

Abstract
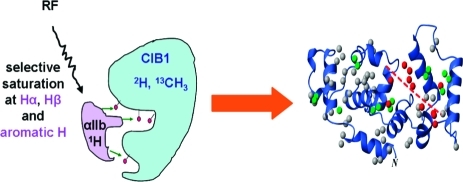
Calcium and integrin binding protein 1 (CIB1) is a specific binding partner for the cytoplasmic domain of the αIIb subunit of the highly abundant platelet integrin αIIbβ3. This protein has been suggested to be involved in the regulation of the activation of αIIbβ3, a process leading to platelet aggregation and blood coagulation. In this work, the solution structure of the deuterated Ca2+-CIB1 protein complexed with an αIIb peptide was first determined through modern RDC-based NMR methods. Next, we generated a complex structure for CIB1 and the αIIb domain (Ca2+-CIB1/αIIb) using the program Haddock, which is based on experimental restraints obtained for the protein–peptide interface from cross-saturation NMR experiments. In this data-driven complex structure, the N-terminal α-helix of the cytoplasmic domain of αIIb is buried in the hydrophobic pocket of the C-lobe of Ca2+-CIB1. The C-terminal acidic tail of αIIb remains unstructured and likely interacts with several positively charged residues in the N-lobe of Ca2+-CIB1. A potential molecular mechanism for the CIB1-mediated activation of the platelet integrin could be proposed on the basis of the model structure of this protein complex. Another feature of this work is that, in the NMR cross-saturation experiments, we applied the selective radio frequency irradiation to the smaller binding partner (the αIIb peptide), and successfully detected the binding interface on the larger binding partner Ca2+-CIB1 through its selectively protonated methyl groups. This ‘reverse’ methodology has a broad potential to be employed to many other complexes where synthetic peptides and a suitably isotope-labeled medium- to large-sized protein are used to study protein–protein interactions.
Introduction
Integrins are cell surface receptors that mediate cell adhesion processes, including cell–cell, cell–extracellular matrix, and cell–pathogen interactions.1,2 In general, integrins are heterodimeric glycoproteins that are formed by a single α and a single β subunit. Both the α and β subunits are single-pass transmembrane proteins, and such proteins are often involved in signal transduction processes. Integrins are known to be involved in bidirectional signal transduction across the plasma membrane, commonly referred to as “inside-out” and “outside-in” signaling.1−3 Both the α and β subunits are made up of a large extracellular ectomembrane domain, a single transmembrane helix, and a small cytoplasmic domain.1,2 It has been reported that the dissociation of the cytoplasmic domains of the α and β subunits is critical for bidirectional transmembrane signaling events.4
The αIIbβ3 integrin heterodimer is exclusively expressed at very high levels in platelets. Because of its role in the formation of blood clots and maintaining hemostasis, the structure of this complex is of particular interest. These two subunits associate in the membrane and in the cytoplasm with a low affinity.2 The interaction between these two parts of the dimeric integrin protein has been hard to detect in aqueous solution5 or studies resulted in different structures, especially for the cytoplasmic domains of αIIbβ3,6−8 probably reflecting the dynamic nature of the cytoplasmic domain of αIIbβ3. In the inside-out signaling pathway, the cytoskeletal protein talin9 has been reported to be able to bind specifically to the cytoplasmic domain of β3 and in turn activate integrin αIIbβ3.
The calcium- and integrin-binding protein 1 (CIB1) was originally discovered as a binding partner for the cytoplasmic domain of αIIb in a yeast two-hybrid screening study.10 CIB1 is now known as a ubiquitous regulatory protein that is made up of four EF-hands11 with a molecular weight of 22 kDa. CIB1 can carry a myristoyl group at its N-terminus, but the function of this myristoyl group in vivo is not yet fully understood since it has been found that CIB1 can interact with αIIb in vitro without the myristoyl group.12−14 The myristoyl group of CIB1 appears to act like a membrane anchor, similar to what has been found for many related calcium-binding proteins.15 CIB1 has four helix–loop–helix EF-hand motifs, but only EF-III and EF-IV have the capacity to bind divalent metal ions.16 In the resting state of most cells, magnesium is present in the cytoplasm at ∼1–2 mM allowing the folded form of Mg2+-CIB1 to be present.17 When the cell is excited, Ca2+ enters the cytoplasm and its concentration rises to micromolar levels; Ca2+ displaces Mg2+ and forms Ca2+-CIB1 in a well-folded conformation.16 Interestingly, both Ca2+-CIB1 and Mg2+-CIB1 can interact with a synthetic peptide representing the αIIb cytoplasmic domain and part of its transmembrane domain, with similar micromolar affinities.12 Solution NMR studies of CIB1 require deuteration and methyl group labeling17,18 because of its relatively large molecular weight (24 kDa including the purification-tag). It has been shown that Ca2+-CIB1 and Mg2+-CIB1 have a similar overall topology with Mg2+-CIB1 having a relatively more exposed hydrophobic pocket.17 NMR chemical shift perturbation studies concerning the interaction of αIIb with both Ca2+- and Mg2+-bound CIB1 suggest that both the Ca2+- and Mg2+-forms interact with αIIb via a hydrophobic pocket in the C-terminal domain of CIB1.17 It has been reported previously that CIB1 is capable of disrupting the association of αIIb and β3 by binding to αIIb and in turn activate the integrin αIIbβ3.14 Furthermore, blocking complex formation between the Wiskott-Aldrich syndrome protein (WASP) and CIB1 has been shown to affect the conformational change of αIIbβ3 and further affects its interaction with fibrinogen.19 In contrast, CIB1 has also been suggested to negatively regulate the activation of integrin αIIbβ3 by competing with talin for binding to αIIbβ3.20 Therefore, structural characterization of the CIB1/αIIb complex is of considerable interest to provide further insight into the mechanism of the regulation of the activation of αIIbβ3 by CIB1 or to determine the role of CIB1 in collaborating with talin in the regulation of αIIbβ3 activation as suggested by Yuan et al.20 Even though the CIB1/αIIb interaction has been suggested to be similar to that of the homologous protein calcineurin B (CnB) with calcineurin A (CnA) in terms of the relative orientation of the proteins,13 no direct experimental results have been reported to date to support this notion. By determining a structure for the Ca2+-CIB1/αIIb complex, we hope to obtain insight into the inside-out signaling of integrin as well as to establish the connections between the Ca2+-signaling pathway and the integrin-signaling pathways.
In this work, we report a structure for the Ca2+-CIB1/αIIb complex. An experimental approach of employing reverse cross-saturation NMR experiments using the labeled methyl groups of Ile/Leu/Val (I/L/V) in an otherwise deuterated CIB1 protein has been implemented to accurately identify the interface for the Ca2+-CIB1/αIIb complex. Moreover, residual dipolar couplings (RDCs) as determined in multiple alignment media were used to provide additional information about the structure of Ca2+-CIB1 in complex with αIIb.
Materials and Methods
Sample Preparation
The CIB1 protein containing different stable isotope labels was prepared as previously described.17,21 The [U–2H,15N,13C]-labeled sample was used to acquire backbone 1DC′N and 1DNH residual dipolar couplings (RDCs). The {[U–15N, 2H, 12C]; Ileδ1-[13CH3]; Leu,Val-[13CH3,12CD3]}-labeled sample was used for the methyl cross-saturation experiments to determine the interface for the interaction between CIB1 and the human platelet αIIb subunit. The 26-residue αIIb synthetic peptide (Ac-L983VLAMWKVGFFKRNRPPLEEDDEEGQ1008-OH) as previously described18 was used; this peptide corresponds to amino acids 983–1008 of the αIIb cytoplasmic domain and part of the transmembrane domain (hereafter referred to as αIIb-L, Figure 1). Another truncated 15-residue synthetic peptide (Ac-L983VLAMWKVGFFKRNR997-NH2) (αIIb-S, Figure 1) was also studied, which corresponds to amino acids 983–997 of the αIIb subunit. These synthetic peptides were both purchased from GenScript Corp. and they were more than 95% pure as determined by mass spectrometry and HPLC. Protein and peptide concentrations were determined by using the extinction coefficients: ε280= 3040 for CIB1, ε280= 5500 for αIIb-L, and ε280= 5500 for αIIb-S.
Figure 1.

The sequences of the αIIb peptides used in this work. αIIb-L represents the human integrin αIIb 983–1008 region, in which the L983, W988, F992, and F993 have been previously identified to be critical for the interaction between Ca2+-CIB1 and αIIb.13 αIIb-L contains part of the transmembrane domain (shaded in gray box) and the full cytoplasmic domain of integrin αIIb. αIIb-S represents αIIb residues 983–997. The R995 (red) has been reported to form a salt bridge with the integrin β3 subunit, which is critical for the association of the cytoplasmic domain of αIIb and β3.7,58,59.
NMR Experiments
NMR spectra were recorded at 37 °C on a Bruker AVANCE 500 MHz or a Bruker AVANCE 700 MHz NMR spectrometer each equipped with a triple resonance inverse cryoprobe with a single axis z-gradient. Each Ca2+-CIB sample, with or without αIIb peptides, contained 0.5–0.7 mM CIB1 in 50 mM HEPES, 100 mM KCl, 0.1 mM 2,2-dimethyl-2-silapentane-5-sulfonic acid (DSS), 10 mM DTT, 10% or 99.9% D2O, pH 7.5 ± 0.05; 2 mM CaCl2 was added for the Ca2+-bound sample and 3 mM MgCl2 for Mg2+-bound sample. The [U–2H,15N,13C]-labeled Ca2+-CIB1 sample complexed with αIIb-L was used to acquire the backbone RDCs (1DC′N and 1DNH) in different alignment media. One of the alignment media used was 14 mg/mL pf1 phage (Asla Lab) for partial alignment of protein molecules.22 A second medium used was the polyethylene glycol ether (PEG) medium containing 5% C12E5/hexanol in 0.96/1 ratio.23 The 1DC′N RDC were measured using the 3D IPAP-J-HNCO (CA) experiment,24 with 1024 × 128 × 40 complex points. The digital resolution was 2.06 Hz/pt in F2 (13C). A scale factor of 4 was used in the measurement of the 1DC′N RDCs. The 1DNH RDCs were measured using the 2D IPAP-HSQC experiment,25 with 1024 × 512 complex points, a digital resolution of 1.1 Hz was obtained after linear prediction and zero filling.
Reverse Methyl Cross-Saturation
Reverse methyl cross-saturation experiments were performed using 500 μM samples of {[U–15N, 2H, 12C]; Ileδ1-[13CH3]; Leu,Val-[13CH3,12CD3]}-labeled CIB1 in 100 mM KCl, 99.9% D2O, 10 mM DTT, 50 mM d18-HEPES (98% pure) with and without 600 μM αIIb-L at pD 7.5 (not corrected for isotope effects). Four scans were used, resulting in a total measurement time of just 40 min. The methyl-based cross-saturation irradiation using an RF (radio frequency) field was applied as suggested,26 which covers from 3.5 to 8.5 ppm with the irradiation centered at 6.0 ppm, a region including the residual water protons, amide protons, aromatic protons, most of the α-protons, and some of the other aliphatic (β and γ) protons of the αIIb-L peptide. Selective irradiation was done by using the adiabatic WURST-2 band with a strength up to 0.17 kHz. The saturation time (Tsat) was set at 1.5 s and the interscan delay was 2 s. On the basis of the spectra with (Tsat = 1.5 s) and without irradiation (Tsat = 0 s), the signal loss was calculated using the intensity (I) ratio for each methyl group: Signal Loss = 1 – [I(Tsat=1.5)/I(Tsat=0)]. Error bars were generated from duplicates of the cross-saturation experiments.
Structure Calculation
A two-stage simulated annealing approach27 based on three sets of 1DNH, 1DC′N RDCs measured in two alignment media (phage pf1 and organic solvent PEG) was implemented for the structure determination of Ca2+-CIB1 in complex with αIIb-L using the program XPLOR-NIH 2.18. This calculation protocol is designed for studies of homologous proteins or the same protein under different conditions.27 Briefly, for stage 1, the temperature was cooled from 200 to 20 K, and a strong unramped force constant at 300 kcal mol–1 rad–2 for the dihedral angle (derived from the chemical shifts of Ca2+-CIB1 in complex with aIIb-L using the program Talos,28 after correction for the deuterium isotope effect29) was used to ensure proper secondary structure of the resulting structure. Also in stage 1, the force constant of dipolar couplings was ramped from 0.05 to 5 kcal mol–1 Hz2–. The obtained structure still has a certain amount of RDC energy; in stage 2, the temperature was cooled from 20 to 2 K and the lowest RDC energy structure obtained from stage 1 was used as a starting model. In stage 2, the force constant for the dihedral angle (derived from the lowest energy structure from stage 1) was ramped down from 300 to 50 kcal mol–1 rad–2 while the force constant for RDC was kept static at 1 kcal mol–1 Hz–2. Other force constants used in the calculation using Xplor-NIH were the same as previously described.17,30 The same approach has been used successfully to determine the structures of various other proteins.30−35 The structure was deposited to the Protein Data Bank with accession code 2LM5. NMR data were deposited to the BMRB with the code 18099.
The structure of αIIb-L was generated based on a recent NMR structure for the transmembrane and cytoplasmic domains of αIIbβ3 (PDB 2KNC).7 In this study, the αIIb subunit adopts an α-helical structure from I966 until N996 with an extended C-terminal tail at R997–E1008. We used the homology modeling program Swiss-Model web-server36 to generate the starting structure for αIIb-L, which has a Q1008 residue13,18 instead of E1008 at the C-terminal end. The structure of αIIb-L (L983-Q1008) obtained from homology modeling is essentially the same as the published αIIb structure in the αIIbβ3 complex.7 The structure of αIIb-L generated in this manner was used in the subsequent docking model studies of the Ca2+-CIB1/αIIb-L complex. In doing so, we assume that the tail of αIIb does not undergo a large conformational change when it binds to Ca2+-CIB1.
Docking Model of the Ca2+-CIB1/αIIb-L Complex
Using the refined solution structure of Ca2+-CIB1 in the complex and the structure of αIIb-L, a docking model for the Ca2+-CIB1/αIIb-L complex was generated using the Haddock web-server.37,38 It is known that the C-terminal extension of Ca2+-CIB1 experiences on/off slow motions from the hydrophobic pocket in the C-lobe of Ca2+-CIB1 in solution, thereby blocking nonspecific interactions.17,18 Hence, in our calculations, the structure of the complexed Ca2+-CIB1 protein was truncated in order to avoid blocking the access of αIIb-L to the hydrophobic pocket of Ca2+-CIB1; the truncated version of Ca2+-CIB1 contains residues 8–178 with the C-terminal tail of the protein (residues 179–191) removed. In the setup, positive methyl cross-saturation results were implemented as interfacial restraints for docking. The significantly affected methyl groups of residues I73, I114, L131, L135, I153, I168, V176, and I177 were set as the active residues, while the slightly affected methyl-containing residues I27, L28, L61, V76, V97, V132, L152, and I156 were set as passive residues in the Haddock calculations. Residues L983, W988, F992, and F993 in αIIb-L have been reported to be critical for the interaction between Ca2+-CIB1 and αIIb-L by a combined approach of site-directed mutagenesis and in vitro fluorescence studies.13 Hence, these four residues were set as the active residues for αIIb-L. Also in the setup, since αIIb-L residues L983–R995 have been determined to be helical and the remainder (N996–Q1008) to be an extended structure,7,39 fragment 995–1008 of αIIb-L was set as fully flexible for the docking model generation. Because Haddock can automatically detect semiflexible fragments,38 CIB1 was not set as fully flexible. RDC restrains that were used for the structure calculation for the Ca2+-CIB1 in complex with αIIb-L were also implemented in the docking model generation. In addition, dihedral angle restraints for CIB1 (8–178) and αIIb-L (983–995) were created based on the lowest RDC energy structure determined for the Ca2+-CIB1 in protein complex with αIIb-L as well as the available αIIb structure (PDB 2KNC) in complex with β3.7 Other parameters were standard default values except that 3000 structures were calculated for the stage of rigid body docking, 400 structures for the subsequent structural analysis stage, and 20 structures were set as the minimal number for clustering.37,38 The lowest interaction energy docking structure cluster was taken as the complex model for Ca2+-CIB1/αIIb-L.
Further Characterization of Calculated Structures
The lowest RDC energy structures of Ca2+-CIB1 (residues 8–191) in complex with αIIb-L were selected for further analysis. Procheck 3.5.440 was used to examine the structures determined for stereochemical quality. An in-house script was used to measure the interhelical angles for the helix pairs in each EF-hand.
Results
The Solution Structure of Ca2+-CIB1 in Complex with αIIb-L Reveals an Enlarged Hydrophobic Pocket Induced by αIIb-L Binding
The solution structure (Figure 2) of the Ca2+-CIB1 protein in complex with αIIb-L was refined based on three sets of backbone RDCs obtained in two alignment media (NH and C′N RDCs in pf1, and NH RDCs in PEG) using the solution structure of Ca2+-CIB1 (PDB 2L4H) as the starting model and a two-stage low-temperature simulated annealing protocol.27 The use of multiple sets of alignment media has been previously suggested because it can avoid the intrinsic degeneracy problem of using RDCs as well as avoid any preference for a minor conformation that could result from the use of a single alignment medium.41,42 Some residues on the flexible loop of CIB1 (residues 137–145) and the C-terminal end (180–191) have reduced RDC values due to local mobility,43,44 and these residues were excluded from the structure calculation.45 A good structural precision has been obtained for the structure calculation; the backbone rmsd of the 10 lowest energy structures is 0.46 Å (Figure 2A). The statistics of the structure determination of the Ca2+-CIB1 protein when complexed with αIIb-L have been summarized in Supporting Information Table S1. The solution structure of the Ca2+-CIB1 protein in the complex also shows a similar overall topology compared with the solution structure of Ca2+-CIB1 alone, with a backbone rmsd of 3.7 Å (Figure 2B). Compared with the Ca2+-CIB1 structure, the N-terminal α-helix (Helix 0, H0) in the new structure has shifted to some extent. The N-terminal α-helix is assumed to carry a myristoyl group in vivo which should associate with the cell membrane. Therefore, binding of αIIb-L could affect the orientation of CIB1 relative to the cell membrane. Moreover, the interhelical angles measured using an in-house script suggest that the opening of the helix pairs in each EF hand changes upon binding aIIb. (Table S2). The two EF-hands in the C-lobe have a bigger interhelical opening than free Ca2+-CIB1 by approximately 20° (Figure 2C). The resultant enlarged hydrophobic pocket in the C-lobe could readily adopt aIIb. The Q factors46 (Table S3 and Figure S1) before and after structural refinement denote a significant improvement in the results of fitting RDCs to the obtained structure of Ca2+-CIB1 complexed with αIIb-L compared with the starting model, that is, the free Ca2+-CIB1 structure (PDB 2L4H).
Figure 2.

The structure of Ca2+-CIB1 in complex with αIIb-L. (A) Superposition of the best 10 structures of the Ca2+-CIB1 protein in complex with αIIb-L; (B) superposition of the solution structures of Ca2+-CIB1 alone (PDB 2L4H, cyan) and in complex with αIIb-L (pink) (backbone rmsd = 3.7 Å). (C) The superimposed structures of the C-lobe of Ca2+-CIB1 alone (cyan) and in complex with αIIb-L (pink) demonstrate the increased opening of helix pairs H6/H7 and H8/H9. The two structures were superimposed based on H7 and H8; thus, the opening is highlighted by the altered orientations of H6 and H9.
Reverse Cross-Saturation Detects the Interface of the Ca2+-CIB1/αIIb-L Complex
Cross-saturation NMR experiments employing backbone amide proton NH resonances were originally developed to detect the interface for protein–protein interactions.47 Compared with the commonly used NMR Chemical Shift Perturbation (CSP) approach, which is widely used for determining the interface of protein/peptide complexes, the cross-saturation method avoids possible artifacts arising from induced conformational changes, where residues that experienced a large chemical shift change can in fact be far away from the actual interface.48 A more recent version of the cross-saturation experiment uses isotope labeled methyl groups rather than backbone amides.26 It has several advantages over the traditional cross-saturation backbone 15N–H method,47 such as high sensitivity, higher efficiency of magnetization transfer, and suitability for studying hydrophobic interactions that are often involved in protein/protein complex formation.26 Traditionally, the cross-saturation approach has been used to detect the binding interface of the smaller partner by irradiating the larger partner because the magnetization will be more efficiently transferred by spin diffusion49 in a larger protein. In our work, we applied the selective irradiation in a reverse manner (referred to as reverse cross-saturation), in a way that is somewhat similar to previous studies.50,51 We saturated the smaller binding partner (αIIb-L, 3 kDa) to detect the interface on the larger binding partner (Ca2+-CIB1, 24 kDa including the purification His-tag) (Figure 3). In our methyl-probe cross-saturation experiments, selective irradiation was employed to cover the spectral region from 3.5 to 8.5 ppm (Figure S2). To avoid artifacts, 99.9% D2O was used as the solvent.26 In the spectral area from 3.5 to 8.5 ppm (Figure S2), methyl labeled but otherwise perdeuterated Ca2+-CIB1 has no signals except for some peaks due to the d18-HEPES buffer (98% pure) used to dissolve the Ca2+-CIB1/αIIb-L complex, while αIIb-L contains aromatic protons, aliphatic proteins (Hα, Hβ, Hγ, etc.) and no amide protons (NH) because of the presence of the D2O solvent (Figure S2). Therefore, the irradiation selectively targets αIIb-L but not the deuterated CIB1. This saturation should then be transferred to the protonated methyl groups involved in the Ca2+-CIB1/αIIb-L interface by spin-diffusion (Figure 3). The interface of CIB1 in its complex with αIIb-L can therefore be accurately mapped using cross-saturation experiments.48 The results for these experiments are shown in Figure 4. In this case, control experiments were carried out on an NMR sample containing only Ca2+-CIB1, and the results show that all the methyl groups were evenly affected to a minimum level (half of all methyls are below 0.1 and the other half are between 0.1 and 0.2 in terms of signal loss) (Figure 4C). Since the irradiation pulse was suggested to be adiabatic,26 the signal loss in the control experiments is probably due to the presence of residual protons of the d18-HEPES buffer (50 mM, 98% pure) (Figure S2; for further discussion see Shimada48,51).
Figure 3.

Schematic diagram illustrating the mechanism of the reverse methyl cross-saturation for the Ca2+-CIB1/αIIb-L complex. Selective RF (Radio Frequency) irradiation was applied to the Hα, Hβ, and aromatic proton spectral region of αIIb-L, and the intensity change of the protonated methyl groups on deuterated Ca2+-CIB1 was monitored.
Figure 4.

Reverse methyl cross-saturation experiment of the Ca2+-CIB1/αIIb-L complex. (A) HSQC spectra of Ca2+-CIB1/αIIb-L showing the effects of saturation on the Ile residues. The spectrum on the left is the 1H, 13C-HSQC with no saturation, and the spectrum on the right includes selective saturation. (B) Signal loss ratio of the intensities of the methyl groups in Ca2+-CIB1 complexed with αIIb-L, with and without the selective irradiation. The signal losses over 30% and 20% were highlighted with gridlines and these results were further utilized in the generation of docking complex structural model for Ca2+-CIB1 and αIIb-L using the program Haddock. (C) Control experiment of methyl cross-saturation with the free Ca2+-CIB1: only a minimum saturation effect was observed on nearly all methyl groups.
To identify the interface between Ca2+-CIB1 and αIIb-L, an identical setup as the one used for the control experiments was used for a sample of Ca2+-CIB1 complexed with 1/1.2 ratio of αIIb-L. The results obtained are illustrated for the superimposed HSQC spectra of the Ile region (Figure 4A1). With irradiation, the I73, I114, and I177 residues show significant signal loss compared to the reference spectrum (Tsat = 0 s) (Figure 4A2 and B), whereas other methyl protonated isoleucines (e.g., I58, I106, I162, and I189) are relatively less affected by the cross-saturation. The significantly affected methyl-containing Ile, Leu, and Val residues with a signal loss ratio above 0.3 include I73, I114, L131, L135, I153, I168, V176, and I177 (Figure 4B), and these were classified as the active residues in the Ca2+-CIB1/αIIb-L interface that make direct contact with the binding partner αIIb-L. Other slightly affected methyl-containing residues with signal loss ratio between 0.2 and 0.3, which include I27, L28, L61, V76, V97, V132, L152, and I156 (Figure 4B), have been classified as passive residues that are close to the interface, but lack direct contact with αIIb-L. The remaining peaks are the nonaffected or marginally affected methyl groups with a signal loss ratio less than 0.2 (Figure 4B), and these are considered not involved in the hydrophobic interactions between the protein and the peptide.
The cross-saturation effects were mapped on the newly determined structure of Ca2+-CIB1 in complex with αIIb-L (Figure 5B). Compared with methyl CSP (chemical shift perturbation) effects (Figure 5A), cross-saturation experiments provide a similar interface for the interactions between Ca2+-CIB1 and αIIb-L, suggesting that the C-domain of Ca2+-CIB1 is the primary binding site for the cytoplasmic domain of αIIb. Small differences between CSP and cross-saturation were also observed; the distribution of active interfacial residues from the cross-saturation experiments (Figure 5B) seems to suggest one possible orientation for the αIIb-L peptide, whereas two possible orientations were suggested by CSP (Figure 5A). Clearly, a majority of the significantly affected methyl groups are found in the C-lobe of Ca2+-CIB1, except for residue I73, which is in the N-lobe.
Figure 5.

Mapping the reverse cross-saturation effect on the structure. (A) Mapping the methyl Chemical Shift Perturbation (CSP) on the structure of Ca2+-CIB1 in complex with αIIb; the methyl CSP values were previously reported;17 (B) mapping the signal loss values from reverse cross-saturation experiments on the structure of Ca2+-CIB1 in complex with αIIb. In both panels, the dotted lines (red) resemble possible orientations of αIIb-L in its complex structure with Ca2+-CIB1.
The previously discussed C-terminal displacement mechanism for CIB118 allows us to interpret our methyl cross-saturation results. The C-terminal extension of CIB1 undergoes a conformational transition between free and bound states as suggested by our earlier backbone relaxation and relaxation dispersion measurements.17,18 The three assigned methyl groups on the C-terminal extension of Ca2+-CIB1, that is, I189δ1, and V190γ1 and γ2, are only marginally affected by the saturation from αIIb-L, which suggests that these three residues are not in direct contact with αIIb-L in the complex. Thus, the cross-saturation results are consistent with the backbone relaxation and relaxation dispersion results regarding the flexibility of the C-terminal extension; the extension remains flexible when the protein binds to αIIb-L.17,18
αIIb-L Has an N-Terminal α-Helix and an Extended C-Terminal Tail
Initial attempts of using isotope-filtered52 or transferred NOESY experiments53 to determine the structure of αIIb-L in complex with Ca2+-CIB1 failed to provide sufficient intermolecular NOEs between αIIb-L and Ca2+-CIB1. Even though NMR titration experiments (results not shown) suggest that the interaction between these two is in slow exchange regime, previous studies12 suggested that αIIb interacts relatively weakly with Ca2+-CIB1 with a dissociation constant (Kd) of 1.4 μM at 37 °C. This exchange behavior seems to have hindered the determination of the solution structure of αIIb-L in complex with Ca2+-CIB1. Isotope-labeled αIIb-L was then prepared through bacterial expression as a fusion protein but the solubility in aqueous solution became a problem, even though the N-terminal acetylated synthetic αIIb-L peptide was quite soluble. Fortunately, a structure for the αIIb subunit (including the transmembrane and cytoplasmic domains) (PDB 2KNC), in complex with β3, has recently been reported.7 This structure of αIIb (PDB 2KNC) covers the fragment of αIIb-L used in our work (residues 983–1008). It is strikingly similar to a structure previously determined in a 45% trifluoroethanol aqueous solution for the synthetic αIIb-L peptide,39 containing a typical α-helical structure (L983–N996) followed by an extended tail (R997–Q1008).
In addition, it should be noted that the C-terminal extension of CIB1 in the crystal structure of Ca2+-CIB1 (1XO5) is folded back into the protein and adopts an α-helical conformation (Figure S3). This implies that the hydrophobic pocket in the C-domain of Ca2+-CIB1 can readily accommodate a binding partner in an α-helical conformation. Therefore, we suggest that it is reasonable to assume that the fragment L983–R997 adopts an α-helical conformation when interacting with Ca2+-CIB1.
The Structure of the Ca2+-CIB1/αIIb-L Complex Reveals a Relative Orientation Similar to CnB/CnA
The solution structure of the Ca2+-CIB1/αIIb-L complex has been determined by data-driven docking. The 20 lowest energy structures of the Ca2+-CIB1/αIIb-L complex are displayed in Figure 6 in both ribbon and surface charge forms. In Figure 6A, the N-terminal helical part of αIIb-L interacts with the C-domain of Ca2+-CIB1, while the negatively charged C-terminal tail of αIIb-L remains unstructured (Figure 6A), possibly interacting with some positively charged residues, for example, R33 and K65 as was suggested by backbone CSP measurements.18 In our docking experiment setup, we solely relied on the methyl cross-saturation results for Ca2+-CIB1 and on previously reported critical residues in αIIb-L;13 thus, these restraints do not reflect any potential electrostatic interactions. Therefore, the relative orientation of these two partners was mostly driven by the enthalpy of the hydrophobic interactions between the αIIb-L helix and the C-domain of Ca2+-CIB1. A related situation is found for the structure of the homologous protein calcineurin B (CnB) complexed with calcineurin A (CnA) (PDB 1TCO) (Figure 6C), in which the C-terminal domain of CnB interacts with the N-terminal of the CnB binding domain of CnA, with the CnB binding domain of CnA adopting an α-helical conformation. Regarding the structure of the acidic tail (P998PLEEDDEEGQ1008) of αIIb, we attempted to dock this portion better by adding R33 and K65 (residues identified by backbone CSP) of CIB1 as heavily affected residues but the tail still remained dynamic probably because no restraints were available for this portion of the αIIb tail.
Figure 6.

Structural model for the Ca2+-CIB1/αIIb-L complex. (A) The superposition of the 20 best structures of the Ca2+-CIB1/αIIb-L complex generated using Haddock. (B) The Ca2+-CIB1 (8–178) protein is represented with a surface structure, while aIIb-L is represented as a ribbon structure. This figure was generated with the program MolMol.63 The surface polarity is scaled with colors, using blue for the positively charged surface and red for the negatively charged surface, while a white color indicates nonpolar patches. (C) The surface charge model for the complex of CnB/CnA (PDB 1TCO), in which CnB is presented with a surface structure and CnA (residues 340–373) is presented as a ribbon structure.
The Model Is Supported by NMR Studies of the Interactions of Ca2+-CIB1 with αIIb-L and αIIb-S
A shorter version of the αIIb peptide (αIIb-S, Ac-L983VLAMWKVGFFKRNR997-NH2) was synthesized to examine the role of the acidic tail of αIIb in its interaction with Ca2+-CIB1. Specifically, the difference between the αIIb-L and αIIb-S peptides resides in the C-terminal fragment P998PLEEDDEEGQ1008, which bears multiple negative charges, making this region of αIIb extremely acidic. In Figure S4A, the superimposed HSQC spectra of Ca2+-CIB1/αIIb-L and Ca2+-CIB1/αIIb-S show small differences with a few methyl groups experiencing minor chemical shift changes. This suggests that the C-terminal fragment P998PLEEDDEEGQ1008 was not involved in specific interactions with Ca2+-CIB1, which could affect the interactions between Ca2+-CIB1 and αIIb-L. The residues (L8, V45, V76, L92, L94, V97, I 106, and L170) with minor chemical shift changes are mainly located in the N-lobe of CIB1. On the other hand, these shifted methyl groups do not center at a specific location in the structure, implying a dynamic interaction between the N-lobe of CIB1 and the acidic tail (P998PLEEDDEEGQ1008). Perhaps this region of αIIb can associate with other protein binding partners. A recent study54 shows that this part of the αIIb cytoplasmic domain can in fact play a role in platelet activation, but it only does so when it is anchored to the inner surface of the membrane.
The Potential Roles of Mg2+and Ca2+ in the Regulation of CIB1/αIIb Interaction
Since Mg2+ ions are present in the cytoplasm with an almost invariant concentration of around 1–2 mM and the structure of Mg2+-CIB1 is overall similar to that of Ca2+-CIB1,16,17 we were interested in the structural differences between the complexes of CIB1/αIIb-L when bound to Ca2+ and Mg2+, in order to further understand the role of Ca2+ in the regulation of the CIB1/αIIb interaction. Therefore, 1H,13C-HSQC spectra for the methyl groups of CIB1 complexed with αIIb-L were recorded in the presence of either Ca2+ or Mg2+ (Figure S4B). Similar to the backbone NMR studies,55 methyl side chain 1H,13C-HSQC spectra for Ca2+- and Mg2+-bound CIB1/αIIb-L show nearly the same pattern with minor deviations except for two residues in the calcium/magnesium binding loop, I168 and L123 (Figure S4B). These results suggest that Ca2+-CIB and Mg2+-CIB1 interact with αIIb in nearly the same manner.
Discussion
In this study, we have obtained a solution structure for the isotope-labeled Ca2+-CIB1 protein, complexed with the synthetic αIIb-L peptide that encompasses the cytoplasmic domain and part of the transmembrane domain of the αIIb subunit of platelet integrin. The structure for the complexed protein could be determined based on RDC restraints. With the use of the available structure of αIIb (PDB 2KNC), a structural model for the Ca2+-CIB1/αIIb-L complex was obtained using data-driven docking based on reverse cross-saturation NMR experiments. The two binding partners in this complex structure adopt a relative orientation with the α-helical N-terminal portion of αIIb-L buried into the C-lobe hydrophobic pocket of Ca2+-CIB1 and the negatively charged and extended C-terminal tail of αIIb-L pointing toward the N-lobe of Ca2+-CIB1 but being relatively dynamic. The superposition of the ordered structural fragment (residues 983–995) and the corresponding fragment of 2KNC provides the orientation of the Ca2+-CIB1/αIIb-L complex relative to the cell membrane (Figure 7A). As expected, the N-terminal extension of Ca2+-CIB1(helix 0, H0) points toward the cell membrane, which is consistent with its role in linking to a membrane-bound myristoyl group (Figure 7A). However, when modeled together with a membrane, a steric clash was observed between the transmembrane helix of αIIb and the eighth helix of Ca2+-CIB1 (H8) (Figure 7A). Moreover, CIB1 would become associated directly with the membrane to a large extent if the αIIb subunit interacts with Ca2+-CIB1 with both of its transmembrane and cytoplasmic domains. Since CIB1 is mainly found as a myristoylated protein in the cytoplasm3,56 and the binding interface on αIIb primarily includes the fragment of 983–995,3 it seems unlikely that Ca2+-CIB1 will penetrate into the cell membrane or be engaged with a significantly large part of the αIIb transmembrane domain. Therefore, a conformational change is likely to happen in αIIb upon binding to Ca2+-CIB1. We propose that the transmembrane helix of αIIb will bend around W988 (the membrane-border residue for αIIb-L) when binding to Ca2+-CIB1 (Figure 7B,C). In this manner, the steric clash and the potential energy barrier caused by an intrusion of CIB1 into the membrane would be avoided.
Figure 7.

Possible conformational changes of αIIb upon binding of Ca2+-CIB1. (A) Superposition of the ordered structural fragment (983–995) of the averaged structure of αIIb-L complexed with Ca2+-CIB1 with the corresponding fragment of 2KNC. A steric clash was observed between 2KNC and the 8th helix of Ca2+-CIB1 (H8, brown color); The N-terminal extension of Ca2+-CIB1 (H0, pink color) points toward the cell membrane, which is consistent with its role of linking the myristoyl group and the Ca2+-CIB1 to a membrane. (B) The side view of (A); (C) a possible bending mechanism was proposed for the conformational change of αIIb upon binding of Ca2+-CIB1, as indicated by the red dashed line with arrows. The letter N in italics indicates the N-terminal of Ca2+-CIB1.
Recent structural studies of the transmembrane domains57,58 as well as the protein fragments including both the transmembrane and cytoplasmic domains of αIIbβ37,8 have shed some light on the molecular mechanism of the regulation of the activation of αIIbβ3. For the structure of the αIIbβ3 transmembrane domains, various approaches gave rise to almost converged structures (see detailed analysis in Yang et al.7 and Metcalf et al.57). However, the structure of the cytoplasmic domains of αIIbβ3 was reported to be diverse.7,8 The importance of the previously suggested salt bridge between αIIb R995 and β3 D7237,59 that was considered for a long time to be critical for the association of αIIbβ3 has recently been brought into question,8,60 consistent with the dynamic nature of the cytoplasmic domains of αIIbβ3. The G991FFKR995 motif on αIIb is critical for the interactions between αIIb and β3 as revealed in the αIIbβ3 complex structure determined using Cys-scanning-mutagenesis/disulfide-cross-linking/rosetta-modeling61 and another αIIbβ3 complex NMR structure that has been determined using lipid bicelles as the solvent (PDB 2K9J).58 On the basis of a comparison with these two structures, Ca2+-CIB1 seems to dissociate the αIIbβ3 heterodimer, which would potentially activate the αIIbβ3 complex14 because the F992 and F993 side chains become buried in the hydrophobic pocket in the Ca2+-CIB1/αIIb-L complex structure (Figure S5). It has also been reported that an F992A mutation in αIIb may partially impair the interaction with β3 although both F992 and F993 in αIIb point away from the β3 subunit in 2KNC.7 Moreover, a potential conformational change of αIIb upon binding to Ca2+-CIB1 (Figure 7) will also disturb the stability of the structure of the αIIbβ3 complex and cause the dissociation of these two subunits. On the basis of the classic studies indicating that the activation of integrin can be caused by the dissociation of the heterodimer,4 Ca2+-CIB1 is highly likely able to disrupt the association of αIIb and β3.
Conclusion
Taken all data together, a mechanism for activation of αIIbβ3 upon interacting with Ca2+-CIB1 can be proposed, that is similar to the action of the cytoskeletal protein talin,9 where Ca2+-CIB1 dissociates the αIIbβ3 heterodimer and consequently activates integrin αIIbβ3 (Figure 8). Alternatively CIB1 can maintain the activated state of αIIbβ3 by covering the binding site in αIIb and preventing its association with β3. Since nearly identical HSQC NMR patterns were observed for Mg2+-CIB1 and for Ca2+-CIB1 upon interaction with αIIb-L (Figure S4B), our data suggest that the same complex can form under various physiological conditions and is not calcium-dependent per se. This notion is supported by previous in vivo studies, in which αIIb was shown to interact with CIB1 in both resting and stimulated platelets with no requirement for a rise in the intracellular calcium concentration.19 In addition, it is possible that protein phosphatase 1 could compete with CIB1 for αIIb, as these two proteins share the same binding motif (K989VGF992) on the αIIb cytoplasmic domain.62
Figure 8.

A structural model for the mechanism of Ca2+-CIB1 regulation of integrin activation. (A) Inactivated αIIbβ3 complex, modified based on Yang et al.7 (B) In the presence of Ca2+-CIB1, the αIIb cytoplasmic subunit interacts with Ca2+-CIB1, with its N-terminal α-helix buried in the hydrophobic pocket of the C-lobe of CIB1 and its C-terminal negatively charged tail being dynamic but likely being in contact with the N-lobe of CIB1. Subsequently, αIIb and β3 are separated by Ca2+-CIB1 and this αIIbβ3 complex dissociates. The integrin αIIbβ3 dimer is activated by Ca2+-CIB1, which is similar to the activation by talin, a cytoskeleton protein, which binds to the β3 cytoplasmic domain.9.
In this work, we have used a reverse cross-saturation NMR approach to detect the interface on Ca2+-CIB1 that recognizes the bound αIIb-L peptide. In these experiments, we selectively irradiated the smaller partner in the protein–protein complex, and the results from our work (Figure 4 and Figure 5) demonstrate that it is possible to identify the hydrophobic residues that are involved in the interactions between CIB1 and αIIb. This represents a powerful way of using the NMR cross-saturation experiment, where normally the larger partner has been irradiated to achieve more effective spin-difussion.26,47 Many protein complexes currently under investigation can be represented by studying a large protein and a synthetic peptide to represent the intact protein partner or a protein domain. Provided that the proteins of interest can be bacterially expressed, deuterated, and selectively protonated on methyl groups, the reverse cross-saturation experiment with methyl detection should become a useful NMR approach for better understanding protein–protein interactions in general.
Acknowledgments
This research has been supported by the Canadian Institutes of Health Research (CIHR). H.J.V. holds a Scientist award from the Alberta Heritage Foundation for Medical Research, now called Alberta Innovates Health Solutions (AHFMR/AIHS). H.H. was the recipient of an AHFMR Studentship award. We thank the late Dr. Deane McIntyre for maintaining the NMR instrumentation and Dr. Hiroaki Ishida for helpful discussions. Dr. Ryan McKay at NANUC (National High Field Nuclear Magnetic Resonance Centre) is gratefully acknowledged for his assistance in attempting to record intermolecular NOEs between Ca2+-CIB1 and αIIb-L.
Supporting Information Available
Table S1 provides the statistics of the NMR-based structure calculation of Ca2+-CIB1 in complex with αIIb-L. The interhelical angle (Table S2) and Q factor (Table S3) analyses for the structures of Ca2+-CIB1 free and in complex with αIIb-L were also included. Figure S1 shows the NH-RDC (pf1) fitting for the structures of Ca2+-CIB1 free and in complex with αIIb-L. Figure S2 shows the 1-D 1H NMR spectra illustrating the reverse cross-saturation approach employed in this work; Figure S3 shows the crystal structure of Ca2+-CIB1 (PDB 1XO5); Figure S4 shows the superposition of 2-D NMR HSQC spectra demonstrating the roles of the acidic tail of αIIb-L, as well as Ca2+ and Mg2+ in the regulation of the CIB1/αIIb interactions; Figure S5 shows the mapping of critical residues in αIIb-L onto the complex structure obtained in this work. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Author Present Address
† Samuel Lunenfeld Research Institute, Mt. Sinai Hospital, Toronto, Canada, M5G 1X5
Supplementary Material
References
- Luo B. H.; Carman C. V.; Springer T. A. Annu. Rev. Immunol. 2007, 25, 619–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R. O. Cell 2002, 110, 673–687. [DOI] [PubMed] [Google Scholar]
- Leisner T. M.; Yuan W. P.; DeNofrio J. C.; Liu J.; Parise L. V. Curr. Opin. Hematol. 2007, 14, 255–261. [DOI] [PubMed] [Google Scholar]
- Kim M.; Carman C. V.; Springer T. A. Science 2003, 301, 1720–1725. [DOI] [PubMed] [Google Scholar]
- Ulmer T. S.; Yaspan B.; Ginsberg M. H.; Campbell I. D. Biochemistry 2001, 40, 7498–7508. [DOI] [PubMed] [Google Scholar]
- Weljie A. M.; Hwang P. M.; Vogel H. J. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 5878–5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Ma Y. Q.; Page R. C.; Misra S.; Plow E. F.; Qin J. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 17729–17734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf D. G.; Moore D. T.; Wu Y.; Kielec J. M.; Molnar K.; Valentine K. G.; Wand A. J.; Bennett J. S.; DeGrado W. F. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 22481–22486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegener K. L.; Partridge A. W.; Han J.; Pickford A. R.; Liddington R. C.; Ginsberg M. H.; Campbell I. D. Cell 2007, 128, 171–182. [DOI] [PubMed] [Google Scholar]
- Naik U. P.; Patel P. M.; Parise L. V. J. Biol. Chem. 1997, 272, 4651–4654. [DOI] [PubMed] [Google Scholar]
- Gifford J. L.; Walsh M. P.; Vogel H. J. Biochem. J. 2007, 405, 199–221. [DOI] [PubMed] [Google Scholar]
- Yamniuk A. P.; Vogel H. J. Protein Sci. 2005, 14, 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry W. T.; Boudignon-Proudhon C.; Shock D. D.; McFadden A.; Weiss J. M.; Sondek J.; Parise L. V. J. Biol. Chem. 2002, 277, 28877–28883. [DOI] [PubMed] [Google Scholar]
- Tsuboi S. J. Biol. Chem. 2002, 277, 1919–1923. [DOI] [PubMed] [Google Scholar]
- Burgoyne R. D. Biochim. Biophys. Acta 2004, 1742, 59–68. [DOI] [PubMed] [Google Scholar]
- Yamniuk A. P.; Nguyen L. T.; Hoang T. T.; Vogel H. J. Biochemistry 2004, 43, 2558–2568. [DOI] [PubMed] [Google Scholar]
- Huang H.; Ishida H.; Yamniuk A. P.; Vogel H. J. J. Biol. Chem. 2011, 286, 17181–17192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamniuk A. P.; Ishida H.; Vogel H. J. J. Biol. Chem. 2006, 281, 26455–26464. [DOI] [PubMed] [Google Scholar]
- Tsuboi S.; Nonoyama S.; Ochs H. D. EMBO Rep. 2006, 7, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W. P.; Leisner T. M.; McFadden A. W.; Wang Z. Y.; Larson M. K.; Clark S.; Boudignon-Proudhon C.; Lam S. C. T.; Parise L. V. J. Cell. Biol. 2006, 172, 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugarinov V.; Kanelis V.; Kay L. E. Nat. Protoc. 2006, 1, 749–754. [DOI] [PubMed] [Google Scholar]
- Hansen M. R.; Mueller L.; Pardi A. Nat. Struct. Biol. 1998, 5, 1065–1074. [DOI] [PubMed] [Google Scholar]
- Ruckert M.; Otting G. J. Am. Chem. Soc. 2000, 122, 7793–7797. [Google Scholar]
- Yang D. W.; Venters R. A.; Mueller G. A.; Choy W. Y.; Kay L. E. J. Biomol. NMR 1999, 14, 333–343. [Google Scholar]
- Cordier F.; Dingley A. J.; Grzesiek S. J. Biomol. NMR 1999, 13, 175–180. [DOI] [PubMed] [Google Scholar]
- Takahashi H.; Miyazawa M.; Ina Y.; Fukunishi Y.; Mizukoshi Y.; Nakamura H.; Shimada I. J. Biomol. NMR 2006, 34, 167–177. [DOI] [PubMed] [Google Scholar]
- Chou J. J.; Li S. P.; Bax A. J. Biomol. NMR 2000, 18, 217–227. [DOI] [PubMed] [Google Scholar]
- Cornilescu G.; Delaglio F.; Bax A. J. Biomol. NMR 1999, 13, 289–302. [DOI] [PubMed] [Google Scholar]
- Venters R. A.; Farmer B. T.; Fierke C. A.; Spicer L. J. Mol. Biol. 1996, 264, 1101–1116. [DOI] [PubMed] [Google Scholar]
- Huang H.; Ishida H.; Vogel H. J. Protein Sci. 2010, 19, 475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J. J.; Li S. P.; Klee C. B.; Bax A. Nat. Struct. Biol. 2001, 8, 990–997. [DOI] [PubMed] [Google Scholar]
- Park C. J.; Lee J. H.; Choi B. S. Nucleic. Acids. Res. 2005, 33, 4172–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennestri M.; Melino S.; Contessa G. M.; Casavola E. C.; Paci M.; Ragnini-Wilson A.; Cicero D. O. J. Biol. Chem. 2007, 282, 667–679. [DOI] [PubMed] [Google Scholar]
- Verdone G.; Corazza A.; Colebrooke S. A.; Cicero D.; Eliseo T.; Boyd J.; Doliana R.; Fogolari F.; Viglino P.; Colombatti A.; Campbell I. D.; Esposito G. J. Biomol. NMR 2009, 43, 79–96. [DOI] [PubMed] [Google Scholar]
- Gifford J. L.; Ishida H.; Vogel H. J. J. Biomol. NMR 2011, 50, 71–81. [DOI] [PubMed] [Google Scholar]
- Arnold K.; Bordoli L.; Kopp J.; Schwede T. Bioinformatics 2006, 22, 195–201. [DOI] [PubMed] [Google Scholar]
- Dominguez C.; Boelens R.; Bonvin A. M. J. J. J. Am. Chem. Soc. 2003, 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- De Vries S. J.; van Dijk M.; Bonvin A. M. J. J. Nat. Protoc. 2010, 5, 883–897. [DOI] [PubMed] [Google Scholar]
- Hwang P. M.; Vogel H. J. J. Mol. Recognit. 2000, 13, 83–92. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Macarthur M. W.; Moss D. S.; Thornton J. M. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar]
- Clore G. M.; Starich M. R.; Bewley C. A.; Cai M. L.; Kuszewski J. J. Am. Chem. Soc. 1999, 121, 6513–6514. [Google Scholar]
- Prestegard J. H.; Bougault C. M.; Kishore A. I. Chem. Rev. 2004, 104, 3519–3540. [DOI] [PubMed] [Google Scholar]
- Meiler J.; Prompers J. J.; Peti W.; Griesinger C.; Bruschweiler R. J. Am. Chem. Soc. 2001, 123, 6098–6107. [DOI] [PubMed] [Google Scholar]
- Fischer M. W.; Losonczi J. A.; Weaver J. L.; Prestegard J. H. Biochemistry 1999, 38, 9013–9022. [DOI] [PubMed] [Google Scholar]
- Jain N. U.; Wyckoff T. J.; Raetz C. R.; Prestegard J. H. J. Mol. Biol. 2004, 343, 1379–1389. [DOI] [PubMed] [Google Scholar]
- Cornilescu G.; Marquardt J. L.; Ottiger M.; Bax A. J. Am. Chem. Soc. 1998, 120, 6836–6837. [Google Scholar]
- Takahashi H.; Nakanishi T.; Kami K.; Arata Y.; Shimada I. Nat. Struct. Biol. 2000, 7, 220–223. [DOI] [PubMed] [Google Scholar]
- Shimada I. Methods Enzymol. 2005, 394, 483–506. [DOI] [PubMed] [Google Scholar]
- Kalk A.; Berendsen H. J. C. J. Magn. Reson. 1976, 24, 343–366. [Google Scholar]
- Takeda M.; Terasawa H.; Sakakura M.; Yamaguchi Y.; Kajiwara M.; Kawashima H.; Miyasaka M.; Shimada I. J. Biol. Chem. 2003, 278, 43550–43555. [DOI] [PubMed] [Google Scholar]
- Shimada I.; Ueda T.; Matsumoto M.; Sakakura M.; Osawa M.; Takeuchi K.; Nishida N.; Takahashi H. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 54, 123–140. [Google Scholar]
- Ikura M.; Clore G. M.; Gronenborn A. M.; Zhu G.; Klee C. B.; Bax A. Science 1992, 256, 632–638. [DOI] [PubMed] [Google Scholar]
- Post C. B. Curr. Opin. Struct. Biol. 2003, 13, 581–588. [DOI] [PubMed] [Google Scholar]
- Koloka V.; Christofidou E. D.; Vaxevanelis S.; Dimitriou A. A.; Tsikaris V.; Tselepis A. D.; Panou-Pomonis E.; Sakarellos-Daitsiotis M.; Tsoukatos D. C. Platelets 2008, 19, 502–511. [DOI] [PubMed] [Google Scholar]
- Yamniuk A. P.; Gifford J. L.; Linse S.; Vogel H. J. Biochemistry 2008, 47, 1696–1707. [DOI] [PubMed] [Google Scholar]
- Yamniuk A. P.; Vogel H. J. Calcium Binding Proteins 2006, 1, 150–155. [Google Scholar]
- Metcalf D. G.; Kulp D. W.; Bennett J. S.; DeGrado W. F. J. Mol. Biol. 2009, 392, 1087–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau T. L.; Kim C.; Ginsberg M. H.; Ulmer T. S. EMBO J. 2009, 28, 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinogradova O.; Velyvis A.; Velyviene A.; Hu B.; Haas T. A.; Plow E. F.; Qin J. Cell 2002, 110, 587–597. [DOI] [PubMed] [Google Scholar]
- Wang W.; Luo B. H. J. Cell. Biochem. 2010, 109, 447–452. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Luo B. H.; Barth P.; Schonbrun J.; Baker D.; Springer T. A. Mol. Cell 2009, 34, 234–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayan K. V.; Liu Y.; Li T. T.; Bray P. F. J. Biol. Chem. 2004, 279, 33039–33042. [DOI] [PubMed] [Google Scholar]
- Koradi R.; Billeter M.; Wuthrich K. J. Mol. Graphics 1996, 14, 51–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.