

Abstract
The transcription factor NF-κB promotes survival of cancer cells exposed to doxorubicin and other chemotherapeutic agents. IκB kinase is essential for chemotherapy-induced NF-κB activation and considered a prime target for anticancer treatment. An IκB kinase inhibitor sensitized human melanoma xenografts in mice to killing by doxorubicin, yet also exacerbated treatment toxicity in the host animals. Using mouse models that simulate cell-selective targeting, we found that impaired NF-κB activation in melanoma and host myeloid cells accounts for the therapeutic and the adverse effects, respectively. Ablation of tumor-intrinsic NF-κB activity resulted in apoptosis-driven tumor regression following doxorubicin treatment. By contrast, chemotherapy in mice with myeloid-specific loss of NF-κB activation led to a massive intratumoral recruitment of interleukin-1β-producing neutrophils and necrotic tumor lesions, a condition associated with increased host mortality but not accompanied by tumor regression. Therefore, a molecular target-based therapy may be steered toward different clinical outcomes depending on the drug’s cell-specific effects.
Keywords: NF-κB, melanoma, neutrophil, efficacy, toxicity
Introduction
Treatment resistance poses a major challenge in cancer chemotherapy. Tumors refractory to chemotherapeutic agents emerge as intrinsic resistance traits are selected for and stabilized in the tumor cell population (1, 2). Tumor cells may also contribute to host cell-mediated resistance mechanisms; for example, tumor-derived factors can mobilize and recruit specific subsets of circulating and stromal cells and influence their function (7–9), thereby playing an active role in the construction of a microenvironment that defies cytotoxic treatment. However, hematopoietic-derived host cells also influence chemotherapy-induced tumor cell death (3, 4) and modulate tumor-specific immune responses triggered by primary treatment (5, 6). Both tumor-intrinsic and host cell-mediated mechanisms of treatment resistance are likely dictated, albeit to varying extents, by the tumor’s inherent genetic properties.
Another obstacle for successful cancer treatment is the toxicity of therapeutic agents that inevitably limits the dose and frequency of treatment. Treatment-induced toxicity, in most cases, manifests as a mixed condition of direct drug-inflicted adversity and secondary inflammation-mediated damage (11). Toxicity either arises from nonspecific “off-target” effects of the drug or occurs due to interference with an essential physiological function of the intended drug target. In the latter circumstances, substantial clinical benefits can be derived from selectively interfering with the disease-related function of the target molecule while sparing other functions whose loss is responsible for adverse side reactions. Cell-specific drug targeting may offer such selectivity if treatment efficacy and toxicity are mechanistically based in different cell types.
The transcription factor NF-κB is activated in response to diverse stress and inflammatory stimuli, and mediates cellular interpretation of the triggering stimuli via regulation of gene expression. NF-κB activation requires the IκB kinase (IKK), which phosphorylates and induces degradation of the NF-κB-bound inhibitor IκB (12). DNA-damaging anticancer drugs such as doxorubicin are potent inducers of NF-κB activity. Doxorubicin is known to activate IKK via a DNA damage-induced signaling cascade involving phosphorylation, sumoylation, and ubiquitination of the IKKγ/NF-κB essential modulator, the IKK regulatory subunit (13, 14). Among genes induced by chemotherapeutic agents in an NF-κB-dependent manner are several inhibitors of apoptosis that can prevent drug-induced cell death (15–17). Therefore, inhibition of NF-κB signaling has been considered a promising way to overcome tumor cell-intrinsic resistance to killing by cytotoxic agents. In fact, pharmacological inhibitors of IKK were found to increase the apoptotic sensitivity of melanoma and other chemoresistant tumor cells to doxorubicin treatment (18–20). Less clear, however, is the effect of NF-κB pathway inhibitors on tumor-host interactions under conditions of chemotherapy as well as its contributions to treatment-induced toxicity.
We suspected that, given the nodal position of NF-κB in the inflammatory signaling network, systemic inhibition of NF-κB signaling might not only sensitize tumor cells to chemotherapy but will also produce pleiotropic effects on inflammation-mediated processes associated with cancer treatment. We investigated this possibility using mouse models of melanoma chemotherapy, as melanoma is amongst the group of highly chemoresistant malignancies for which very few effective treatment options are available so far (10).
Results
Role of NF-κB signaling in chemoresistance of human melanoma cells
NF-κB is constitutively activated in many cancers, and aberrant NF-κB activation has been linked to maintenance of the malignant state and resistance to cytotoxic chemotherapy (21). To investigate the link between NF-κB and melanoma chemoresistance, we first examined NF-κB expression and activation in five independently established human melanoma cell lines: A375, MM455, MM608, Hs944T, and Roth. The NF-κB proteins RelA and p50, which form a heterodimer, were abundantly expressed in all cell lines (Supplementary Fig. S1A). NF-κB activation entails IκB degradation and nuclear translocation of the liberated NF-κB dimer. The amount of nuclear RelA in untreated and doxorubicin-treated cells varied considerably among the melanoma lines, and the pattern of NF-κB activation ranged from constitutive to inducible to nearly undetectable (Supplementary Fig. S1B). Hs944T and Roth were competent and defective, respectively, in tumor-intrinsic NF-κB activation in response to doxorubicin (Fig. 1A), and chosen as representing two contrasting subtypes of melanoma lines for further analysis.
Figure 1. Doxorubicin-induced NF-κB activation and NF-κB-mediated chemoresistance in Hs944T and Roth human melanoma cells.
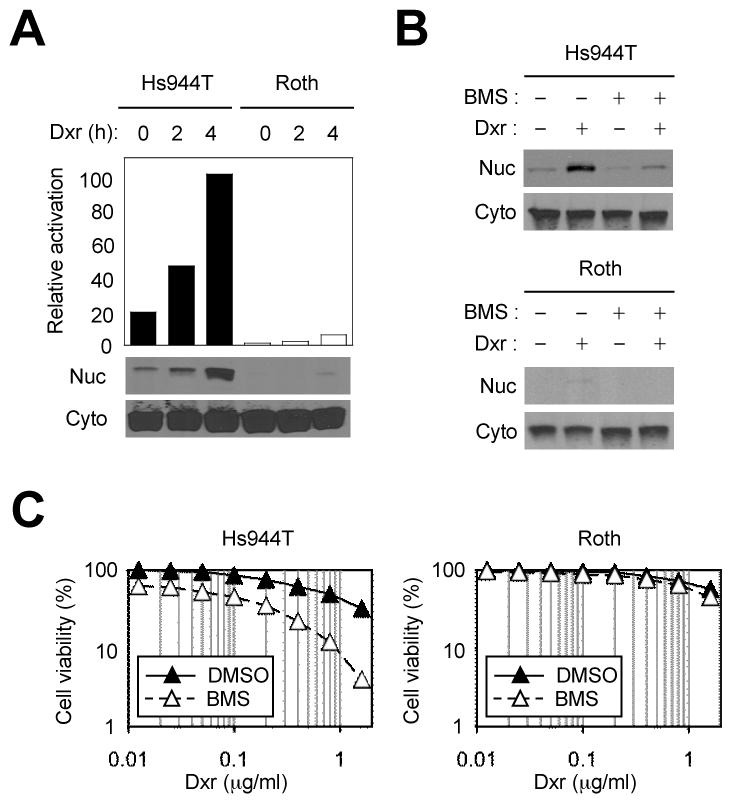
A, Cytoplasmic (Cyto) and nuclear (Nuc) extracts from Hs944T and Roth cells treated with doxorubicin (Dxr; 2 μg/ml) were analyzed by immunoblotting with anti-RelA antibody. Values in the plot represent relative amounts of nuclear RelA. B, Hs944T and Roth cells were treated with doxorubicin and BMS (75 μM; added 1 h prior to doxorubicin treatment) as indicated. Cyto and Nuc extracts were prepared 2 h after doxorubicin treatment and analyzed as in A. C, Hs944T and Roth cells were treated with doxorubicin and BMS as in B. Cell viability was determined by MTT assay 24 h after doxorubicin treatment.
We sought to set up an experimental model where a poorly performing chemotherapeutic regimen for melanoma is converted to a highly effective one by a chemosensitizing agent. Doxorubicin seemed suitable for this purpose; melanoma cells are highly resistant to doxorubicin in vitro, (22) and treatment of melanoma with doxorubicin has been largely ineffective in clinical trials (23, 24). We used BMS-345541 (BMS), a small-molecule inhibitor of IKK, to determine the consequence of NF-κB blockade in melanoma cell response to doxorubicin. BMS efficiently blocked doxorubicin-induced NF-κB activation in Hs944T and other NF-κB-competent cells (Fig. 1B and Supplementary Fig. S1C) and sensitized them to killing by doxorubicin (Fig. 1C; and Supplementary Fig. S1D and S1E). BMS treatment alone was cytotoxic to Hs944T cells, albeit to a lesser degree than in conjunction with doxorubicin (Supplementary Fig. S1D and S1E). These results show that NF-κB-dependent resistance to apoptosis is frequently co-opted by melanoma and can be effectively reduced by IKK inhibitor treatment. With Roth cells, wherein neither constitutive nor doxorubicin-induced NF-κB activation was detected, BMS did not exhibit similar cytotoxic or sensitizing effects (Fig. 1C). Therefore, the IKK inhibitor does not seem to act on other, NF-κB-independent mechanisms of apoptotic resistance.
Effects of IKK inhibitor and doxorubicin treatment on melanoma xenografts in mice
Drug sensitivity of cancer cells in vitro does not always mirror tumor response to treatment in vivo. To investigate the response of tumors derived from Hs944T and Roth cells to BMS and doxorubicin in the animal host, we established subcutaneous melanoma xenografts in Rag2−/−γc−/− mice, a strain devoid of T cells, B cells, and NK cells shown to support engraftment of human melanoma cells (25). Under our treatment regimen, administration of doxorubicin alone did not induce remission in mice bearing Hs944T or Roth tumors or delay the growth of the xenografts. However, BMS alone was effective against Hs944T tumors in mice and dramatically sensitized Roth tumors to doxorubicin chemotherapy (Fig. 2A and 2B), suggesting that NF-κB blockade by BMS could subvert melanoma chemoresistance in vivo independently of whether the engrafted cells responded to the inhibitor in vitro. Almost complete tumor regression occurred in animals where BMS exhibited therapeutic efficacy, either alone or in conjunction with doxorubicin. However, combined treatment of tumor-bearing mice with BMS and doxorubicin caused severe side effects: mice carrying Hs944T and Roth tumors became lethargic, ataxic, and emaciated (Fig. 2C). These signs of illness were most visible and intense when tumor size was rapidly decreasing.
Figure 2. The efficacy and toxicity of doxorubicin and IKK inhibitor treatment in mice harboring Hs944T and Roth tumors.

Hs944T and Roth cells were infected with lentivirus expressing luciferase (Luc) and injected s.c. into RAG-2−/−γc−/− mice (n=3). After tumors grew to 100 mm3 in volume, mice were administered i.p. with doxorubicin (Dxr; 2 mg/kg) and BMS (125 mg/kg) as indicated on day 0, 2, 4, 6, and 8. A, Tumors were visualized in pseudocolor by bioluminescence imaging on day 10. Representative images from each group are shown along with the reference scale of bioluminescence depicted on the right. B and C, Tumor sizes (B) and the severity of illness (C) were determined on day 10. Triangles and circles indicate values in individual mice. Severity of illness was expressed in scores representing decrease in liveliness (DIL) and grade of emaciation (GOE). D, Tumor sections from the indicated groups were analyzed by TUNEL staining (upper) and immunostaining with Ly6G-specific antibody (lower). The number within each image indicates relative fluorescence intensity (TUNEL and Ly6G signal, respectively) and represents mean ± standard deviation from three independent areas. Apoptotic nuclei (green) and neutrophils (red) are shown together with the counter staining of DNA (blue). Scale bar, 100 μm.
We examined histological features of the tumors obtained immediately following treatment but before their complete destruction. Under the treatment conditions that induced tumor regression, areas of apoptotic nuclei emerged in Hs944T tumors and greatly expanded in Roth tumors (Fig. 2D, upper). The discrepancy between the in vitro (Fig. 1C) and in vivo effects (Fig. 2A, 2B, and 2D) of BMS on Roth cells suggests that the antiapoptotic function of NF-κB can be induced by chemotherapy independently of the tumor cell’s inherent ability to activate NF-κB in response to the cytotoxic agent.
Inflammatory cytokines could serve as NF-κB-activating stimuli for tumor cells under chemotherapy; tissue injury and ensuing inflammatory responses are inevitable in tumors exposed to a cytotoxic agent and likely create a cytokine-rich microenvironment at the tumor site. Both Hs944T and Roth cells were indeed able to activate NF-κB in response to interleukin (IL)-1β (Supplementary Fig. S2A), a cytokine whose production has been associated with chemotherapy-induced tumor cell death (5). We noted that inhibition of NF-κB signaling produced mixed effects on gene expression in melanoma cells. BMS suppressed basal and IL-1β-induced expression of CXCL8 (encoding the chemokine IL-8) but not CXCL5 (encoding another chemokine, ENA-78); rather, basal CXCL5 expression was markedly elevated by IKK inhibitor treatment (Supplementary Fig. S2B). Since both these genes encode neutrophil chemoattractants, we examined neutrophil recruitment in Hs944T and Roth tumors in vivo. Intriguingly, areas of Ly6G+ neutrophil infiltration were greatly increased in tumors from BMS-treated mice regardless of doxorubicin treatment (Fig. 2D, lower). Of note, BMS-induced neutrophil recruitment in Roth tumors was dissociated from tumor regression. Therefore, we postulated that intratumoral neutrophil recruitment was not mechanistically linked to the antitumor efficacy of BMS, but is indicative of inflammatory responses in tumor tissue that might contribute to or serve as a surrogate marker for BMS toxicity.
Experimental models simulating cell-selective inhibition of NF-κB activation
Our results from the melanoma xenograft experiments revealed mixed effects of NF-κB pathway inhibition on melanoma chemotherapy: sensitization to treatment at the cost of severe host toxicity. In all likelihood, the chemosensitizing effect was due to inhibition of tumor-intrinsic NF-κB activation. The origin of toxicity was not as apparent, but possibly hinted at by the observation that BMS treatment promoted a neutrophilic inflammatory milieu within the tumor. Related to this finding were previous reports showing that mice with myeloid cell-specific deficiency of IKKβ, a catalytic subunit of IKK, failed to limit inflammatory responses upon endotoxemia and bacterial infection and were prone to inflammation-mediated damage (26, 27). Of note, myeloid cells represent the major type of hematopoietic cells spared in the Rag2−/−γc−/− mice we used as hosts for our melanoma xenografts. Therefore, under the condition of doxorubicin-induced tumor injury and inflammation, BMS might have caused toxicity, at least in part, through interference with the anti-inflammatory mechanism mediated by myeloid NF-κB signaling.
We sought to verify the hypothesis that efficacy and toxicity of IKK inhibitor treatment arise from inhibition of NF-κB signaling in different target cells—tumor cells and host myeloid cells, respectively. To this end, we employed B16 murine melanoma cells and C57BL/6 hosts to construct syngeneic tumor models in which NF-κB activation was specifically blocked in only one of the two cell types. B16 cells expressed RelA and p50 (Supplementary Fig. S3), and are competent to induce their nuclear translocation in response to doxorubicin (Fig. 3A). To ablate NF-κB activation in B16 melanoma, we generated a B16 derivative stably expressing the IκBα “super-repressor,” a mutant IκBα refractory to IKK phosphorylation and degradation (Fig. 3B, upper). The resultant cell line, B16SR, exhibited impaired NF-κB activation following doxorubicin treatment (Fig. 3B, lower) and defects in doxorubicin-induced expression of NF-κB-responsive antiapoptotic genes (Fig. 3C; Supplementary Fig. S4A and S4B). Compared to control cells (B16GFP), B16SR cells were highly sensitive to killing by doxorubicin in vitro (Fig. 3D). Silencing of RelA expression in B16 cells with siRNA produced a similar chemosensitizing effect (Supplementary Fig. S5A and S5B). To selectively suppress myeloid NF-κB activation in host mice, we established B16 tumors in mice whose IKKβ gene was floxed and deleted by Cre recombinase expressed under the control of the lysozyme M promoter (IKKβΔM). Myeloid cells from these mutant mice have been shown to have defects in NF-κB activation and NF-κB-dependent gene transcription (28, 29).
Figure 3. Doxorubicin-induced NF-κB activation and NF-κB-mediated chemoresistance in B16 mouse melanoma cells.
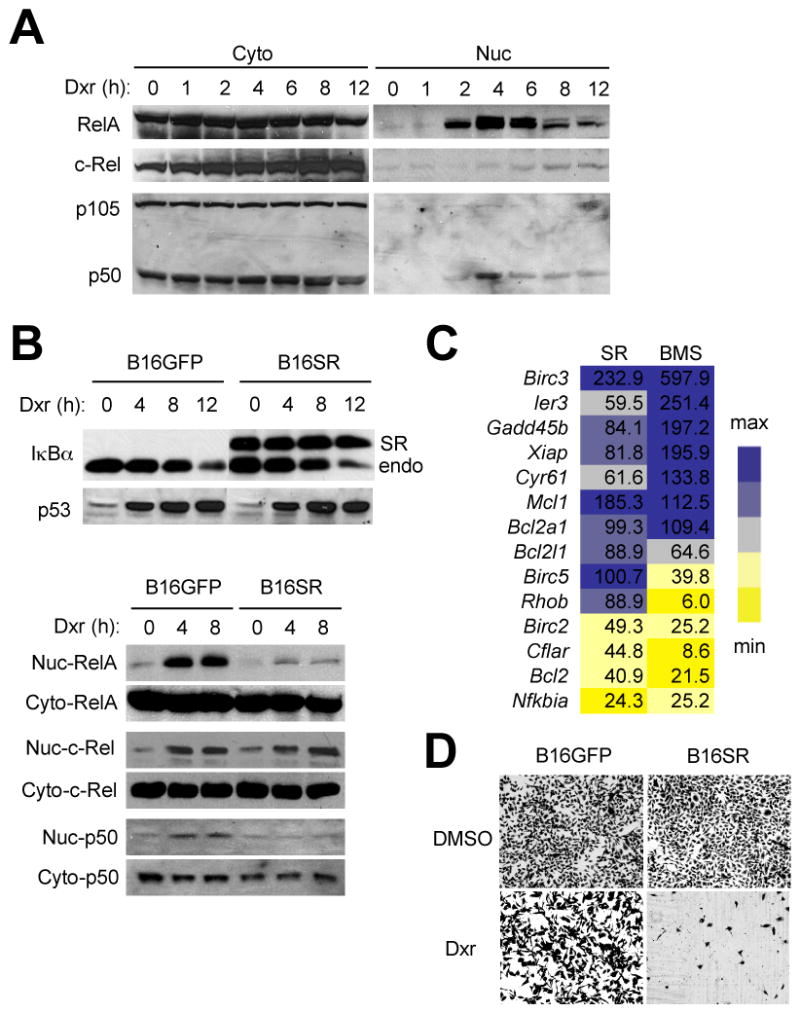
A, Cytoplasmic (Cyto) and nuclear (Nuc) extracts from B16 cells treated with doxorubicin (Dxr; 2 μg/ml) were analyzed by immunoblotting with antibodies against the proteins indicated on the left. B, B16 cells stably expressing GFP (B16GFP) and an IκBα “super-repressor” (B16SR) were treated with doxorubicin (2 μg/ml). Whole cell lysates (upper), and Cyto and Nuc extracts (lower) were prepared and analyzed as in A. Anti-IκBα antibody detects both transfected (SR) and endogenous IκBα (endo). C, B16SR and BMS-preincubated B16 cells (SR and BMS, respectively) were treated with doxorubicin (2 μg/ml) for 4 h. Total RNA was isolated and analyzed by quantitative real-time polymerase chain reaction using primers specific to the genes indicated on the left. Values represent percent mRNA relative to that in B16GFP (SR) and DMSO-preincubated B16 (BMS) cells. Expression is relative to that of Ppia (encoding cyclophilin). D, B16GFP and B16SR cells were treated with DMSO and doxorubicin (0.5 μg/ml) as indicated. Shown are viable adherent cells stained with crystal violet 24 h after treatment.
Chemosensitization via inhibition of tumor-intrinsic NF-κB signaling
The growth of B16GFP and B16SR tumors in C57BL/6 mice was comparable. However, administration of doxorubicin resulted in rapid regression of B16SR tumors whereas B16GFP tumor growth was largely refractory to the treatment (Fig. 4A). In neither group did the host animals exhibit signs of illness similar to those of BMS-treated animals. Histologically, B16SR tumor sections from doxorubicin-treated mice exhibited karyopyknosis (Fig. 4B) and widespread areas of apoptotic nuclei (Fig. 4C, upper). Apoptotic cells signal to promote an anti-inflammatory milieu in the surrounding tissue and recruit phagocytes for their engulfment and clearance. Accordingly, the apoptotic B16SR tissues were infiltrated with F4/80+ macrophages but not Ly6G+ neutrophils (Fig. 4C, middle and lower). None of these histological features were detected in B16GFP tumors under doxorubicin treatment. The findings from the analysis of B16SR tumors indicate that melanoma chemoresistance is mainly attributable to tumor-intrinsic NF-κB signaling, and its targeted inhibition may substantially increase therapeutic efficacy in melanoma treatment.
Figure 4. Increased sensitivity to doxorubicin treatment and apoptotic tumor regression resulting from ablation of tumor-intrinsic NF-κB activity.
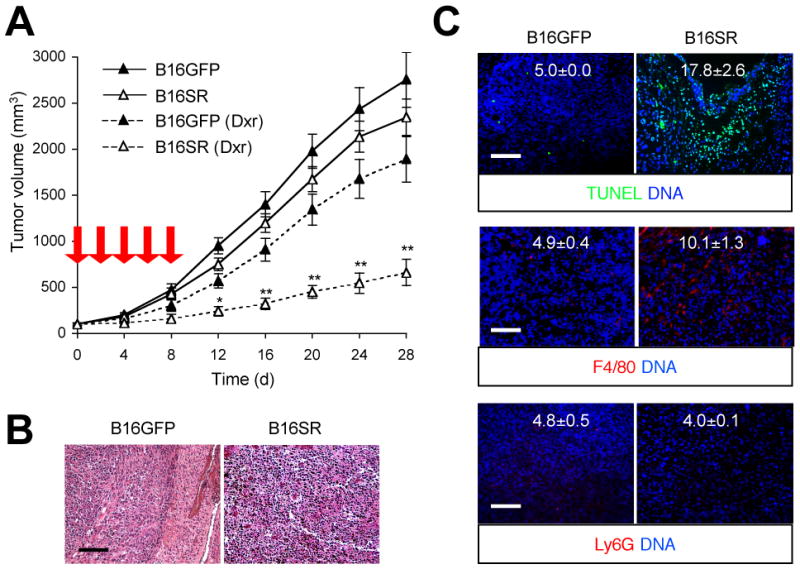
B16GFP and B16SR cells were injected s.c. into C57BL/6 mice (n=8). After tumors grew to 100 mm3 in volume, mice were treated with DMSO (solid line) and doxorubicin (Dxr; dotted line) as in Figure 2. A, Tumor sizes were determined at the indicated time points. Red arrow, time of doxorubicin treatment. **, P < 0.01; *, P < 0.05. B and C, B16GFP and B16SR tumor sections were prepared on day 28 and analyzed by H&E staining (B), TUNEL staining (C, upper), and immunostaining with F4/80- and Ly6G-specific antibodies (C, middle and lower, respectively). The number within each image indicates relative fluorescence intensity (TUNEL, F4/80, and Ly6G signal, respectively) and represents mean ± standard deviation from three independent areas (C). Apoptotic nuclei (green), macrophages (red), and neutrophils (red) are shown together with the counter staining of DNA (blue). Scale bar, 100 μm.
Host toxicity due to inhibition of NF-κB signaling in myeloid cells
We next examined how myeloid cell-specific deficiency of NF-κB signaling affects the response of B16 tumors and the tumor-bearing animals to chemotherapy. Growth of B16 tumors in both WT and IKKβΔM hosts was unabated during and after doxorubicin treatment (Fig. 5A, left), indicating that myeloid NF-κB signaling did not exert a critical influence on the treatment resistance of B16 tumors. Meanwhile, IKKβΔM host animals developed severe illness and eventually succumbed, resulting in a drastically increased mortality (Fig. 5A, middle). This increase in lethality was accompanied by incidents of local tumor necrosis (Fig. 5A, right), a condition reminiscent of tumor lysis syndrome (30). Even before gross necrotic lesions occurred, tumor sections obtained from doxorubicin-treated IKKβΔM mice displayed hyperpigmented regions of micronecrosis and karyolysis, but few apoptotic areas (Fig. 5B; and Fig. 5C, upper). In marked contrast to apoptotic B16SR tumor tissues, these necrotic tumor areas were heavily infiltrated with Ly6G+ neutrophils but not F4/80+ macrophages (Fig. 5C, middle and lower; and Fig. 5D). Therefore, perturbed NF-κB signaling in myeloid cells during cytotoxic cancer therapy may engender an intratumoral tissue environment prone to necrotic injury and neutrophilic inflammatory responses and ultimately result in adverse, potentially lethal toxic effects in the host.
Figure 5. Increased host mortality with necrotic tumor lesions and intratumoral neutrophil infiltration in doxorubicin-treated mice defective in myeloid NF-κB signaling.

B16 cells were injected s.c. into WT and IKKβΔM mice (n=34 and 18, respectively). After tumors grew to 100 mm3 in volume, mice were treated with doxorubicin as in Figure 2. A, Tumor sizes (left) and host survival (middle) were determined at the indicated time points. Red arrow, time of doxorubicin treatment. **, P < 0.01. The incidence of necrotic tumor lesions in each group was determined and expressed as percent animals with varying scores (right). Necrotic lesions were scored according to the degree of maximum necrosis throughout the treatment period in each host animal. B and C, Tumor sections were prepared on day 20 and analyzed as in Figure 4 by H&E staining (B), TUNEL staining (C, upper), and immunostaining with F4/80- and Ly6G-specific antibodies (C, middle and lower, respectively). The number within each image indicates relative fluorescence intensity (TUNEL, F4/80, and Ly6G signal, respectively) and represents mean ± standard deviation from three independent areas (C). Scale bar, 100μm. D, Percentage of CD11b+Ly6G+ cells in tumors of the indicated host mice was determined by flow cytometry.
IKKβ and NF-κB exert a negative control on the processing of pro-IL-1β into its mature functional form. Myeloid cells lacking IKKβ, hence deprived of this control, show excessive production of bioactive IL-1β upon lipopolysaccharide challenge (26). Similar disregulation might have occurred in IKKβΔM mice under doxorubicin treatment. B16 tumor sections from IKKβΔM hosts indeed included intratumoral areas intensely stained for IL-1β (Fig. 6A). IL-1β was almost always detected at or near the necrotic tumor lesions. Immunoblot analysis showed that both unprocessed and mature IL-1β were present in the tumors (Fig. 6B). IL-1β was not detected in tumors from wild-type hosts by either immunostaining or immunoblotting. We identified Ly6G+ neutrophils as the principal source of intratumoral IL-1β expression in IKKβΔM mice (Fig. 6C and 6D). Consistent with an earlier report (26), IKKβ-deficient neutrophils exhibited enhanced production of fully processed IL-1β upon inflammatory challenge in vitro (Fig. 6E). Therefore, tumor necrosis in doxorubicin-treated IKKβΔM mice is not only associated with a recruitment of IL-1β-producing neutrophils, but likely also with a higher rate of IL-1β production by the recruited neutrophils. We found that B16 melanoma cells were directly responsive to IL-1β in vitro as were the human melanoma cells (Supplementary Fig. S2A and S2B), inducing NF-κB activity and the expression of neutrophil-specific chemokine genes (Supplementary Fig. S6A and S6B). Thus, it seems that IL-1β production by intratumoral neutrophils establishes a self-sustaining or even self-amplifying state by maintaining neutrophil influx via tumor-derived chemoattractants.
Figure 6. Inflammatory responses and necrotic tumor lesions driven by neutrophil-derived IL-1β.

A–D, Tumors were prepared and analyzed as in Figure 5 by immunostaining (A, C), immunoblotting (B), and flow cytometry (D) with antibodies against IL-1β, F4/80, and Ly6G. E, Whole cell lysates from WT and IKKβΔM neutrophils treated with lipopolysaccharide (LPS; 100 ng/ml) were analyzed by immunoblotting with IL-1β- and actin-specific antibodies. F–H, IKKβΔM mice were administered with doxorubicin (2 mg/kg) alone on day 0, 2, 4, 6, and 8 or in conjunction with anakinra (25 mg/kg) on day 3, 5, 7, 9, 11, and 13. Tumor sections were analyzed as in Figure 5 by H&E staining (F), immunostaining with Ly6G-specific antibodies (G, upper), and TUNEL staining (G, lower). The number within each image indicates relative fluorescence intensity (Ly6G and TUNEL signal, respectively) and represents mean ± standard deviation from three independent areas (G). Scale bar, 100 μm. Serum amounts of IL-1β in the indicated mice were determined on day 14 by ELISA (H).
We tested whether IL-1β production by intratumoral neutrophils is essential for the formation of necrotic lesions and inflammatory infiltration of the tumors. Administration of anakinra, an IL-1 antagonist, in doxorubicin-treated IKKβΔM hosts substantially prevented the occurrence of hyperpigmented necrotic lesions and neutrophil infiltration in the tumors (Fig. 6F; and Fig. 6G, upper). Anakinra exerted similar effects on tumors in BMS-treated wild-type mice (Supplementary Fig. S7). Under anakinra treatment, tumor cell apoptosis was preserved or moderately enhanced in IKKβΔM mice (Fig. 6G, lower). Following doxorubicin treatment, IKKβΔM host animals had higher amounts of circulating IL-1β compared to wild-type animals. The serum concentration of IL-1β was greatly reduced in both wild-type and IKKβΔM mice by anakinra treatment (Fig. 6H). Therefore, IL-1β overproduction and resulting inflammation likely caused both local (intratumoral) and systemic responses in IKKβΔM mice during melanoma chemotherapy. Some reversal of the inflammatory events notwithstanding, anakinra-administered IKKβΔM animals suffered even higher mortality under doxorubicin treatment than the anakinra-untreated group. This suggests that IL-1 signaling, whereas potentially contributing to treatment-associated toxicity via inflammation-mediated mechanisms, may play some indispensable protective role in IKKβΔM mice during cytotoxic cancer therapy.
Discussion
Tumor cells exposed to cytotoxic agents incur injury and invoke damage-induced signaling cascades. Our findings illustrate that inhibition of NF-κB signaling in doxorubicin-treated individuals can lead damaged tumor cells to two alternative fates: apoptotic demise or proinflammatory necrosis (Fig. 7). The mode of tumor cell death and the pattern of intratumoral infiltration in doxorubicin-treated mice differed markedly depending on the cell type in which NF-κB signaling is targeted: apoptosis and macrophage recruitment (tumor cell-restricted inhibition) versus necrosis and neutrophil recruitment (myeloid cell-restricted inhibition). These histological parameters were associated with, and therefore predictive of, the rate of tumor regression and the severity of host toxicity. Necrotic lesions with massive neutrophilic infiltration characterized the histology of tumors in doxorubicin-treated IKKβΔM mice. Given the observed effect of anakinra, IL-1β appears to be instrumental for intratumoral neutrophil recruitment and other pathological features in this setting. IL-1β was found to induce the expression of neutrophil-recruiting chemokines in the melanoma cell lines tested even in the presence of the IKK inhibitor. Importantly, the neutrophils themselves turned out to be the major source of IL-1β in the necrotic tumor areas. IL-1β production by newly recruited neutrophils presumably requires activation by biochemical stimuli released from doxorubicin-injured tumor cells, as unstimulated IKKβ-deficient neutrophils do not produce significant amounts of IL-1β in vitro. A role for such “danger” signals in neutrophil activation under inflammatory conditions was recently demonstrated (31, 32). Taken together, we postulate that neutrophil-derived IL-1β and tumor-derived chemokines constitute a self-sustaining cycle of local signaling that maintains or amplifies intratumoral necrosis and inflammatory responses.
Figure 7. Clinical outcomes resulting from inhibition of tumor-intrinsic and myeloid NF-κB activation in melanoma chemotherapy.
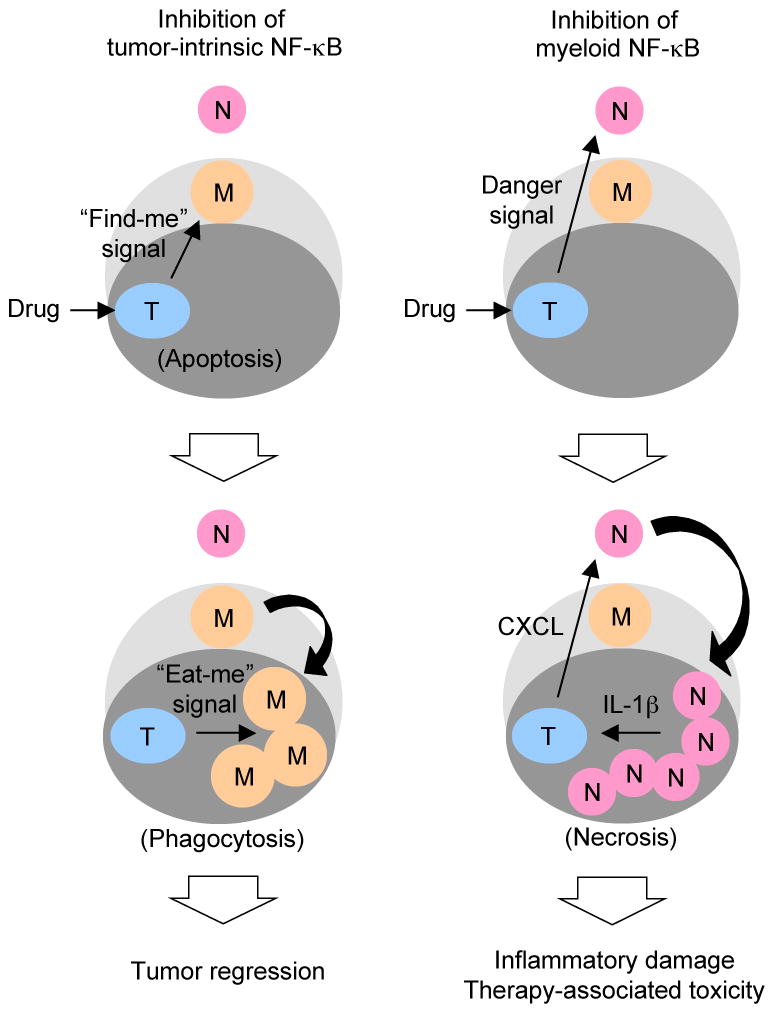
Dark and light gray areas denote tumor and tumor stromal tissues, respectively. The outer unshaded area indicates the circulation. Tumor stromal macrophages (M) and circulating neutrophils (N) are recruited to tumor tissues (T) depending on the cell type in which NF-κB activation is inhibited and the mode of tumor cell death.
Different enzymatic mechanisms of pro-IL-1β cleavage appear to operate in distinct subsets of myeloid cells: caspase-1-dependent and -independent (33). Therefore, there is a possibility that pro-IL-1β processing in neutrophils can be specifically inhibited, thus preventing its adverse clinical effects, without interfering with any protective function that IL-1β from other subtypes of myeloid cells may serve. Notably, it was demonstrated that dendritic cells link chemotherapy-induced tumor cell death to tumor-specific CD8+ T cell-mediated immune responses through IL-1β production (5). Tumor-derived chemokines responsible for intratumoral neutrophil recruitment may be considered as another group of therapeutic targets for the prevention of neutrophil-mediated treatment toxicity.
The pro-tumor function of NF-κB is not merely confined to promoting chemoresistance and other mechanisms of tumor persistence. NF-κB activity in premalignant and malignant cells is critically required for the development and growth of many different forms of cancer (34–37). Some melanoma cells tested in this and other studies, as well as many other tumor cells of different origin, are “addicted” to NF-κB signaling to such an extent that treatment with an IKK inhibitor or other means of NF-κB blockade was sufficient to induce their apoptosis and regression (18, 38–40). While these findings offer new opportunities in cancer treatment, it is becoming increasingly clear that the IKK-NF-κB signaling axis also serves important physiological functions whose loss could result in undesired pathological conditions. For example, systemic ablation of IKKβ expression in mice perturbs myeloid homeostasis: increased proliferation of IKKβ-deficient myeloid progenitors leads to neutrophilia and neutrophil-mediated tissue inflammation (41, 42). Importantly, we demonstrate these adverse events can be suppressed by eliminating IL-1 receptor 1-mediated signaling in the IKKβ mutant mice. Therefore, long-term treatment with an NF-κB-blocking agent either alone or in conjunction with other cytotoxic agents may give rise to clinical adversities resulting from excessive production and activity of IL-1β. Based on the target cell-specific effects of NF-κB pathway inhibition identified in the current study, we thus suggest that antitumor therapeutic modalities that are built upon their ability to block NF-κB activation be specifically delivered to or activated in tumor cells. Such selectivity will prevent or alleviate adverse clinical events that result from disregulation of NF-κB signaling in host myeloid cells.
Materials and Methods
Melanoma and primary cells
A375, MM455, MM608, Hs944T, and Roth cells are independently established cell lines from human melanoma obtained from the sources described previously (43, 44). These cell lines were authenticated by DNA fingerprinting based on polymerase chain reaction amplification and DNA sequencing of specific loci (44). B16 melanoma cells are of C57BL/6 mouse origin and were obtained from the American Type Culture Collection; no validation was performed. Neutrophils were isolated from mouse bone marrow as described (45).
Animals
Rag2−/−γc−/− and C57BL/6 mice were obtained from Taconic and The Jackson Laboratory, respectively. IKKβΔM (IKKβfl/fl-LysMCre) mice were described previously (28). All animal studies were conducted under IACUC-approved protocols.
Preparation of BMS
BMS, N1-(1,8-Dimethylimidazo[1,2-a]quinolin-4-yl)-ethane-1,2-diamine, was prepared according to the procedure described in the PCT patent application (WO 02/060386 A2) beginning with commercially available starting materials, 3-fluoro-4-nitrotoluene and ethyl 4-methyl-5-imidazolecarboxylate. The purity of the BMS preparation used was greater than 96%, as assessed by high-performance liquid chromatography.
Human and mouse melanoma models and treatment
Hs944T and Roth cells were transduced with a lentivirus expressing luciferase under the control of the cytomegalovirus promoter according to established methods (46). 5 × 107 transduced cells were injected s.c. into the right hind legs of 8-week-old Rag2−/−γc−/− mice. B16 and its derivative cells were similarly injected into 8-week-old C57BL/6 mice. After tumors grew to 100 mm3 in volume, mice were administered i.p. doxorubicin (2 mg/kg; Sigma) alone or together with BMS (125 mg/kg) on day 0, 2, 4, 6, and 8. Complete tumor regression occurred with 125 mg/kg of BMS, either as such or in conjunction with doxorubicin; lower BMS doses tested were far less effective and produced inconsistent results.
Monitoring and measurement of tumor growth and host toxicity
Luciferase-expressing tumors were visualized by bioluminescence-based imaging (IVIS 200; Caliper) on day 10. Tumor size was manually measured with a caliper. Toxicity of treatment was monitored, and expressed in scores ranging from 0 to 3 representing decrease in liveliness (0, normal; 1, mildly lethargic; 2, lethargic and/or ataxic; 3, severely lethargic and/or ataxic) and grade of emaciation, which was based on the extent of body weight loss (0, normal; 1, less than 10%; 2, equal to or greater than 10% and less than 20%; 3, equal to or greater than 20%). These criteria were adapted from a previously described guideline (47).
Histology and immunofluorescence
Tumor tissue sections were prepared and analyzed as described (48). Antibodies against the following markers were used: F4/80 (MCA497B; Serotec), Ly6G/Gr-1 (553125; BD Biosciences) and IL-1β (AF-401-NA; R&D Systems). TUNEL staining was performed using the In Situ Cell Death Detection kit (Roche). Relative fluorescence intensity was determined using the ImageJ software (National Institutes of Health).
Protein and RNA analysis
Whole cell lysates were prepared and analyzed as described (48). To prepare cytoplasmic and nuclear extracts, 4 × 106 cells were resuspended in 0.5 ml of buffer L1 (50 mM Tris-chloride, pH 8.0, 2 mM ethylenediaminetetraacetic acid, 0.1% Nonidet P-40, 10% glycerol, 25 mM β-glycerophosphate, and protease inhibitors), incubated 5 min at 4°C, and centrifuged 5 min at 4,500× g. Cytoplasmic supernatants were stored, and pelleted nuclei were rinsed by resuspending and centrifuging in 0.5 ml of buffer L1, and extracted further in buffer L2 (20 mM 4-[2-hydroxyethyl]-1-piperazineethanesulfonic acid-potassium, pH 7.6, 150 mM sodium chloride, 2 mM ethylenediaminetetraacetic acid, 1% Triton X-100, 0.1% sodium dodecyl sulfate, 10% glycerol, 25 mM β-glycerophosphate, and protease inhibitors). Lysed nuclei were centrifuged at 16,000× g, and the supernatant was collected for use as the nuclear fraction. The purity of cytoplasmic and nuclear extracts was verified by immunoblotting with antibodies against various cytoplasmic and nuclear marker proteins (Supplementary Fig. S8). Antibodies against the following proteins were used in immunoblot analysis: IκBα (sc-371), RelA (sc-372), RelB (sc-226), c-Rel (sc-71), p105/p50 (sc-1190), p100/p52 (sc-298) and p53 (sc-6243; all from Santa Cruz Biotechnology); IL-1β (AF-401-NA; R&D Systems); and actin (A4700; Sigma). Total RNA was isolated using Trizol (Invitrogen), and analyzed using quantitative real-time polymerase chain reaction as described (29). Serum IL-1β was assayed by ELISA (R&D Systems).
Flow cytometry
Myeloid marker and IL-1β expression in tumor-associated cells were analyzed by flow cytometry using FACSCanto (BD) and the FlowJo software (Tristar). Antibodies against the following markers were used: CD11b (12-0112; eBioscience), IL-1β (17-7114; eBioscience) and Ly6G (clone 1A8; 551460; BD Biosciences).
Statistical analysis
Data values are expressed as mean ± SEM. P values were obtained with the unpaired, two-tailed Student’s t-test, and the Log-rank test. P < 0.05 was considered significant.
Supplementary Material
Significance.
Our findings show that the IκB kinase-NF-κB signaling pathway is important for both promoting treatment resistance and preventing host toxicity in cancer chemotherapy; however, the two functions are exerted by distinct cell type-specific mechanisms, and can therefore be selectively targeted to achieve an improved therapeutic outcome.
Acknowledgments
Grant support: This work was supported by the Cutaneous Biology Research Center-Wellman Center for Photomedicine Collaborative Project (J.M. Park).
We thank D. Fisher and T. Lawrence for discussion.
Abbreviations
- BMS
BMS-345541
- IκB
inhibitor of nuclear factor κB
- IKK
inhibitor of nuclear factor κB kinase
- NF-κB
nuclear factor κB
References
- 1.Savage P, Stebbing J, Bower M, Crook T. Why does cytotoxic chemotherapy cure only some cancers? Nat Clin Pract Oncol. 2009;6:43–52. doi: 10.1038/ncponc1260. [DOI] [PubMed] [Google Scholar]
- 2.Cotter TG. Apoptosis and cancer: the genesis of a research field. Nat Rev Cancer. 2009;9:501–7. doi: 10.1038/nrc2663. [DOI] [PubMed] [Google Scholar]
- 3.Paulus P, Stanley ER, Schäfer R, Abraham D, Aharinejad S. Colony-stimulating factor-1 antibody reverses chemoresistance in human MCF-7 breast cancer xenografts. Cancer Res. 2006;66:4349–56. doi: 10.1158/0008-5472.CAN-05-3523. [DOI] [PubMed] [Google Scholar]
- 4.DeNardo DG, Brennan DJ, Rexhapaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discovery. 2011;1:54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1β-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–8. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 6.Ma Y, Aymeric L, Locher C, Mattarollo SR, Delahaye NF, Pereira P, et al. Contribution of IL-17-producing γδ T cells to the efficacy of anticancer chemotherapy. J Exp Med. 2011;208:491–503. doi: 10.1084/jem.20100269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–40. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–58. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Takahashi H, Lin WW, Descargues P, Grivennikov S, Kim Y, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–6. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soengas MS, Lowe SW. Apoptosis and melanoma chemoresistance. Oncogene. 2003;22:3138–51. doi: 10.1038/sj.onc.1206454. [DOI] [PubMed] [Google Scholar]
- 11.Laskin DL, Sunil VR, Gardner CR, Laskin JD. Macrophages and tissue injury: agents of defense or destruction? Annu Rev Pharmacol Toxicol. 2011;51:267–88. doi: 10.1146/annurev.pharmtox.010909.105812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 13.Huang TT, Wuerzberger-Davis SM, Wu ZH, Miyamoto S. Sequential modification of NEMO/IKKγ by SUMO-1 and ubiquitin mediates NF-κB activation by genotoxic stress. Cell. 2003;115:565–76. doi: 10.1016/s0092-8674(03)00895-x. [DOI] [PubMed] [Google Scholar]
- 14.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-κB signaling in response to genotoxic stimuli. Science. 2006;311:1141–6. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- 15.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–3. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 16.Wang CY, Guttridge DC, Mayo MW, Baldwin AS., Jr NF-κB induces expression of the Bcl-2 homologue A1/Bfl-1 to preferentially suppress chemotherapy-induced apoptosis. Mol Cell Biol. 1999;19:5923–9. doi: 10.1128/mcb.19.9.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I. p53 stabilization is decreased upon NF-κB activation: a role for NF-κB in acquisition of resistance to chemotherapy. Cancer Cell. 2002;1:493–503. doi: 10.1016/s1535-6108(02)00068-5. [DOI] [PubMed] [Google Scholar]
- 18.Hideshima T, Neri P, Tassone P, Yasui H, Ishitsuka K, Raje N, et al. MLN120B, a novel IκB kinase β inhibitor, blocks multiple myeloma cell growth in vitro and in vivo. Clin Cancer Res. 2006;12:5887–94. doi: 10.1158/1078-0432.CCR-05-2501. [DOI] [PubMed] [Google Scholar]
- 19.Tapia MA, González-Navarrete I, Dalmases A, Bosch M, Rodriguez-Fanjul V, Rolfe M, et al. Inhibition of the canonical IKK/NF-κB pathway sensitizes human cancer cells to doxorubicin. Cell Cycle. 2007;6:2284–92. doi: 10.4161/cc.6.18.4721. [DOI] [PubMed] [Google Scholar]
- 20.Schön M, Wienrich BG, Kneitz S, Sennefelder H, Amschler K, Vöhringer V, et al. KINK-1, a novel small-molecule inhibitor of IKKβ, and the susceptibility of melanoma cells to antitumoral treatment. J Natl Cancer Inst. 2008;100:862–75. doi: 10.1093/jnci/djn174. [DOI] [PubMed] [Google Scholar]
- 21.Baud V, Karin M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panneerselvam M, Bredehorst R, Vogel CW. Resistance of human melanoma cells against the cytotoxic and complement-enhancing activities of doxorubicin. Cancer Res. 1987;47:4601–7. [PubMed] [Google Scholar]
- 23.Vorobiof DA, Rapoport BL, Mahomed R, Karime M. Phase II study of pegylated liposomal doxorubicin in patients with metastatic malignant melanoma failing standard chemotherapy treatment. Melanoma Res. 2003;13:201–3. doi: 10.1097/00008390-200304000-00015. [DOI] [PubMed] [Google Scholar]
- 24.Smylie MG, Wong R, Mihalcioiu C, Lee C, Pouliot JF. A phase II, open label, monotherapy study of liposomal doxorubicin in patients with metastatic malignant melanoma. Invest New Drugs. 2007;25:155–9. doi: 10.1007/s10637-006-9002-y. [DOI] [PubMed] [Google Scholar]
- 25.Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466:133–7. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, et al. NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell. 2007;130:918–31. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fong CH, Bebien M, Didierlaurent A, Nebauer R, Hussell T, Broide D, et al. An antiinflammatory role for IKKβ through the inhibition of “classical” macrophage activation. J Exp Med. 2008;205:1269–76. doi: 10.1084/jem.20080124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park JM, Greten FR, Li ZW, Karin M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science. 2002;297:2048–51. doi: 10.1126/science.1073163. [DOI] [PubMed] [Google Scholar]
- 29.Park JM, Greten FR, Wong A, Westrick RJ, Arthur JS, Otsu K, et al. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis--CREB and NF-κB as key regulators. Immunity. 2005;23:319–29. doi: 10.1016/j.immuni.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 30.Howard SC, Jones DP, Pui CH. The tumor lysis syndrome. N Engl J Med. 2011;364:1844–54. doi: 10.1056/NEJMra0904569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonald B, Pittman K, Menezes GB, Hirota SA, Slaba I, Waterhouse CC, et al. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science. 2010;330:362–6. doi: 10.1126/science.1195491. [DOI] [PubMed] [Google Scholar]
- 33.Netea MG, Simon A, van de Veerdonk F, Kullberg BJ, Van der Meer JW, Joosten LA. IL-1β processing in host defense: beyond the inflammasomes. PLoS Pathog. 2010;6:e1000661. doi: 10.1371/journal.ppat.1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118:285–96. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 35.Meylan E, Dooley AL, Feldser DM, Shen L, Turk E, Ouyang C, et al. Requirement for NF-κB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462:104–7. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKβ- and JNK1-dependent inflammation. Cancer Cell. 2010;17:89–97. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang J, Splittgerber R, Yull FE, Kantrow S, Ayers GD, Karin M, et al. Conditional ablation of Ikkb inhibits melanoma tumor development in mice. J Clin Invest. 2010;120:2563–74. doi: 10.1172/JCI42358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang J, Amiri KI, Burke JR, Schmid JA, Richmond A. BMS-345541 targets inhibitor of κB kinase and induces apoptosis in melanoma: involvement of nuclear factor κB and mitochondria pathways. Clin Cancer Res. 2006;12:950–60. doi: 10.1158/1078-0432.CCR-05-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature. 2009;459:717–21. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dougan M, Li D, Neuberg D, Mihm M, Googe P, Wong KK, et al. A dual role for the immune response in a mouse model of inflammation-associated lung cancer. J Clin Invest. 2011;121:2436–46. doi: 10.1172/JCI44796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu LC, Enzler T, Seita J, Timmer AM, Lee CY, Lai TY, et al. IL-1β-driven neutrophilia preserves antibacterial defense in the absence of the kinase IKKβ. Nat Immunol. 2011;12:144–50. doi: 10.1038/ni.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mankan AK, Canli O, Schwitalla S, Ziegler P, Tschopp J, Korn T, et al. TNF-α-dependent loss of IKKβ-deficient myeloid progenitors triggers a cytokine loop culminating in granulocytosis. Proc Natl Acad Sci U S A. 2011;108:6567–72. doi: 10.1073/pnas.1018331108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsao H, Goel V, Wu H, Yang G, Haluska FG. Genetic interaction between NRAS and BRAF mutations and PTEN/MMAC1 inactivation in melanoma. J Invest Dermatol. 2004;122:337–41. doi: 10.1046/j.0022-202X.2004.22243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang G, Rajadurai A, Tsao H. Recurrent patterns of dual RB and p53 pathway inactivation in melanoma. J Invest Dermatol. 2005;125:1242–51. doi: 10.1111/j.0022-202X.2005.23931.x. [DOI] [PubMed] [Google Scholar]
- 45.Lowell CA, Fumagalli L, Berton G. Deficiency of Src family kinases p59/61hck and p58c-fgr results in defective adhesion-dependent neutrophil functions. J Cell Biol. 1996;133:895–910. doi: 10.1083/jcb.133.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abrahamsson AE, Geron I, Gotlib J, Dao KH, Barroga CF, Newton IG, et al. Glycogen synthase kinase 3β missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci U S A. 2009;106:3925–9. doi: 10.1073/pnas.0900189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Montgomery CA., Jr Oncological and toxicological research: Alleviation and control of pain and distress in laboratory animals. Cancer Bulletin. 1990;42:230–7. [Google Scholar]
- 48.Kim C, Sano Y, Todorova K, Carlson BA, Arpa L, Celada A, et al. The kinase p38α serves cell type-specific inflammatory functions in skin injury and coordinates pro- and anti-inflammatory gene expression. Nat Immunol. 2008;9:1019–27. doi: 10.1038/ni.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.