequence, peptide length, and cysteine placement within the sequence [Flemer S, Hondal RJ. Unpublished Results ]. Also, we were interested in using this deprotection approach in tandem with disulfide formation. To address these issues and to illustrate a broader understanding of the utility of this method, three peptide systems were constructed encompassing a wide diversity of length, sequence, and disulfide architecture. The first system bore the identical sequence (VTGGCA) as the small test peptide previously studied on Acm- and Mob-protected cysteines [6], except that in this case additional peptides were built bearing S-protection corresponding to the acid-stable Cys derivatives outlined in Table 1. A second oxytocin template [19] was constructed in order to merge a standard DTNP deprotection of identically-protected cysteine pairs with subsequent disulfide formation to yield the native cyclized sequence. Lastly, an apamin peptide template [20] was designed to highlight the use of this deprotection methodology in an orthogonal fashion within a two disulfide containing system.
The linear syntheses of the VTGGCA and oxytocin sequences were carried out in a routine fashion with no difficulties. Syntheses of the apamin sequences were surprisingly challenging, however, given the presumed ease with similar systems whose construction has been described in the literature [21]. Early attempts at synthesis of this sequence met with crude product profiles fraught with deletion sequences, as evidenced by HPLC. These deletion-prone syntheses were dismayingly reproducible, even when using very low-loading resins and employing very high-end coupling conditions (ie: HATU). The key to overcoming this obstacle was finally discovered to be the use of a Glu7-Thr8 pseudoproline dipeptide sequence [22] in order to disrupt undesired secondary-structure formation during construction. The installation of this specialized dipeptide during the synthesis of the apamin constructs proved successful in overcoming the majority of deletion problems inherent in the linear sequence.
DTNP-deprotection assays of the VTGGCA test systems were carried out on equimolar aliquots of protected peptide at increasing intervals of DTNP concentration in both neat TFA and 2% thioanisole/TFA. Following ether precipitation of each reaction, comparison of HPLC chromatogram peak areas of the crude deprotection isolates allowed for deblocking efficiency to be plotted as a function of increasing DTNP concentration for each protecting group assayed (Figure 3). In the case of these simple test sequences, the deprotected species were measured as their 5-Npys conjugates.
Figure 3.

Graphic representation of the effectiveness of DTNP-mediated deprotection in VTGGCA test peptide systems (1–6) containing a selection of cysteine S-protectants, showing comparisons of deprotection effectiveness in the presence and absence of thioanisole.
As illustrated in Figure 3, a diverse spectrum of applicability was found for this deprotection methodology toward these cysteine S-protectants. The benzyl-templated series show differing extents of lability, highly dependent upon the identity of the electron-donating substituent at the para position on the phenyl ring, with effectiveness of deprotection noted as Mob > Meb > Bzl. The Bzl functionality showed no lability whatsoever even at higher concentrations of DTNP, while the Meb group demonstrated only partial (~16%) removal. In the presence of thioanisole, the Mob group was completely removed when treated with ~2 eq. (20 mM) DTNP. However, in the absence of thioanisole, the Mob functionality was much more robust, showing only partial removal, even at greatly increased DTNP concentrations.
Deprotection assays of the remaining Cys S-protectants revealed additional instances in which thioanisole played a vital role in deprotection efficiency as well as those in which it did not. The S-deprotection profile of the tBu group, for instance, showed equal efficiency to the DTNP conditions whether thioanisole was used or not. This finding stands to reason due to the existing efficient Npys-Cl tBu-removal vector for cysteine [23], based upon both reagents’ installation of the identical electron-deficient 5-Npys moiety onto the Cys thiol. In contrast to the thioanisole-independence of the tBu deprotection protocol, the DTNP-lability of the Acm and StBu functionalities were more highly thioanisole-dependent. Acm deprotection followed the general trend as that of Mob, although it was found to be much more robust. The DTNP deprotection profile of the StBu group was quite remarkable in that there existed an absolute requirement for thioanisole. As illustrated in Figure 3, StBu protection was exceedingly robust in the absence of thioanisole, showing no lability even at elevated DTNP concentrations. However, in the presence of thioanisole, complete StBu deprotection was carried out at stoichiometric DTNP concentrations. Indeed, the StBu group highlighted the squarest orthogonality profile yet observed using this methodology.
It was found that the previously-utilized DTNP conditions did not translate to the expected extent of deprotection in the case of oxytocin test sequences 7–10. In fact, even at higher DTNP concentrations, MALDI analysis indicated that while one Cys was consistently converted to its corresponding 5-Npys conjugate, the other partner was sluggish to deprotect and subsequently stalled, showing incomplete global deprotection under the previously-used conditions (data not shown). As a result, higher DTNP concentrations with increased temperature and time were required to effect dual deprotection of these constructs in most instances. It was found that the ease of bis-deprotection followed the trend Mob > tBu > StBu > Acm, with full deprotection of the bis-Acm pair unattainable. Surprisingly, although the StBu protection was most easily removed on the single cysteine containing test peptide 6, it was one of the more robust protecting groups in the bis-Cys containing oxytocin template.
It was hoped that in carrying out these dual DTNP deprotections on the oxytocin sequences, spontaneous disulfide formation might occur due to the highly favorable disposition of the two cysteine residues within the sequence. It was found, however, that the species which emerged in all cases was the bis-Npys adduct, with less than 1% formation of the cyclized native peptide. We envisioned that it might be possible to induce disulfide cyclization if, after isolation of the bis-Npys intermediate, one equivalent of thiol reductant (ie: DTT) was added to a NH4HCO3-buffered aqueous solution of this intermediate to carry out reduction of one of the Npys moieties, leading to spontaneous cyclization of the liberated cysteine thiol upon its Npys-conjugated partner to afford the native oxytocin species. After some optimization, we settled on a method of incubating the crude deprotection isolate in a 100 mM NH4HCO3 buffer to a final optimal peptide concentration of 1 mM. To this briskly-stirred mixture, a 1 eq. bolus of DTT was added followed by immediate HPLC analysis of the resulting deep yellow solution. Identical HPLC profiles from various time points after the initial addition of DTT indicated that the collapse of the hemi-reduced intermediate into the native oxytocin was virtually instantaneous (Figure 4).
Figure 4.

Stepwise HPLC comparison of the deprotective profile of oxytocin systems 7–10 (bis-Acm system used as example). Chromatogram on left shows bis-Acm-protected oxytocin peptide (1). Middle chromatogram shows crude HPLC profile following treatment with DTNP/thioanisole of bis-Npys oxytocin intermediate (2). Chromatogram on right is crude HPLC profile following treatment with 1 eq. DTT, showing peaks for free 5-Npys moiety (3) and cyclized native oxytocin (4).
Since the apamin constructs were templates for highlighting this method’s utility in stepwise disulfide construction, the DTNP deprotection procedure was merely a component of a larger process. As illustrated in Table 2, peptides 11–13 were designed to have two differentially-protected cysteine pairs. The Cys3,15 pair, possessing trityl S-protection during synthesis, provide native thiol functionality upon peptide cleavage. Alternatively, the Cys1,11 pair was designed to bear the more robust protection protocol (Mob, Acm, tBu) whose removal was to be effected by the DTNP-mediated process. The cyclization of the Cys3,15 free thiol pair was achieved by incubation of an aqueous solution of each peptide with CLEAR-OX resin [18]. Following isolation and purification of this intermediate, the DTNP deprotection protocol was then carried out as previously described. In addition to the protected Cys pairs requiring different concentrations of DTNP to effect full deprotection, apamin constructs 11–13 required elevated reaction temperatures of 37–50 °C and longer reaction times in order to drive the dual deprotection to completion, similar to those required for the bis-deprotection of oxytocin systems 7–10, except that in this case the bis-Acm pair on the apamin construct could be fully deprotected. This finding again highlighted the differences in deprotection effectiveness using this methodology when applied to different peptide sequences.
Once the DTNP deprotection chemistry had been carried out on each apamin template, HPLC and MALDI data again suggested that the crude isolates consisted exclusively of the bis-Npys species. In similar fashion to the previous disulfide closure on oxytocin models 7–10, the addition of 1 eq. DTT to each peptide dissolved in aqueous buffer induced the singular collapse of these intermediates into native apamin structure (Figure 5a). Care had to be taken in these apamin conversions, however, as we found that initial peptide isolate concentrations exceeding 2 mM began to show competing intermolecular disulfide formation (data not shown). Co-injection of the experimentally-derived apamin constructs with an authentic sample yielded a single peak without splitting or shouldering, confirming that disulfide scrambling had not occurred (see Supplemental Information).
Figure 5.

a: Stepwise HPLC comparison of the deprotective profile of apamin systems 11–13. Spectrum on left shows C1,11 Protected)-C3,13(SH) apamin peptide (bis-Acm system used as example) (1). Middle spectrum illustrates crude HPLC profile following treatment with Clear-OX resin, showing peak for disulfide-bonded intermediate (2). Chromatogram on right is crude HPLC profile following treatment with DTNP/thioanisole followed by 1 eq. DTT, showing peaks for free 5-Npys moiety (3) and cyclized native apamin (4). 5b: Stepwise HPLC comparison of the deprotective profile of StBu/tBu apamin system 14. Chromatogram on left shows StBu/tBu-protected apamin peptide (1). Middle chromatogram illustrates crude HPLC profile following treatment with DTNP and 1 eq. DTT, showing peaks for free 5-Npys moiety (2), native apamin (3), and desired cyclized StBu intermediate (4). Chromatogram on right is crude HPLC profile of purified cyclized StBu intermediate following treatment with DTNP/thioanisole followed by 1 eq. DTT, showing peaks for free 5-Npys moiety (2) and cyclized native apamin (5).
As a final testament to the selectivity of this approach and to its applicability toward stepwise disulfide formation, an apamin-based model system was constructed bearing tBu-protection on the Cys3,15 pair and StBu-protection at the Cys1,11 pair (14). Since these two protecting groups were shown to be orthogonal in the absence of thioanisole, a two-step approach toward sequential disulfide formation was carried out (Figure 5b). The peptide was carried through one iteration of (tBu) deprotection/cyclization via treatment with DTNP/TFA in the absence of thioanisole followed by treatment of the crude isolate in 100 mM ammonium bicarbonate with 1 eq. DTT. HPLC and MALDI analysis of the resulting solution showed that both of the tBu protectants were indeed removed in the presence of the StBu groups and the Cys3,15 disulfide was selectively formed [m/z: 2207.7 (M+H)]. Following isolation and purification of the hemi-cyclized intermediate, further treatment with DTNP (this time in the presence of thioanisole) removed both StBu protectants, and the second disulfide bond was formed as previously described. Most likely due to the underlying architecture of construct 14, some difficulties were encountered in the DTT-cyclization step following the first (tBu) deprotection. It was found that, in addition to the desired single-disulfide intermediate produced by DTT-mediated collapse of the bis-Npys intermediate, a minor yet significant amount of native apamin was also present in the crude reduction mixture. This was likely the result of unintentional over-reduction of the intermediate, in which excess DTT partially reduced the StBu protecting groups still present within the peptide framework and subsequent collapse of the apamin construct into its conformationally-favored native structure may have resulted. Since it is well known that Cys(StBu) is readily reduced with exogenous thiol at slightly basic conditions, this over-reduction phenomenon is not surprising.
Conclusions
An expansion on previous work pertaining to the effectiveness of DTNP/TFA deprotection on two common cysteine S-protecting groups has been carried out. Specifically, the acid-stable protecting groups listed in Table 1 were assayed to determine their respective lability to these same conditions in both the presence and the absence of thioanisole. DTNP assays carried out on Cys test peptides bearing these six protecting groups in the presence as well as the absence of thioanisole showed a wide diversity of deprotective effectiveness. In some cases, in particular that of the StBu-protected Cys peptide, thioanisole additive accelerated the deprotection significantly. These protecting group lability trends, once determined, were applied to more complex peptide models bearing multiple Cys residues, carrying out protecting group removal in tandem with stepwise disulfide closure.
The continued development of new protecting groups or application of new conditions for more facile removal of existing protecting groups is a crucial consideration when designing chemical syntheses of increasingly-complex peptides. Due to the high reactivity of its sidechain thiol, blocking protocol for cysteine has enhanced importance in approaches toward iterative assembly of multiple disulfide-containing peptide systems. The method expanded upon here adds an essential, milder deprotection vector for many commercially-available cysteine protectants. It is hoped that this methodology can be applied in greater detail to amino acid constructs similar in reactivity to cysteine (ie: selenocysteine) but whose current protection scheme is limited by architecture and available deprotection methodologies.
Supplementary Material
Footnotes
These studies were supported by National Institutes of Health Grant GM094172 to RJH.
HPLC chromatograms and MALDI mass spectra of all peptide intermediates and final products are contained in the Supplemental Information section associated with this article.
REFERENCES
- 1.Annis I, Hargittai B, Barany G. Disulfide bond formation in peptides. Methods Enzymol. 1997;289:198–221. doi: 10.1016/s0076-6879(97)89049-0. [DOI] [PubMed] [Google Scholar]
- 2.Moroder L, Besse D, Musiol H-J, Rudolph-Böhner S, Siedler F. Oxidative folding of cystine-rich peptides vs. regioselective cysteine pairing strategies. Biopolymers. 1996;40:207–234. doi: 10.1002/(sici)1097-0282(1996)40:2<207::aid-bip2>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 3.Andreu D, Albericio F, Solé NA, Munson MC, Ferrer M, Barany G. In: Molecular Biology - Peptide Synthesis Protocols. Pennington MW, Dunn BM, editors. Vol 35. Totowa NJ: Humana Press; 1994. [Google Scholar]
- 4.Isidro-Llobet A, Alvarez M, Albericio F. Amino acid protecting groups. Chem. Rev. 2009;109:2455–2504. doi: 10.1021/cr800323s. [DOI] [PubMed] [Google Scholar]
- 5.Moroder L, Musiol H-J, Schaschke N, Chen L, Hargittai B, Barany G. In: Houben-Weyl - Synthesis of Peptides and Peptidomimetics. Goodman M, Felix A, Moroder L, Toniolo C, editors. Vol E22a. Stuttgart: Thieme; 2003. [Google Scholar]
- 6.Harris KM, Flemer S, Hondal RJ. Studies on deprotection of cysteine and selenocysteine side-chain protecting groups. J. Pept. Sci. 2007;13:81–93. doi: 10.1002/psc.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grassetti DR, Murray JF., Jr The use of 2,2’-dithiobis-(5-nitropyridine) as a selective reagent for the detection of thiols. J. Chromatog. 1969;41:121–123. doi: 10.1016/0021-9673(64)80109-6. [DOI] [PubMed] [Google Scholar]
- 8.du Vigneaud V, Audrieth LF, Loring HS. The reduction of cystine in liquid ammonia by metalic sodium. J. Am. Chem. Soc. 1930;52:4500–4504. [Google Scholar]
- 9.Erickson BW, Merrifield RB. Acid stability of several benzylic protecting groups used in solid phase peptide synthesis; rearrangement of O-benzyltyrosine to 3-benzyltyrosine. J. Am. Chem. Soc. 1973;95:3750–3756. doi: 10.1021/ja00792a046. [DOI] [PubMed] [Google Scholar]
- 10.Tamamura H, Otaka A, Takada W, Terakawa Y, Yoshizawa H, Masuda M, Ibuka T, Murakami T, Nakashima H, Waki M, Matsumoto A, Yamamoto N, Fujii N. Solution-phase synthesis of an anti-human immunodeficiency virus peptide, T22 ([Tyr5,12, Lys7]-Polyphemusin II), and the modification of Trp by the p-methoxybenzyl group of Cys during trimethylsilyl trifluoromethanesulfonate deprotection. Chem. Pharm. Bull. 1995;43:12–18. doi: 10.1248/cpb.43.12. [DOI] [PubMed] [Google Scholar]
- 11.Veber DF, Milkowski JD, Varga SL, Denkewalter RG, Hirschmann R. Acetamidomethyl; a novel thiol protecting group for cysteine. J. Am. Chem. Soc. 1972;94:5456–5461. doi: 10.1021/ja00770a600. [DOI] [PubMed] [Google Scholar]
- 12.Nishimura O, Kitada C, Fujino M. New method for removing the S-p-methoxybenzyl and S-t-butyl groups of cysteine residues with mercuric trifluoroacetate. Chem. Pharm. Bull. 1978;26:1576–1585. [Google Scholar]
- 13.Moroder L, Gemeiner M, Goehring W, Jaeger E, Thamm P, Wunsch E. New synthesis of somatostatin according to the S-tert-butylthiocysteine procedure. Biopolymers. 1981;20:17–37. doi: 10.1002/bip.1981.360200103. [DOI] [PubMed] [Google Scholar]
- 14.Kellenberger C, Hietter H, Luu B. Regioselective formation of the three disulfide bonds of a 35-residue insect peptide. Peptide Res. 1995;8:321–327. [PubMed] [Google Scholar]
- 15.Barlos K, Gatos D, Hatzi O, Koch N, Koutsogianni S. Synthesis of the very acid-sensitive Fmoc-Cys(Mmt)-OH and its application in solid-phase peptide synthesis. Int. J. Peptide Protein Res. 1996;47:148–153. doi: 10.1111/j.1399-3011.1996.tb01338.x. [DOI] [PubMed] [Google Scholar]
- 16.Han Y, Barany G. Novel S-xanthenyl protecting groups for cysteine and their applications for the Nα-9-Fluorenylmethyloxycarbonyl (Fmoc) strategy of peptide synthesis. J. Org. Chem. 1997;62:3841–3848. [Google Scholar]
- 17.Munson MC, Garcia-Echeverria C, Albericio F, Barany G. S-2,4,6- trimethoxybenzyl (Tmob): a novel cysteine protecting group for the N-α-(9- fluorenylmethoxycarbonyl) (Fmoc) strategy of peptide synthesis. J. Org. Chem. 1992;57:3013–3018. [Google Scholar]
- 18.Darlak K, Wiegandt-Long D, Czerwinski A, Darlak M, Valenzuela F, Spatola AF, Barany G. Facile preparation of disulfide-bridged peptides using the polymer-supported oxidant CLEAR-OX. J. Peptide Res. 2004;63:303–312. doi: 10.1111/j.1399-3011.2004.00153.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee HJ, Macbeth AH, Pagani JH, Young WS. Oxytocin: the great facilitator of life. Progress in Neurobiology. 2009;88:127–151. doi: 10.1016/j.pneurobio.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Habermann E. Apamin. Pharmacol. Ther. 1984;25:255–270. doi: 10.1016/0163-7258(84)90046-9. [DOI] [PubMed] [Google Scholar]
- 21.Fiori S, Pegoraro S, Rudolph-Böhner S, Cramer J, Moroder L. Synthesis and conformational analysis of apamin analogs with natural and non-natural cystine/selenocystine connectivities. Biopolymers (Peptide Science) 1999;53:550–564. doi: 10.1002/(SICI)1097-0282(200006)53:7<550::AID-BIP3>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Martin F, White P, Steinauer R, Cote S, Tulla-Pucche J, Albericio F. The synergy of ChemMatrix resin and pseudoproline building blocks renders RANTES, a complex aggregated chemokine. Biopolymers (Peptide Science) 2006;84:566–575. doi: 10.1002/bip.20564. [DOI] [PubMed] [Google Scholar]
- 23.Pastuszak JJ, Chimiac A. tert-Butyl group as thiol protection in peptide synthesis. J. Org. Chem. 1981;46:1868–1873. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.