

Abstract
RNA healing and sealing enzymes drive informational and stress response pathways entailing repair of programmed 2’,3’ cyclic PO4/5’-OH breaks. Fungal, plant and phage tRNA ligases use different strategies to discriminate the purposefully broken ends of the anticodon loop. Whereas phage ligase recognizes the tRNA fold, yeast and plant ligases do not and are instead hard-wired to seal only the tRNA 3’-OH, 2’-PO4 ends formed by healing of a cyclic phosphate. tRNA anticodon damage inflicted by secreted ribotoxins such as fungal γ-toxin underlies a rudimentary innate immune system. Yeast cells are susceptible to γ-toxin because the sealing domain of yeast tRNA ligase is unable to rectify a break at the modified wobble base of tRNAGlu(UUC). Plant and phage tRNA repair enzymes protect yeast from γ-toxin because they are able to reverse the damage. Our studies underscore how a ribotoxin exploits an Achilles’ heel in the target cell’s tRNA repair system.
INTRODUCTION
“RNA repair” is a mechanism to rectify purposeful breaks in tRNAs and mRNAs incurred during RNA processing and cellular stress. Examples include virus-mediated tRNA repair to evade a host antiviral response (Amitsur et al., 1987), splicing of intron-containing tRNAs (Abelson et al., 1998), and mRNA splicing in the unfolded protein response (Sidrauski et al., 1996). Breakage of the target RNA is triggered by a site-specific endonuclease that generates 2’,3’ cyclic phosphate and 5’-OH ends. For the damage to be repaired, both broken ends must be healed before they can be sealed by an RNA ligase.
Two pathways of tRNA repair have been well delineated: yeast-type and phage-type. The yeast-type pathway is catalyzed by Saccharomyces cerevisiae Trl1, a multifunctional tRNA ligase composed of separable healing and sealing domains (Fig. S1A and 4B) (Apostol et al., 1993; Sawaya et al., 2003). The C-terminal healing domain consists of a polynucleotide kinase module that converts the tRNA 5’-OH end to a 5’-PO4 and a cyclic phosphodiesterase (CPD) module that hydrolyzes the 2’,3’ cyclic phosphate end to a 3’-OH, 2’-PO4. The N-terminal ligase domain then joins the healed ends to form a tRNA with a 2’-PO4, 3’-5’ phosphodiester at the splice junction. The 2’-PO4 is ultimately removed by a phosphotransferase, Tpt1 (Spinelli et al., 1997). Plants have a trifunctional tRNA ligase homologous to yeast Trl1 (Englert and Beier, 2005). Arabidopsis thaliana tRNA ligase (AtRNL; Fig. S1A and 4B) is a true ortholog of Trl1, insofar as it provides all the essential tRNA splicing functions in vivo when expressed in yeast trl1Δ cells (Wang et al., 2006a).
Fig. 4. RNA sealing enzymes dictate sensitivity or resistance to γ-toxin.
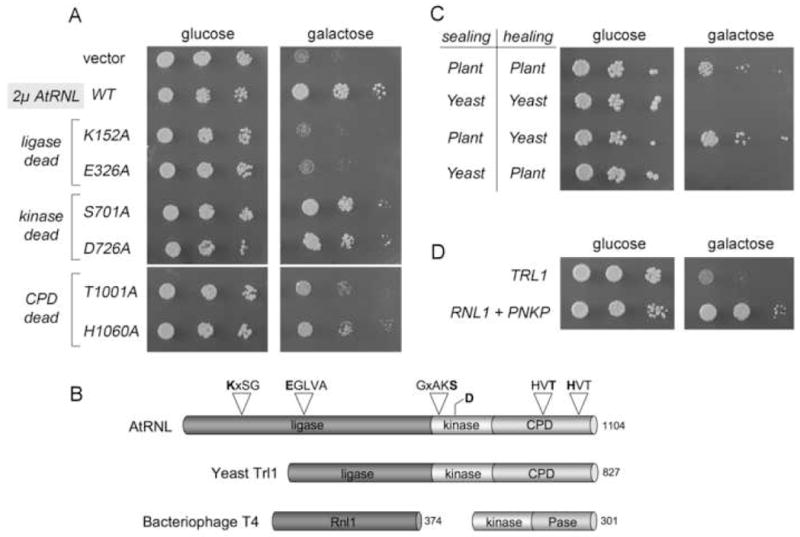
(A) Growth of TRL1 cells harboring pGAL-γ-toxin and 2μ TPI1-AtRNL plasmids encoding wild type plant tRNA ligase or the indicated mutants was assessed by spotting serial 5-fold dilutions to minimal synthetic agar medium containing glucose or galactose. (B) The tRNA ligases of plant (AtRNL) and yeast (Trl1) are composed of three discrete catalytic domains: an N-terminal ligase module; a central 5’-OH polynucleotide kinase module; and a C-terminal RNA 2’,3’ cyclic phosphodiesterase (CPD) module. The positions of the covalent adenylylation motif (KxxG) and the metal-binding motif (EGxxx) at the ligase active site, the P-loop motif GxxK(S/T) at the kinase active, and the two HxT motifs that comprise the CPD active site are depicted above the plant polypeptide. The sites of enzyme-inactivating alanine mutations in the AtRNL active sites are highlighted in bold. The phage T4 tRNA repair system consists of separate sealing (Rnl1) and healing (Pnkp) enzymes. (C) The effects of galactose-induced expression of γ-toxin in trl1Δ cells with 2μ plasmids expressing the indicated combinations of healing and sealing domains of plant and yeast tRNA ligase was tested by spotting serial 5-fold dilutions of the cultures to glucose and galactose-containing medium. (D) Growth of trl1Δ cells carrying pGAL-γ-toxin and CEN plasmids expressing either TRL1 or T4 RNL1 and PNKP was assessed on glucose and galactose medium. The plates were incubated at 30°C for 2.5 (glucose) and 5 days (galactose).
The phage-type tRNA repair pathway is catalyzed by the T4 enzymes polynucleotide kinase-phosphatase (Pnkp) and RNA ligase 1 (Rnl1) (Amitsur et al., 1987). Pnkp is a bifunctional enzyme that removes the 2’,3’ cyclic phosphate to form a 3’-OH, 2’-OH end and phosphorylates the 5’-OH end to yield a 5’-PO4. Rnl1 then joins the 3’-OH and 5’-PO4 termini to form a standard 3’-5’ phosphodiester at the repair junction. Pnkp consists of an N-terminal kinase domain and a C-terminal phosphatase domain (Fig. S1A). The T4 kinase domain includes a nucleotide-binding P-loop motif “GxGKT” that is also present in the kinase module of yeast Trl1 and plant AtRNL. The structure and mechanism of the phage T4 phosphatase domain differ from the yeast and plant 3’ end healing modules; the latter belong to the “2H” phosphoesterase superfamily, named for the pair of “HxT” peptides that comprise the CPD active site (Fig. S1A).
The ligase components of the two repair systems are ATP-dependent sealing enzymes that act via covalent lysyl-AMP intermediates. Ligase adenylylation occurs at a conserved KxxG motif (Fig. S1A) that, together with several other conserved peptides, comprises the active site of yeast/plant and phage RNA ligases (Wang and Shuman, 2005; El Omari et al., 2005; Wang et al., 2006a). Notwithstanding these shared features, the yeast- and phage-type ligases have quite different chemical constraints on the end configurations that can be sealed. Whereas T4 Rnl1 seals at a 3’-OH, 2’-OH end, the yeast and plant ligases cannot; rather, they require the 3’-OH, 2’-PO4 end generated by the CPD component of the yeast/plant repair system (Schwer et al., 2004; Keppetipola et al., 2007).
A crucial, and largely uncharted, aspect of tRNA repair concerns whether and how the healing and sealing enzymes achieve specificity for broken tRNA substrates. Although T4 Rnl1 is used widely as a reagent for RNA end-modification, its activity in sealing generic RNA substrates is relatively feeble (Bullard and Bowater, 2006). T4 Rnl1 displays a strong preference for sealing tRNA substrates in vitro (Wang et al., 2007). tRNA specificity is conferred by a C-terminal domain of unique tertiary structure (El Omari et al., 2005; Wang et al., 2007) that is not present in yeast or plant tRNA ligases.
These insights to the structure and specificity of the phage RNA ligase raise new questions that are addressed presently. Do yeast and plant tRNA ligases display inherent preference for sealing tRNAs? What structural elements of the tRNA molecule are recognized? What advantage (if any) is conferred on the yeast and plant repair systems by packaging the healing and sealing modules in a single polypeptide? Which of the modules contribute to substrate specificity? The results of this inquiry illuminate fundamental differences in the determinants of target specificity by the yeast-type and phage-type RNA repair systems, as well as distinct domain requirements for optimal tRNA repair by the yeast and plant tRNA ligases. We discuss how the properties of these RNA repair systems help confine their action to RNA molecules that are broken purposefully in the pathways they serve, thereby avoiding potentially deleterious ligation of other RNAs.
In addition to the examples of programmed RNA damage cited above, recent studies highlight a slew of target-specific endoribonuclease toxins in bacteria and fungi and the role of such “ribotoxins” in defending the organism against non-self species or viruses (reviewed in Masaki and Ogawa 2002; Lacadena et al., 2007) and as mediators of programmed cell death (Nariya and Inouye, 2008). Secreted ribotoxins comprise an RNA-based innate immune system in which killing or growth arrest of the “foreign” cell that takes up the toxin relies on that cell’s inability to repair the toxin-induced damage to essential RNAs. tRNAs figure prominently as the specific targets of secreted bacterial and fungal ribotoxins, exemplified by the bacterial anticodon nucleases colicin D and colicin E5 and the Kluyveromyces lactis γ-toxin (Ogawa et al., 1999; Tomita et al., 2000; Graille et al., 2004; Lin et al., 2005; Lu et al., 2005,2008; Jablonowski et al., 2006). Other studies have revealed a promising therapeutic niche for enzymatic and chemical ribotoxins. For example, the induction of damage to tRNAs in human tumor cells by cytotoxic ribonuclease Onconase exemplifies a new paradigm for a cancer chemotherapy (Saxena et al., 2002; Sushani and Sirdeshmukh, 2006). tRNA damage has also been invoked as a factor in the antitumor action of bleomycin (Abraham et al., 2003).
A deeper knowledge of RNA repair is pertinent to understand the biological efficacy of ribotoxins in eukaryal cells and how to usefully modulate ribotoxin action. One can envision adjuvant therapies whereby inhibition of RNA repair might enhance ribotoxin killing of the desired target (e.g., tumor cells or fungal pathogens) while maneuvers that enhance RNA repair in bystander cells and tissues might spare them from cytotoxicity. Here we provide proof-of-principle for the ability of a heterologous tRNA repair enzyme to protect a eukaryal cell from a tRNA-specific nuclease.
RESULTS
Preferential tRNA repair by yeast Trl1 and the benefits of domain fusion
To better delineate the specificity of end-healing and end-sealing during yeast tRNA repair/splicing, we compared the activities of purified recombinant full-length Trl1 and its component ligase and kinase-CPD domains (Fig. S1B) in an in vitro tRNA splicing system (Englert and Beier, 2005). The broken tRNA substrate was generated by treating a 32P-labeled intron-containing pre-tRNA with a tRNA splicing endonuclease, which led to quantitative release of a linear intron and the formation of two “half-tRNA” molecules (Fig 1A). Reaction of the broken RNA with yeast Trl1 resulted in joining of the half-tRNAs to yield a mature spliced tRNA (Fig. S2A and 1D). The fact that Trl1 catalyzed little or circularization of the intron attests to its specificity for tRNA repair. The yield of spliced tRNA was proportional to input Trl1 (Fig. S2C), such that 6.6 fmol of tRNAs were spliced per fmol of enzyme. To gauge whether there is an advantage to the fusion of healing and sealing domains, we compared the tRNA splicing activity of native Trl1 with that of a 1:1 mixture of the purified ligase and kinase-CPD domains (Fig. S2A). The specific activity of the domain mixture (0.58 fmol tRNA spliced per fmol enzyme) was an order of magnitude lower than that of native Trl1 (Fig. S2C).
Fig. 1. Effect of tRNA structural alterations on tRNA repair in vitro.
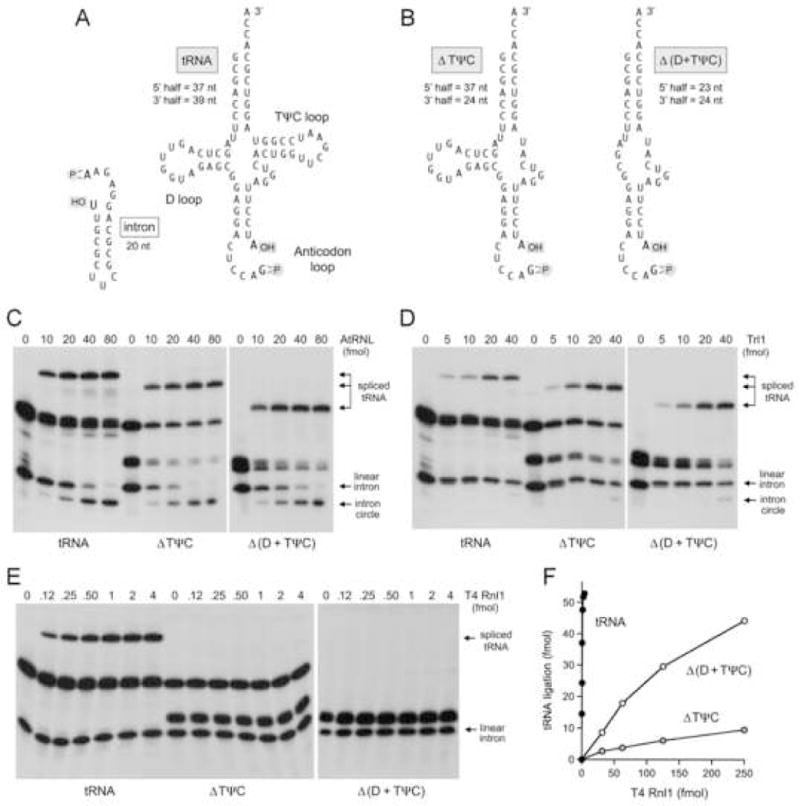
(A) The primary and secondary structures of the cleaved pre-tRNA and the excised intron substrates for in vitro RNA repair are shown. (B) Mutated derivatives of the RNA substrate in which the ΔTψC loop was deleted or both the D-loop and the TψC loop were deleted are shown. The sizes of the fragments generated by splicing endonuclease cleavage are indicated. (C–E) Reaction mixtures contained 140 fmol 32P-labeled pre-cleaved tRNA or the ΔTψC or Δ (D+ TψC) variants and the indicated amounts of RNA repair enzymes: either AtRNL (panel C), Trl1 (panel D), or 200 fmol T4 Pnkp plus T4 Rnl1 as specified (panel E). (F) The titration of T4 Rnl1 with the mutated substrates was extended to higher enzyme concentrations. The yields of spliced product are plotted as a function of input Rnl1.
We considered the prospect that substrate recognition by one of the domains might assist in delivering the other module to the tRNA, thereby rationalizing selection for the native fusion protein. To test this idea, we titrated each domain of Trl1 in the presence of a saturating level of the other domain (Fig. S2B). The specific activity of the isolated Lig domain was 0.69 fmol of tRNA spliced per fmol of enzyme (Fig. S2D). By contrast, the specific activity of the isolated kinase-CPD domain was 66 fmol tRNA spliced per fmol enzyme (Fig. S2D), a value two orders of magnitude higher than that of the ligase domain. These results suggest that in the context of the native Trl1, the kinase-CPD domain recognizes the broken tRNA ends and recruits the covalently tethered ligase domain. It is noteworthy that there is no apparent discrimination against intron circularization when the splicing reactions are constituted with excess Trl1 ligase domain (Fig. S2B), signifying that the ligase domain is the source of the tRNA preference.
AtRNL has a different substrate recognition strategy
Full-length AtRNL and its component ligase and kinase-CPD domains (Fig. S1B) were tested in a similar fashion for tRNA repair in vitro. Native AtRNL catalyzed tRNA splicing and intron circularization, without apparent preference for the broken tRNA versus the intron (Fig. S3A and 1C). The specific activity of full-length AtRNL was 1.4 fmol of tRNA sealed per fmol of enzyme (Fig. S3B). The specific activity of a 1:1 mixture of the isolated plant ligase and kinase-CPD domains was 0.39 fmol of tRNA spliced per fmol of enzyme (Fig. S3). Thus, the domain fusion was advantageous for the plant tRNA ligase, albeit less so than for yeast Trl1. An instructive result emerged when we titrated each domain of AtRNL in the presence of a saturating level of the other domain (Fig. S3C). The specific activity of the isolated plant ligase domain (7.2 fmol tRNA spliced per fmol enzyme) was 50-fold greater than that of the isolated kinase-CPD domain (0.13 fmol/fmol). These findings – which are opposite to those obtained for the yeast tRNA ligase – suggest that the ligase domain of native AtRNL recognizes the RNA and brings the covalently tethered kinase-CPD domain along for the ride.
A comparison of the specific activities of the isolated domains of Trl1 and AtRNL suggested that plant ligase is more active in vitro than yeast ligase, while yeast kinase-CPD is more active than plant kinase-CPD, at least when each is paired with an excess of its own partner domain. We explored this matter further in tests of tRNA repair by 1:1 cross-species mixtures of the healing and sealing domains (Fig. S4A). The combination of plant ligase plus yeast kinase-CPD spliced 7.9 fmol tRNA per fmol enzyme (Fig. S4B). By contrast the mixture of yeast ligase and plant kinase-CPD had 25-fold lower specific activity (0.32 fmol/fmol) (Fig. S4). The winning combination of separate plant sealing and yeast healing domains was at least as active in vitro as the native Trl1 fusion protein and 14-fold more active than the separated yeast domains.
Plant, yeast and phage tRNA repair systems recognize different tRNA structural elements
To examine whether tRNA tertiary structure elements other than the broken anticodon stem-loop are recognized by the respective tRNA repair systems, we prepared mutated pre-tRNA molecules in which either the TψC loop was deleted (ΔTψC) or the D loop and the TψC loop were both deleted (Fig. 1B). The 32P-labeled ΔTψC and Δ (D+TψC) pre-tRNAs were incised in vitro by splicing endonuclease at the exon-intron junctions within the anticodon loop to yield the 5’ and 3’ half-tRNAs depicted in Fig. 1B, along with the same 20-mer intron that was excised from the wild-type tRNA substrate. Electrophoretic analysis of the broken tRNAs confirmed that endonuclease cleavage of the mutated RNAs was accurate and complete (Fig. 1C, lanes 0). The instructive findings were that AtRNL and Trl1 were adept at repairing the broken ΔTψC and Δ (D+TψC) substrates to yield spliced products of the appropriate size (Fig. 1C and D). Splicing efficiency was undiminished by deletion of the D and TψC loops. We conclude that the yeast and plant tRNA repair systems do not require the native tRNA tertiary structure to achieve substrate recognition.
By contrast, the ΔTψC and Δ (D+TψC) mutations exerted profoundly different effects on the bacteriophage T4 RNA repair system. Splicing reactions were constituted with saturating levels of purified Pnkp and varying amounts of purified Rnl1. The yield of spliced product saturated at 2–4 fmol input Rnl1, with no detectable formation of intron circles (Fig. 1E). From the titration profile, we calculated that T4 Rnl1 sealed 72 fmol of tRNA per fmol of enzyme (Fig. 1F). No spliced products were generated by T4 Rnl1 when it was reacted in parallel with the broken ΔTψC and Δ (D+TψC) substrates (Fig. 1E). By extending the analysis to much higher levels of Rnl1 (Fig. 1F), we determined that deletion of the TψC loop suppressed specific activity by >1000-fold, whereas deleting both the D loop and TψC loop elicited a 300-fold decrement in repair compared to the wild-type tRNA. We surmise that T4 RNA ligase, unlike the yeast and plant enzymes, specifically recognizes the native tRNA fold.
A heterologous RNA repair system protects yeast cells from ribotoxin growth arrest
The yeast Kluyveromyces lactis harbors a cytoplasmic episome (the “killer” plasmid) that encodes a secreted protein toxin known as zymocin (Stark and Boyd, 1986; Jablonowski and Schaffrath, 2007). Zymocin is a heterotrimer of α, β and γ subunits that arrests growth of the non-self yeast species Saccharomyces cerevisiae by exposure from without (Fig. 2A). The α and β subunits interact with the target cell surface to effect the transport of the β subunit (the immediate toxin) into the cytoplasm of the target cell. Induced expression in S. cerevisiae of only the γ-toxin polypeptide suffices to inhibit cell growth (Butler et al., 1991). γ-toxin is an anticodon nuclease that specifically cleaves tRNAGlu(UUC) at a single phosphodiester 3’ of the modified wobble base mcm5s2U (5-methoxycarbonylmethyl-2-thiouridine) of the UUC anticodon (Fig. 2B) and thereby affects protein synthesis by depleting the pool of functional tRNAGlu(UUC) (Lu et al., 2005; Jablonowski et al., 2006). The mcm5s2U modification is required for tRNA cleavage by γ-toxin; yeast mutants that lack any of the enzymes responsible for synthesizing the mcm5 moiety are resistant to exogenous zymocin and endogenous γ-toxin (Lu et al., 2005; Huang et al., 2005; Jablonowski et al., 2006).
Fig. 2. K. lactis zymocin is a secreted ribotoxin that targets tRNAGlu.
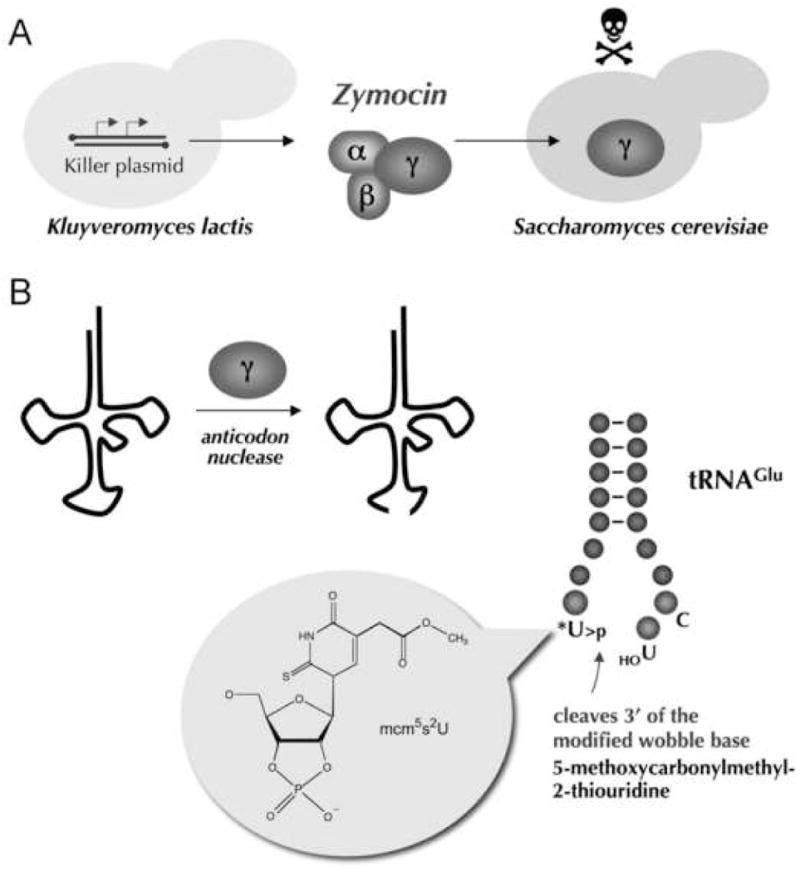
(A) K. lactis contains a killer plasmid that encodes a secreted heterotrimeric toxin, zymocin. The α and β subunits of zymocin interact with the surface of S. cerevisiae cells and mediate the uptake of the γ subunit into the cytoplasm to arrest growth of the target cell. (B) γ-toxin is a tRNA anticodon nuclease that incises tRNAGlu(UUC) immediately 3’ of the modified mcm5s2U wobble base to yield a 2’,3’ cyclic phosphate terminus. The structure of the mcm5s2U>p product is shown.
γ-toxin’s nuclease activity generates 2’,3’ cyclic phosphate and 5’-OH termini at the break in the tRNA anticodon loop (Lu et al., 2005) (Fig. 2B). Given that that such termini are the natural substrates for fungal, plant and phage tRNA repair systems, we embraced γ-toxin-induced growth arrest as a means to address the following questions: Can a heterologous tRNA repair enzyme protect S. cerevisiae from γ-toxin? Why doesn’t endogenous Trl1 protect S. cerevisiae from γ-toxin?
We used S. cerevisiae strains that contained a galactose-inducible γ-toxin expression cassette (Fig. 3A). In the presence of glucose, γ-toxin expression is suppressed and wild-type TRL1 yeast cells grow normally. In the presence of galactose, γ-toxin is produced and growth of wild-type TRL1 cells is inhibited (Fig. 3B). The yeast trm9Δ mutant lacks the methyltransferase that adds the terminal methyl group of the mcm5 moiety of the wobble uridine (Kalhor and Clarke, 2003); therefore, trm9Δ cells are resistant to γ-toxin (Jablonowski et al., 2006) (Fig. 3B). We initially considered the possibility that the normal level of Trl1 expression might not suffice to repair broken tRNAGlu. Thus, the first experiment was to increase Trl1 expression by placing the TRL1 gene on a multicopy 2μ plasmid under the control of the strong yeast TPI1 promoter. In parallel, we constructed a 2μ vector with plant AtRNL driven by the TPI1 promoter. The plasmids were introduced into a trl1Δ strain bearing the inducible γ-toxin cassette. Whereas 2μ TRL1 had no impact on the sensitivity of the yeast cells to growth inhibition on galactose, expression of the plant tRNA ligase rescued yeast growth under toxin-inducing conditions (Fig. 3B). It is apparent from the colony size that the salutary effect of the plant tRNA repair enzyme does not match that of ablating Trm9 (Fig. 3B). This is entirely reasonable and underscores the old adage that “an ounce of prevention is worth a pound of cure”. Loss of wobble base modification is tantamount to complete prevention of ribotoxin “disease”, because no tRNAGlu damage ever occurs in trm9Δ cells (Lu et al., 2005; Jablonowski et al., 2006). It is remarkable that plant tRNA ligase alleviates the pathology to the extent seen in the face of unrelenting tRNA damage caused by constitutive expression of γ-toxin.
Fig. 3. Plant tRNA ligase preventsγ-toxin induced growth arrest.
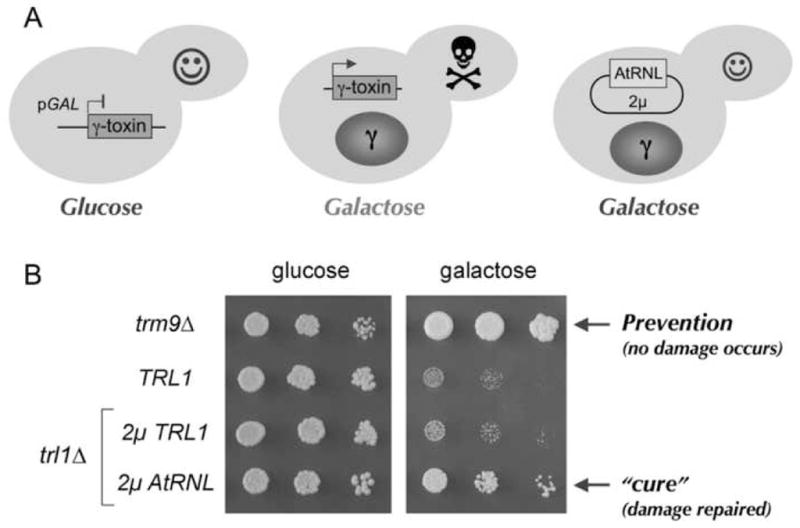
(A) Schematic depiction of yeast cells harboring pGAL-γ-toxin in the presence of glucose or galactose as the carbon source. The right-most cell also expresses plant ligase. (B) Serial dilutions (5-fold) of trm9Δ cells, a wild type TRL1 strain, or trl1Δ cells carrying 2μ plasmids pTPI1-TRL1 or pTPI1-AtRNL were spotted on minimal synthetic agar medium containing 2% (w/v) glucose or 2% (w/v) galactose. The plates were photographed after incubation at 30 C for 2.5 days (glucose) or 5 days (galactose).
Which activities of the plant tRNA ligase are required for γ-toxin resistance?
The observation that plant tRNA ligase rescues cells from γ-toxin growth arrest while yeast tRNA ligase does not suggests that there are intrinsic differences in the ability of the plant and yeast systems to repair the broken tRNAGlu substrates. To probe the roles of the three catalytic activities of AtRNL, we tested a collection of mutant AtRNL alleles bearing lethal alanine mutations in the active sites that specifically ablate the ligase (K152A or E326A), kinase (S701A or D726A) or CPD (T1001A or H1060A) functions (Wang et al., 2006a) (Fig. 4B). The wild type, kinase-dead, ligase-dead, and CPD-dead AtRNL proteins were expressed from 2μ plasmids in TRL1 cells bearing the γ-toxin cassette and tested in parallel for growth on glucose and galactose media (Fig. 4A). None of the AtRNL-Ala mutations affected growth on glucose because tRNA splicing activity is provided by the endogenous Trl1 enzyme. However, these mutations had disparate effects on yeast growth on galactose, depending on which catalytic activity was affected. The 5’ kinase activity of AtRNL was clearly not required to confer γ-toxin resistance, insofar as the kinase-dead alleles were as effective as wild-type AtRNL in rescuing growth (Fig. 4A). We surmise that the endogenous level of yeast Trl1 kinase suffices to heal the 5’-OH end of broken tRNAGlu. By contrast, the two ligase-dead AtRNL mutants failed to protect against γ-toxin and their growth on galactose phenocopied that of a strain bearing the empty 2μ vector (Fig. 4A). Thus, the ligase activity of AtRNL is essential for toxin resistance. CPD-dead versions of AtRNL provided a diminished level of toxin resistance compared to wild-type AtRNL, albeit more than the vector control or the ligase-dead mutants (Fig. 4A).
Additional experiments to identify determinants of toxin resistance versus sensitivity entailed pairwise coexpression of the yeast and plant healing and sealing domains in same-species and cross-species fashion (Fig. 4C). The instructive findings were that: (i) the yeast ligase plus plant kinase-CPD failed to restore growth on galactose; and (ii) plant ligase plus yeast kinase-CPD rescued cells from toxin-induced growth arrest (Fig. 4C). Indeed, the cross-species combination of plant sealing and yeast healing domains led to even better growth in galactose medium than the plant/plant domain pair. These results echo the hierarchy of in vitro tRNA repair activities of the cross-species domain combinations (Fig. S4). The salient conclusion is that differences in the yeast versus plant ligase component are the critical determinant of sensitivity versus resistance to γ-toxin in vivo.
Phage tRNA repair enzymes protect cells from γ-toxin
Previous studies showed that the two-component phage T4 RNA repair system could substitute for yeast Trl1 in vivo (Schwer et al., 2004). Here we introduced CEN plasmids expressing RNL1 and PNKP into a yeast trl1Δ strain bearing the γ-toxin cassette and tested growth under toxin-off and toxin-on conditions. Whereas control TRL1 cells did no thrive on medium containing galactose, the phage tRNA repair system promoted cell growth (Fig. 4D). The colony size of the RNL1 PNKP strain on galactose-containing medium was similar to that of the 2μ AtRNL strain (not shown). These results suggest that the plant and phage tRNA repair systems can rectify the toxin-induced damage, but the endogenous yeast system cannot.
RNA repair spares tRNAGlu from damage by zymocin
S. cerevisiae cells expressing either the plant or phage tRNA repair systems were also resistant to exogenous zymocin (Fig. 5A). To bolster the argument that RNA repair alleviates ribotoxicity by rectifying damage to the tRNA target, we compared the levels of intact tRNA in zymocin-treated versus control yeast cultures (Fig. 5B). tRNAGlu(UUC) (the toxin target) and tRNAGly (an internal control) cDNAs were synthesized by reverse transcription of total cellular RNA. The cDNAs were then amplified by PCR with oligonucleotide primers on either side of the anticodon loop; the RT-PCR products were resolved by agarose gel electrophoresis (Fig. 5B). In this procedure, only intact tRNAGlu(UUC) will be amplified. Consistent with previous reports (Lu et al., 2005; Jablonowski et al., 2006), we found that tRNAGlu(UUC) was depleted from wild-type yeast cells treated with zymocin, an effect that was blocked by the trm9Δ mutation (Fig. 5B). Expression of full-length plant tRNA ligase at high gene dosage or the phage tRNA repair enzymes in single-gene dosage prevented zymocin depletion of tRNAGlu; increased dosage of the yeast TRL1 gene did not (Fig. 5B). Pairwise coexpression of the yeast and plant healing and sealing domains underscored the correlation between toxin resistance and preservation of intact tRNAGlu (Fig. 4C and 5B). Whereas tRNAGlu was depleted by zymocin in cells expressing yeast ligase plus plant kinase-CPD, expression of plant ligase plus yeast kinase-CPD alleviated this effect (Fig. 5B). Thus, RNA repair is an antidote to cytotoxic RNA damage.
Fig. 5. Growth arrest of S. cerevisiae by zymocin is relieved by tRNA repair.
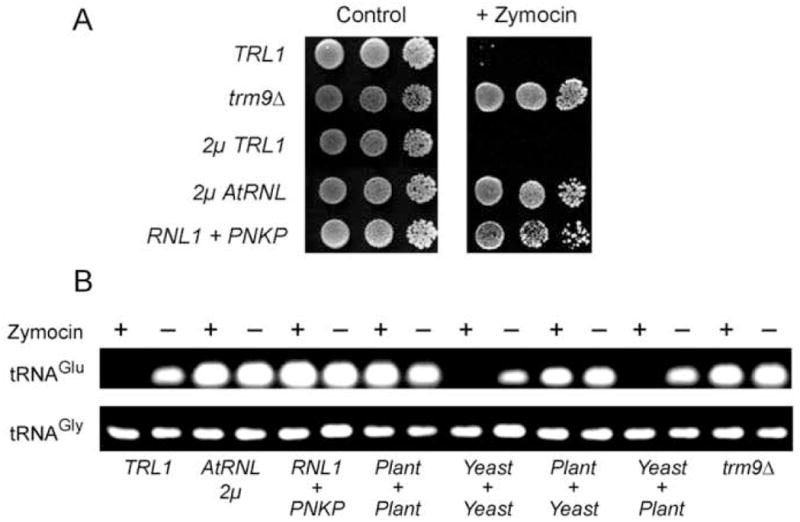
(A) Serial 10-fold dilutions of the indicated S. cerevisiae strains were spotted on YPD agar plates (control) or YPD plates containing 45% (v/v) zymocin. (B) RT-PCR analysis of tRNA levels in yeast cells grown in the presence or absence of zymocin. The PCR products were analyzed by agarose gel electrophoresis and stained with ethidium bromide. In strains expressing split domains of tRNA ligase, the source of the sealing domain (ligase) is indicated above the + sign and the source of the healing domain (kinase-CPD) is specified below.
DISCUSSION
Specificity of RNA Repair Systems
Yeast, plant and phage RNA healing and sealing enzymes are essential components of informational and stress response pathways entailing the repair of programmed RNA breaks. Although the target specificity of these pathways resides with the endoribonucleases that create the damage, there has been little attention paid to whether and how the repair enzymes discriminate the purposefully broken RNA ends from the vast excess of other RNA termini available in the cell. Here we show that the bacteriophage and eukaryal tRNA repair systems have evolved cleverly distinct strategies to direct sealing to the “appropriate” ends and thereby avoid indiscriminate RNA end-joining that could have toxic consequences. The plant and yeast sealing enzymes evince no specificity for the native tRNA fold. This is not a problem, because their sealing activity is restricted to breaks in which one partner has a 3’-OH, 2’-PO4 terminus. Because there is no enzymatic pathway known to us in which an RNA 3’-OH, 2’-OH could be directly phosphorylated to a 3’-OH, 2’-PO4, this means that eukaryal tRNA ligases are unable to catalyze “recombination” events at the unprocessed 3’ ends of native RNAs. The route to forming a sealable substrate for plant/yeast ligases is via hydrolysis of an RNA 2’,3’ cyclic phosphate with scission of the O3’–P bond. This is precisely what is accomplished when the CPD domain of eukaryal tRNA ligases acts at a cyclic phosphate end. Consequently, the yeast and plant tRNA repair systems are hard-wired so that only the purposefully broken RNAs (which have cyclic phosphate ends) will be joined.
Because phage Rnl1 is unfettered by requirements for unusual 3’-OH ends, it might in principle be capable of catalyzing all sorts of RNA rearrangements at native RNA ends, or at the 3’ and 5’ ends of degradation or processing intermediates. Whereas such ligation events might have been useful in promoting genetic diversification in a primordial RNA-protein world, it is clearly to be avoided in modern life, insofar as unprogrammed post-transcriptional scrambling of RNA sequences could create open reading frames for toxic peptides (especially in eukarya, if 5’ capped RNA fragments generated during 3’ to 5’ decay were reattached to other processed RNAs with 5’-PO4 ends). One might quibble that avoidance of random RNA rearrangements is not a concern in a T4 phage-infected bacterium, which is destined for a lytic death. But, recall that the prrC+ host bacterium will kill itself prematurely through tRNA restriction and thereby thwart phage replication – unless Rnl1 repairs the tRNA damage in a timely fashion (Amitsur et al., 1987). Thus, the phage RNA ligase (through its C-terminal domain) recognizes the native tRNA fold and thereby avoids nonspecific ligations.
A fungal ribotoxin exposes an Achilles heel of yeast tRNA ligase
Determinants of the sensitivity of S. cerevisiae to K. lactis zymocin have been identified through the classical approach of isolating zymocin resistant mutants. Two main classes of resistance loci have been described: (i) genes that affect the target cell surface and its interaction with and transport of extracellular zymocin and (ii) genes that control wobble base modification of the tRNA target of γ-toxin cleavage (Jablonowski and Schaffrath, 2007). The strong specificity of γ-toxin for cleaving adjacent to the modified mcm5s2U base of the tRNAGlu anticodon loop echoes the theme described originally for the bacterial tRNA anticodon nuclease colicin E5, which cleaves tRNAs containing queosine at the wobble position (Ogawa et al., 1999). The E. coli strain targeted by the colicin E5 ribotoxin has no endogenous tRNA repair enzymes. However, S. cerevisiae clearly does in Trl1. Our studies here show that the ligase domain of Trl1 is incapable of repairing the damage to tRNAGlu inflicted by γ-toxin, notwithstanding that the Trl1 kinase-CPD domain is able to heal the broken tRNAGlu ends.
The physiological tRNA substrates for Trl1 are intron-containing pre-tRNAs that have been broken in the anticodon by tRNA splicing endonuclease. Anticodon nucleoside modifications flanking the splice site are introduced after completion of tRNA splicing by Trl1 and Tpt1 (Spinelli et al., 1997), which means that Trl1 does not normally act at termini with elaborate base modifications. S. cerevisiae tRNAGlu has no intron and is thus not dependent on Trl1 for its maturation. Because Trl1 is capable of sealing a “generic” broken tRNA substrate lacking base modifications via recognition of the termini of the cleaved anticodon stem loop, we surmise that the Trl1 ligase domain is hindered from sealing γ-toxin-incised tRNAGlu by the presence of the bulky mcm5s2U base at the 3’-OH, 2’-PO4 end. By cleaving only the modified tRNA at a site that cannot be repaired by Trl1, the K. lactisγ-toxin exploits the unique vulnerability of the tRNA repair machinery of the target cell. Indeed, S. cerevisiae depends on the mcm5 group of the wobble nucleoside for optimal fitness at physiologic tRNA levels (Esberg et al., 2006; Begley et al., 2007); consequently, cells that fail to synthesize mcm5U, though viable under laboratory conditions and zymocin-resistant, have pleiotrophic defects (Frohloff et al., 2001) that could render them unfit in the wild, and hence less of a threat to K. lactis.
Induction of tRNA damage as a mechanism of cell attrition, or in response to stress (Lee and Collins, 2005), is an emerging theme. In addition to the several tRNA ribotoxins for which the targets are known, there are numerous putative anticodon nuclease homologs in diverse bacterial species (Blanga-Kanfi et al., 2006), the function, substrates, and regulation of which are tabula rasa. Here we demonstrated that RNA repair enzymes can protect against cytotoxic RNA damage in vivo in a eukaryote. The capacity of the plant AtRNL to rectify zymocin-induced tRNA depletion is a unique property of its ligase domain, which is apparently adept at sealing the broken tRNA with a mcm5s2U–3’OH, 2-PO4 end. The structural features of the yeast and plant ligases that dictate their differential sealing of the modified break remain to be determined. The plant ligase domain is larger than the yeast counterpart (Fig. S1A); perhaps structural elements unique to AtRNL are the source of its broader substrate repertoire.
Validation that RNA repair can be an antidote to toxic RNA damage sets the stage for broader screening of yeast, plant, phage and other RNA repair enzymes as antagonists of a spectrum of toxic endoribonucleases that target ribosomal, messenger, and transfer RNAs and leave broken 2,’3’ cyclic phosphate and 5’-OH ends in their wake. Based on the present findings, it is likely that different RNA repair systems, and cross-system combinations of repair modules, will afford varying degrees of protection depending on which toxin-target system is used and in what organism. Such studies will illuminate new principles of “RNome integrity” and suggest useful ways to titrate RNA damage.
EXPERIMENTAL PROCEDURES
tRNA repair in vitro
The procedures for the purification of yeast, plant and phage tRNA repair enzymes and for the preparation of broken tRNA substrates are described in Supplemental Material published online. Repair reaction mixtures (10 μl) containing 50 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 2 mM DTT, 20 μM ATP, 20 μM GTP, 140 fmol radiolabeled cleaved tRNA substrate, and enzymes as specified were incubated for 30 min at 37°C. The products were analyzed by denaturing PAGE, visualized by autoradiography and quantified by scanning the gel with a phosphorimager.
Inducible expression of γ-toxin in budding yeast
The endogenous tRNA ligase was replaced in S. cerevisiae trl1Δ cells with the plant and bacteriophage repair systems. Alternatively, plasmids encoding plant or bacteriophage repair enzymes were introduced into TRL1 yeast cells. The various strains were then transformed with pLF16 (UASGAL1-γ-toxin LEU2 CEN) for induced expression of γ-toxin (Jablonowski et al. 2006) and Leu+ transformants were selected on glucose-containing medium lacking leucine (and other nutrients when necessary for plasmid maintenance). Cultures were grown in selective liquid media at 30°C, adjusted to A600 of 0.1 and then diluted serially in 5-fold increments in water. Aliquots (3 μl) were spotted to glucose and galactose-containing agar plates, which were incubated at 30°C.
Assay of tRNA depletion by zymocin
S. cerevisiae cells were inoculated in 50 ml of YPD liquid medium at an A600 of 0.2 and grown at 30 C for 6 h in the absence or presence of 50% (v/v) zymocin. The zymocin preparation was a cell-free filtrate of culture supernatants of K. lactis strain AWJ137 grown for 48 h in YPD medium. Total RNA was isolated from equal numbers (2 x 107) of zymocin-treated and control cells. Equal amounts of RNA from treated/control pairs (1–4 μg) were used as template for cDNA synthesis primed by random hexamer deoxyribonucleotides. After first strand cDNA synthesis, 1/20 of the reaction mixtures was subjected to 20 cycles of PCR amplification by Taq DNA polymerase primed by DNA oligonucleotide pairs specific for tRNAGlu and tRNAGly. The RT-PCR products were analyzed by electrophoresis through a 2% agarose gel and visualized by staining the gel with ethidium bromide.
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM42498. R.S. acknowledges technical assistance by Patrick Studte.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abelson J, Trotta CR, Li H. tRNA splicing. J Biol Chem. 1998;273:12685–12688. doi: 10.1074/jbc.273.21.12685. [DOI] [PubMed] [Google Scholar]
- Abraham AT, Lin JJ, Newton DL, Rybak S, Hecht SM. RNA cleavage and inhibition of protein synthesis by bleomycin. Chemistry & Biology. 2003;10:45–52. doi: 10.1016/s1074-5521(02)00306-x. [DOI] [PubMed] [Google Scholar]
- Amitsur M, Levitz R, Kaufman G. Bacteriophage T4 anticodon nuclease, polynucleotide kinase, and RNA ligase reprocess the host lysine tRNA. EMBO J. 1987;6:2499–2503. doi: 10.1002/j.1460-2075.1987.tb02532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostol BL, Westaway SK, Abelson J, Greer CL. Deletion analysis of a multifunctional yeast tRNA ligase polypeptide: identification of essential and dispensable functional domains. J Biol Chem. 1991;266:7445–7455. [PubMed] [Google Scholar]
- Begley U, Dyavaiah M, Patil A, Rooney JP, DiRenzo D, Young CM, Conklin DS, Zitomer RS, Begley TJ. Trm9-catayzed tRNA modifications link translation to the DNA damage response. Mol Cell. 2007;28:860–870. doi: 10.1016/j.molcel.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanga-Kanfi S, Amitsur M, Azem A, Kaufmann G. PrrC-anticodon nuclease: functional organization of a prototypal bacterial restriction RNase. Nucleic Acids Res. 2006;34:3209–3219. doi: 10.1093/nar/gkl415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard DR, Bowater RP. Direct comparison of nick-joining activity of the nucleic acid ligases from bacteriophage T4. Biochem J. 2006;398:135–144. doi: 10.1042/BJ20060313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AR, Porter M, Stark MJ. Intracellular expression of Kluyveromyces lactis toxin gamma subunit mimics treatment with exogenous toxin and distinguishes two classes of toxin-resistant mutant. Yeast. 1991;7:617–625. doi: 10.1002/yea.320070610. [DOI] [PubMed] [Google Scholar]
- El Omari K, Ren J, Bird LE, Bona MK, Klarmann G, LeGrice SFJ, Stammers DK. Molecular architecture and ligand recognition determinants for T4 RNA ligase. J Biol Chem. 2005;281:1573–1579. doi: 10.1074/jbc.M509658200. [DOI] [PubMed] [Google Scholar]
- Englert M, Beier H. Plant tRNA ligases are multifunctional enzymes that have diverged in sequence and substrate specificity from RNA ligases of other phylogenetic origins. Nucleic Acids Res. 2005;33:388–399. doi: 10.1093/nar/gki174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esberg A, Huang B, Johansson MJO, Byström AS. Elevated levels of two tRNA species bypass the requirement for elongator complex in transcription and exocytosis. Mol Cell. 2006;24:139–148. doi: 10.1016/j.molcel.2006.07.031. [DOI] [PubMed] [Google Scholar]
- Frohloff F, Fichtner L, Jablonowski D, Breunig KD, Schaffrath R. Saccharomyces cerevisiae elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J. 2001;20:1993–2001. doi: 10.1093/emboj/20.8.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graille M, Mora L, Buckingham RH, van Tilbeurgh H, de Zamaroczy M. Structural inhibition of the colicin D tRNase by the tRNA-mimicking immunity protein. EMBO J. 2004;23:1474–1482. doi: 10.1038/sj.emboj.7600162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B, Johansson MJO, Byström AS. An early step in wobble tRNA modification requires the elongator complex. RNA. 2005;11:424–436. doi: 10.1261/rna.7247705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonowski D, Shaffrath R. Zymocin, a composite chitinase and tRNase killer toxin from yeast. Biochem Soc Trans. 2007;35:1533–1537. doi: 10.1042/BST0351533. [DOI] [PubMed] [Google Scholar]
- Jablonowski D, Zink S, Mehlgarten C, Daum G, Schaffrath R. tRNAGlu wobble uridine methylation by Trm9 identifies elongator’s key role for zymocin-induced cell death in yeast. Mol Microbiol. 2006;59:677–688. doi: 10.1111/j.1365-2958.2005.04972.x. [DOI] [PubMed] [Google Scholar]
- Kalhor HR, Clarke S. Novel methyltransferase for modified uridine residues at the wobble position of tRNA. Mol Cell Biol. 2003;23:9283–9292. doi: 10.1128/MCB.23.24.9283-9292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppetipola N, Nandakumar J, Shuman S. Reprogramming the tRNA splicing activity of a bacterial RNA repair enzyme. Nucleic Acids Res. 2007;35:3624–3630. doi: 10.1093/nar/gkm110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacadena J, Alvarez-Garcia E, Carreras-Sangrà N, Herrero-Galán E, Alegre-Cebollada J, García-Ortega L, Oñaderra M, Gavilanes JG, Martínez del Pozo A. Fungal ribotoxins: molecular dissection of a family of natural killers. FEMS Microbiol Rev. 2007;31:212–237. doi: 10.1111/j.1574-6976.2006.00063.x. [DOI] [PubMed] [Google Scholar]
- Lee SR, Collins K. Starvation-induced cleavage of the tRNA anticodon loop in Tetrahymena thermophila. J Biol Chem. 2005;280:42744–42749. doi: 10.1074/jbc.M510356200. [DOI] [PubMed] [Google Scholar]
- Lin YL, Elias Y, Huang RH. Structural and mutational studies of the catalytic domain of colicin E5: a tRNA-specific ribonuclease. Biochemistry. 2005;44:10494–10500. doi: 10.1021/bi050749s. [DOI] [PubMed] [Google Scholar]
- Lu J, Huang B, Esberg A, Johanson M, Byström AS. The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. RNA. 2005;11:1648–1654. doi: 10.1261/rna.2172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Esberg A, Huang B, Byström AS. The Kluyveromyces lactisγ-toxin, a ribonuclease that recognizes the anticodon stem loop of tRNA. Nucleic Acids Res. 2008;36:1072–1080. doi: 10.1093/nar/gkm1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki H, Ogawa T. The modes of action of colicins E5 and D, and related cytotoxic tRNases. Biochimie. 2002;84:433–438. doi: 10.1016/s0300-9084(02)01425-6. [DOI] [PubMed] [Google Scholar]
- Nariya H, Inouye M. MazF, an mRNA interferase, mediates programmed cell death during multicellular Myxococcus development. Cell. 2008;132:55–66. doi: 10.1016/j.cell.2007.11.044. [DOI] [PubMed] [Google Scholar]
- Ogawa T, Tomita K, Ueda T, Watanabe K, Uozumi T, Masaki H. A cytotoxic ribonuclease targeting specific tRNA anticodons. Science. 1999;283:2097–2100. doi: 10.1126/science.283.5410.2097. [DOI] [PubMed] [Google Scholar]
- Sawaya R, Schwer B, Shuman S. Genetic and biochemical analysis of the functional domains of yeast tRNA ligase. J Biol Chem. 2003;278:43298–43398. doi: 10.1074/jbc.M307839200. [DOI] [PubMed] [Google Scholar]
- Saxena SK, Sirdeshmukh R, Ardelt W, Mikulski SM, Shogen K, Youle RJ. Entry into cells and selective degradation of tRNAs by a cytotoxic member of the RNase A family. J Biol Chem. 2002;277:15142–15146. doi: 10.1074/jbc.M108115200. [DOI] [PubMed] [Google Scholar]
- Schwer B, Sawaya R, Ho CK, Shuman S. Portability and fidelity of RNA-repair systems. Proc Natl Acad Sci USA. 2004;101:2788–2793. doi: 10.1073/pnas.0305859101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Cox JS, Walter P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell. 1996;87:405–413. doi: 10.1016/s0092-8674(00)81361-6. [DOI] [PubMed] [Google Scholar]
- Spinelli SL, Consaul SA, Phizicky EM. A conditional lethal yeast phosphotransferase mutant accumulates tRNA with a 2’-phosphate and an unmodified base at the splice junction. RNA. 1997;3:1388–1400. [PMC free article] [PubMed] [Google Scholar]
- Stark MJR, Boyd A. The killer toxin of Kluyveromyces lactis: characterization of the toxin subunits and identification of the genes which encode them. EMBO J. 1986;5:1995–2002. doi: 10.1002/j.1460-2075.1986.tb04455.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sushani AN, Sirdeshmukh R. Transfer RNA cleavages by Onconase reveal unusual cleavage sites. J Biol Chem. 2006;281:12201–12209. doi: 10.1074/jbc.M504488200. [DOI] [PubMed] [Google Scholar]
- Tomita K, Ogawa T, Uozumi T, Watanabe K, Masaki H. A cytotoxic ribonuclease which specifically cleaves four isoaccepting arginine tRNAs at their anticodon loops. Proc Natl Acad Sci USA. 2000;97:8278–8283. doi: 10.1073/pnas.140213797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Shuman S. Structure-function analysis of yeast tRNA ligase. RNA. 2005;11:966–975. doi: 10.1261/rna.2170305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Schwer B, Englert M, Beier H, Shuman S. Structure-function analysis of the kinase-CPD domain of yeast tRNA ligase (Trl1) and requirements for complementation of tRNA splicing by a plant Trl1 homolog. Nucleic Acids Res. 2006a;34:517–527. doi: 10.1093/nar/gkj441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Schwer B, Shuman S. Structure-guided mutational analysis of T4 RNA ligase 1. RNA. 2006b;12:2126–2134. doi: 10.1261/rna.271706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LK, Nandakumar J, Schwer B, Shuman S. The C-terminal domain of T4 RNA ligase 1 confers specificity for tRNA repair. RNA. 2007;13:1235–1244. doi: 10.1261/rna.591807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.