

Chemokine production, the chemokine monomer-dimer equilibrium, and differences in GAG-interactions collectively influence in vivo tissue-specific patterns of chemokine gradient formation and neutrophil recruitment.
Keywords: gradient, microenvironment, leukocyte trafficking
Abstract
Chemokines exert their function by binding the GPCR class of receptors on leukocytes and cell surface GAGs in target tissues. Most chemokines reversibly exist as monomers and dimers, but very little is known regarding the molecular mechanisms by which the monomer-dimer equilibrium modulates in vivo function. For the chemokine CXCL8, we recently showed in a mouse lung model that monomers and dimers are active and that the monomer-dimer equilibrium of the WT plays a crucial role in regulating neutrophil recruitment. In this study, we show that monomers and dimers are also active in the mouse peritoneum but that the role of monomer-dimer equilibrium is distinctly different between these tissues and that mutations in GAG-binding residues render CXCL8 less active in the peritoneum but more active in the lung. We propose that tissue-specific differences in chemokine gradient formation, resulting from tissue-specific differences in GAG interactions, are responsible for the observed differences in neutrophil recruitment. Our observation of differential roles played by the CXCL8 monomer-dimer equilibrium and GAG interactions in different tissues is novel and reveals an additional level of complexity of how chemokine dimerization regulates in vivo recruitment.
Introduction
Recruitment of circulating neutrophils from the bloodstream to the site of injury or infection forms the first line and a critical component of host defense [1]. Chemokine CXCL8 (also known as IL-8) plays an important role in this process [2]. Chemokines produced as a consequence of infection/injury in the tissue migrate to the vasculature, where they bind cell surface GAGs, which function as directional cues for neutrophil homing [3]. Precise spatial and temporal control of this process is essential to mount an effective defense, while at the same time, preventing a runaway inflammatory response that could result in destruction of healthy tissue and in severe cases, death [4]. Currently, very little is known regarding the structural basis and the molecular mechanisms by which CXCL8 regulates neutrophil recruitment, and such knowledge is essential to understand how the complex in vivo milieu influences neutrophil recruitment in health and disease.
CXCL8 reversibly exists as monomers and dimers, and so, its recruitment profile would be influenced not only by the monomer-dimer equilibrium constant but also by the binding interactions of monomer and dimer to receptors on neutrophils and to GAGs on cell surface and interstitial space in the target tissue. Animal, ex vivo, and cell-based studies have shown that CXCL8 and related chemokines bind to cell surface and soluble GAGs on endothelial cells and the ECM and that these interactions regulate recruitment [5–11]. The structure of the CXCL8 monomer is similar to that of the monomer in the dimer, except that the last six C-terminal residues are unstructured in the monomer and structured in the dimer [12, 13]. As these C-terminal residues are involved in GAG binding, differences in the monomer and dimer structures will influence their GAG-binding properties. GAG–chemokine interactions are complex, and multiple factors, such as the distribution of lysines and arginines on the chemokine, length and nature of GAG, and binding affinities and kinetic constants, have been shown to influence the type, duration, and strength of the chemokine gradient in the tissue, ECM, and vasculature [9–11, 14–16]. At this time, even whether the CXCL8 monomer or dimer is the high-affinity ligand for GAGs is unclear, as both have been reported to bind with higher affinity compared with the other [8, 17]. Our in vitro chemotaxis studies of different CXCL8 variants using a microfluidic device showed recruitment profiles that could not be simply correlated to the total amount of CXCL8 and were sensitive to the steepness of the gradient [18]. In a recent study, CXCL8 GAG mutants were actually more active in a mouse lung model, and the authors suggest that the kinetics of CXCL8–GAG interactions influences spatiotemporal gradient formation and neutrophil recruitment [11].
Studying the in vivo role of WT monomers and dimers in isolation is challenging, as the very process of monomer-dimer equilibrium will interfere in the process. We have tackled this bottleneck by designing a trapped monomer that cannot associate and a trapped dimer that cannot dissociate and have shown that their functions must reflect those of the native monomer and dimer [19–21]. Using these variants, we showed recently that CXCL8 monomers and dimers are active in a mouse lung model and that the ability of WT to reversibly exist as monomers and dimers regulates neutrophil recruitment [18]. This study was the very first to show that monomeric and dimeric forms of a chemokine are present in vivo, and each could be active in isolation.
To better understand in vivo molecular mechanisms and the role of the tissue architecture and microenvironment in modulating neutrophil recruitment, we have now expanded upon our previous studies and have characterized the recruitment profiles of the CXCL8 monomer, dimer, and WT in a mouse peritoneum model and of GAG mutants in the peritoneum and lung. We observe that the recruitment profiles are distinctly different between the peritoneum and the lung and most interestingly, that the GAG mutants are less active than WT in the peritoneum but more active in the lung. These data indicate that the role of the monomer-dimer equilibrium and of the GAG interactions of monomers and dimers is tissue-specific and that these tissue-specific differences lead to differences in chemokine gradient formation and neutrophil recruitment. These observations are quite novel and provide important insights into the molecular mechanisms underlying the complex relationship among chemokine synthesis, chemokine dimerization, and the tissue microenvironment. Further, our findings provide critical knowledge to better interpret the results of lung and peritoneum animal model studies of chemokine-mediated leukocyte trafficking in health and disease.
MATERIALS AND METHODS
Design and synthesis of human CXCL8 variants
The trapped CXCL8 monomer was designed by substituting Leu-25 at the dimer interface with the non-natural amino acid N-methyl Leu, which disrupts H-bonding interactions and further introduces steric bulk about the twofold symmetry point [12]. The trapped human CXCL8 dimer was designed by mutating Arg-26 at the dimer interface to Cys-26. This results in the formation of a disulfide bridge between two monomers in the dimer [21]. The trapped monomer was synthesized by solid-phase peptide synthesis, and the WT, trapped dimer, and inactive R6K mutant were expressed recombinantly.
Cloning, expression, and purification of WT CXCL8 GAG mutants
Recombinant WT GAG mutants—K20A, R60A, K64A, K67A, and R68A—were generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA), expressed and purified as discussed previously [22]. The purity and MW of the mutants were confirmed using analytical HPLC and MALDI-mass spectrometry.
Neutrophil recruitment in mice
Eight- to 10-week-old female BALB/c mice were purchased from Harlan Sprague-Dawley (Houston, TX, USA) and housed under pathogen-free conditions in the animal research facility, in accordance with NIH and university guidelines for animal care. Animal work was approved by The University of Texas Medical Branch's Institutional Animal Care and Use Committee (Galveston, TX, USA). Different groups of mice were inoculated i.p. with different doses (from 0.1 to 10 μg) of various CXCL8 variants, and mice inoculated with PBS served as control. At 6 h postinoculation, mice were sacrificed by cervical dislocation. The peritoneal cavity was flushed with 1.5 ml cold HBSS to collect infiltrated neutrophils. PF (100 μl) was cytospun onto glass microscope slides and stained with H&E (Hema 3 stain, Fisher Scientific, Waltham, MA, USA), and the percentage of neutrophils was determined. At least 200 cells were counted for each slide. Total leukocytes in PF were determined by staining with Turks blood diluting fluid and counting using a hemocytometer. Neutrophil recruitment in the lung was carried out as described previously [18].
Statistical analysis
Statistical analysis was carried by one-way and two-way ANOVA to determine statistical significance for comparing the recruitment of the CXCL8 monomer, dimer, and WT and recruitment of GAG mutants, respectively. Unless otherwise indicated, results are expressed as mean ± sem.
MPO assay
MPO is a neutrophil granule enzyme, and its activity has been shown to correlate with neutrophil levels. Briefly, cells from PF were resuspended in 50 mM phosphate buffer, pH 6.0, containing 0.5% hexadecyltrimethylammonium bromide. After sonication and centrifugation, MPO activity in the supernatants was measured using o-dianisidine (Sigma-Aldrich, St. Louis, MO, USA) and H2O2 as substrates. The change in absorbance at 460 nm was measured at 1-min intervals for 7–8 min. The data show that measured MPO activity levels correlate with neutrophil levels obtained by differential leukocyte counts.
RESULTS AND DISCUSSION
In this study, we first characterized how the reversible monomer-dimer equilibrium of the chemokine CXCL8 regulates neutrophil recruitment in the mouse peritoneum. We measured neutrophil recruitment at different doses using three CXCL8 variants: a “trapped” monomeric CXCL8 variant, which cannot dimerize; a trapped dimeric CXCL8 variant, which cannot dissociate; and WT CXCL8, which can reversibly exist as monomers and dimers. We have previously determined the structures of our trapped monomer and dimer and have verified that the activities of the trapped monomer and dimer reflect those of the WT monomer and dimer [19–21]. A schematic of the structures is shown (Fig. 1). With the use of these elegant CXCL8 variants, we designed experiments that can provide crucial insights into not only whether WT exists as monomer, dimer, or both in the context of the in vivo milieu of the peritoneum but also how their differential interactions with their receptors and GAGs in different in vivo environments dictate overall recruitment patterns.
Figure 1. Schematic of CXCL8 monomer and dimer.

Structures of the CXCL8-trapped monomer (A) and WT dimer (B) are shown. The two monomers within the dimer structure are shown in different colors, and the structures reveal that the structure of the monomer is similar to the monomer structure in the dimer.
A mouse model is ideally suited for characterizing the molecular mechanisms by which human chemokines recruit circulating neutrophils to different tissues. Unlike human neutrophils, which express two receptors—CXCR1 and CXCR2—mouse neutrophils express only CXCR2 [23, 24]. Various in vitro assays, using neutrophils and transfected cells, have shown that human CXCL8 is active against mouse CXCR2 and has similar functional profiles as mouse keratinocyte-derived chemokine and MIP-2 [23]. Similarly, several in vivo studies, including ours, have shown that human CXCL8 recruits neutrophils at nanomolar concentrations [11, 18].
We measured peritoneal neutrophil recruitment at four different doses (0.1, 0.25, 1, and 10 μg) for all three CXCL8 variants (Fig. 2). The WT is more active than the monomer or dimer at all doses except at the 1-μg dose, where it has similar activity as the monomer. As a result of the reversible monomer-dimer equilibrium, the WT alone can exist as a monomer, dimer, or monomer and dimer. The higher WT recruitment activity observed at low and high doses cannot be explained, as a result of the individual activities of the WT monomer or dimer, or as a sum of monomer and dimer activities. These observations suggest synergy between monomer and dimer and most importantly, indicate that a mixture of trapped monomer and trapped dimer will not capture the WT activity. This is particularly striking for the 0.1-μg data; the dimer is inactive at this dosage. Indeed, at this dose, the actual amounts of monomeric and dimeric forms of WT CXCL8 will be lower in vivo (say 0.08 μg monomer and 0.02 μg dimer) than the amounts used for individual monomer and dimer experiments (0.1 μg); however, the WT still shows higher activity than either. The trafficking pattern of the WT shows no resemblance to the monomer or dimer, and the recruitment profiles of the monomer and dimer are also strikingly different (Fig. 2 and Supplemental Fig. 1). The monomer is quite active at all doses, although it shows lower activity at the highest dose, resulting in a bell-shaped-like curve. On the other hand, the dimer is barely active at low doses but shows robust and higher activity than the monomer at the highest dose. We also measured neutrophil levels using a MPO activity assay and observe the same pattern as obtained by manual differential leukocyte counts (Fig. 3).
Figure 2. Neutrophil recruitment activity of CXCL8 variants.

Total neutrophils recruited into the peritoneum are plotted for each of the variants at different doses. Data are presented as mean ± sem and are from 10 to 19 animals from a total of two to four independent experiments with four to six animals/group for each experiment. One-way ANOVA was used to determine statistical significance. The recruitment of the WT compared with the monomer and dimer is significant at all doses (P<0.01), except at 1 μg between WT and monomer. The recruitment of WT and monomer compared with the control was significant at all doses (P<0.01) and of the dimer compared with control was significant only at the 10 μg dosage. Inset: The R6K mutation renders CXCL8 completely inactive, and its recruitment at 10 μg dose is similar to that of control PBS, indicating that recruitment as a result of spurious contamination or bacterial byproducts can be ruled out.
Figure 3. Recruitment of neutrophils using MPO assay.
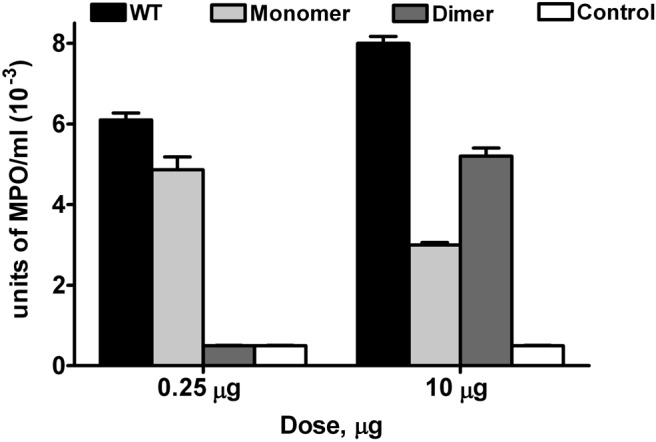
MPO is expressed predominantly only in neutrophils, and the measured MPO activity correlates with the neutrophil levels shown in Fig. 2. Each data set represents an average of two to three experiments using four to six animals/group. One-way ANOVA was used to determine statistical significance. P is at least <0.01 between monomer and dimer, monomer and WT, and dimer and WT at both doses.
We carried out control experiments to confirm that the recruitment profiles are caused directly by exogenously administered CXCL8 variants and not a result of endogenously expressed cytokines and chemokines or spurious contaminants, such as bacterial LPS. WT and trapped dimer are expressed recombinantly; we therefore tested the recruitment activity of a known, inactive mutant R6K, which was also expressed recombinantly and purified using the same protocol and observed no recruitment (Fig. 2). We also investigated the levels of endogenous mouse chemokines and cytokines using a Bioplex assay and saw no differences in these levels between mice injected with CXCL8 and those injected with buffer (data not shown).
We have previously studied neutrophil recruitment of the CXCL8 variants in the lung [18], which allows the comparison of neutrophil recruitment patterns in different tissue microenvironments (Fig. 4). The recruitment profile of the WT is appreciably different in the two tissue environments, whereas the individual profiles of the monomer and dimer are similar but not identical. The WT at low doses was significantly active in the peritoneum but completely inactive in the lung. Recruitment remains high in the peritoneum at higher doses, and the recruitment profile is therefore shallow, with no more than twofold change across the dosage range. Recruitment in the lung has a very different profile as a result of low activity at low doses and substantial increase in activity at the highest dose. Further, although there is evidence of synergy between WT monomer and dimer in peritoneal recruitment, the WT recruitment in the lung is always intermediate between monomers and dimers, indicating additive, not synergistic, interactions.
Figure 4. Neutrophil recruitment profiles in the peritoneum versus lung.

Neutrophil recruitment mediated by CXCL8 WT, monomer, and dimer in the peritoneum is compared with the previously reported recruitment in the lung [9]. The recruitment profile of the WT is distinctly different between the peritoneum and lung, indicating differential roles for the monomer-dimer equilibrium in regulating neutrophil recruitment.
The individual recruitment profiles of the trapped monomer and dimer provide crucial insights into how the WT monomer and dimer regulate recruitment. The trapped monomer shows low activity at low and high doses, resulting in a bell-shaped-like recruitment profile in both tissues; however, the profile is shallow and symmetric in the peritoneum and skewed in the lung (Fig. 4). The difference in the monomer recruitment profiles is mainly a result of the difference in its activity in the peritoneum and lung at low doses. As the monomer is the high-affinity CXCR2 ligand, reduced recruitment at higher doses in both tissues cannot be a result of reduced receptor affinity. At higher doses, more of the monomer is likely to exist in the soluble form in systemic circulation, which may result in less-effective gradients in the vasculature and in the tissue. Therefore, the lower recruitment at these doses could be a result of desensitization of the CXCR2 receptor and/or inefficient gradient formation. The dimer was inactive at low doses (up to 1 μg) in the peritoneum and lung but highly active at 10 μg, which is especially striking in the lung (Fig. 4). Cell-based studies have shown that the dimer, if anything, is only slightly less active than the monomer for receptor binding and activation [19], and so, lack of activity at lower doses and substantial activity at the higher dose of the dimer cannot be a result of differences in receptor function and must be a result of differences in GAG-binding interactions.
Recruitment is a consequence of multiple interconnected factors, such as (i) the nature of the chemokine gradient that includes the steepness and range/distance, (ii) tissue architecture and local concentration and type of the GAG, and (iii) the monomer-dimer equilibrium, and differences in GAG-binding interactions of the monomer and dimer. Therefore, to better define the role of GAG interactions between the tissues in defining the chemokine gradient, we characterized the recruitment activity of a panel of GAG mutants in the peritoneum and lung.
Previous structure-function studies have shown that the residues K20, R60, K64, K67, and R68 mediate GAG binding [14]; we therefore generated single Ala mutants K20A, R60A, K64A, K67A, and R68A on the WT background. We initially characterized their activity in the peritoneum model and then tested selected mutants in the lung model. We characterized peritoneal recruitment at three different doses: 0.25, 1, and 10 μg, and observe that all of the mutants were less active than WT at all doses, with the exception of K67A at the 0.25-μg dose, which was as active as WT (Fig. 5). As none of the GAG-binding residues are involved in dimer interactions, reduced activity of all of the mutants must come from altered GAG interactions and not from altered monomer-dimer equilibrium. We did not characterize the recruitment of the GAG mutants in the monomer and dimer background but anticipate reduced recruitment on the basis of WT data. As discussed earlier, WT recruitment is predominantly from the monomeric form at low doses and from monomeric and dimeric forms at the 10-μg dose, and the monomer-dimer equilibrium regulates recruitment at all doses (Fig. 2).
Figure 5. Neutrophil recruitment activity of CXCL8 GAG mutants in the peritoneum.

Recruitment activity of the GAG mutants was measured at three different doses: 0.25 μg (A), 1 μg (B), and 10 μg (C). Data are presented as mean ± sem and represent four to six animals/group. Two-way ANOVA with post-test was used to determine statistical significance. Reduced recruitment of all of the mutants was significant (P<0.01), except for K67A at the 0.25-μg dose.
Recruitment profiles of the GAG mutants also indicate that all of the GAG-binding residues are not equivalent and that the binding interactions of K64 are most crucial, an observation that is especially evident in the 10-μg data. Analysis of the CXCL8 structure reveals that K64 lies in the middle of the helix; its interactions could therefore be an important anchor point for binding interactions and gradient formation. Nevertheless, the overall reduced recruitment at all doses indicates that reduced GAG interactions result in less-than-optimal gradient formation, in turn, effecting reduced recruitment in the peritoneum.
We then characterized the recruitment of the K64A and R60A mutants at 1 and 10 μg doses in the lung model (Fig. 6). In stark contrast to the peritoneum, these mutants were actually more active than the WT, an effect that is especially evident at the 1-μg dose. In the lung, WT is essentially a monomer at the 1-μg dose and a dimer at 10 μg, suggesting that GAG interactions of the monomer result in more optimal gradient formation for recruitment. Our results are also in agreement with the higher recruitment activities previously observed for R68A and for a K64A/K67A/R68A triple mutant in the lung [11]. These authors had interpreted the differences in recruitment as being a result of differences in binding kinetics, resulting in differences in gradient formation that favor recruitment in the lung.
Figure 6. Neutrophil recruitment activity of CXCL8 GAG mutants in the lung.

Recruitment activity of the GAG K64A and R60A mutants was measured at two different doses: 1 μg (A) and 10 μg (B). Data are presented as mean ± sem and represent four to six animals/group. Two-way ANOVA with post-test was used to determine statistical significance. Both mutants showed higher activity at the 1-μg (P<0.01) and 10-μg (P<0.05) doses.
The contrasting role of GAG binding in the lung and peritoneum must be a direct consequence of differences in the nature of the gradient, which is dependent on the architecture and the GAG makeup of these tissues. The lung alveoli consist of an epithelial layer, extracellular matrix, and an endothelial layer lining the capillaries. The peritoneum also consists of three layers: mesothelial cells lining the peritoneal cavity, followed by connective tissue, and endothelial cells lining the capillaries. Chemokines produced in alveolar spaces/peritoneal cavity traverse these layers to reach the circulation, and recruited neutrophils traverse the reverse route. However, the architecture and microenvironment of lung and peritoneal tissue are distinctly different, although the gross structural features are similar. For instance, the peritoneal lining provides a larger and less-convoluted surface area compared with the alveoli, and the distribution of GAGs, such as heparan sulfate and dermatan sulfate, could be different between the lung and peritoneum [25–27]. GAGs are found on the abluminal and luminal faces of epithelial/mesothelial and endothelial cells, are also expressed in connective tissue, and are further cleaved by endogenous proteases and so, can exist in the free-soluble and immobilized cell surface-bound forms. Therefore, these tissue-specific differences most likely result in differential gradient formation for the different CXCL8 variants, resulting in observed, distinct recruitment profiles.
In summary, our results indicate that total neutrophil recruitment is not simply correlated to the amount of chemokine expressed at the site of infection. The CXCL8 monomer-dimer equilibrium and differences in tissue/organ microenvironment and architecture influence the nature of gradient formation, which coordinates and regulates neutrophil recruitment. Most importantly, our finding that the CXCL8 monomer-dimer equilibrium plays differential roles in regulating neutrophil recruitment between tissues is novel, revealing an additional level of complexity in the orchestration of in vivo recruitment by chemokine dimerization. Leukocyte recruitment into the lung and peritoneum are popular animal models for studying chemokines' role in inflammatory/infectious diseases and for studying mechanisms underlying leukocyte trafficking [28, 29]. Therefore, inter-relationships among chemokine production, chemokine dimerization, and differences in gradient formation between tissues must be taken into consideration in the design and interpretation of such studies.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the NIH AI-069152 to K.R. We thank Meena Shanmugasundaram for technical support.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- ECM
- extracellular matrix
- GAG
- glycosaminoglycan
- PF
- peritoneal fluid
- WT
- wild type
REFERENCES
- 1. Nathan C. (2006) Neutrophils and immunity: challenges and opportunities. Nat. Rev. Immunol. 6, 173–182 [DOI] [PubMed] [Google Scholar]
- 2. Murphy P. M. (1997) Neutrophil receptors for interleukin-8 and related CXC chemokines. Semin. Hematol. 34, 311–318 [PubMed] [Google Scholar]
- 3. Colditz I. G., Schneider M. A., Pruenster M., Rot A. (2007) Chemokines at large: in-vivo mechanisms of their transport, presentation and clearance. Thromb. Haemost. 97, 688–693 [PubMed] [Google Scholar]
- 4. Pinheiro da Silva F., Soriano F. G. (2009) Neutrophils recruitment during sepsis: critical points and crossroads. Front. Biosci. 14, 4464–4476 [DOI] [PubMed] [Google Scholar]
- 5. Li Q., Park P. W., Wilson C. L., Parks W. C. (2002) Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell 111, 635–646 [DOI] [PubMed] [Google Scholar]
- 6. Wang L., Fuster M., Sriramarao P., Esko J. D. (2005) Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat. Immunol. 6, 902–910 [DOI] [PubMed] [Google Scholar]
- 7. Kuschert G. S., Coulin F., Power C. A., Proudfoot A. E., Hubbard R. E., Hoogewerf A. J., Wells T. N. (1999) Glycosaminoglycans interact selectively with chemokines and modulate receptor binding and cellular responses. Biochemistry 38, 12959–12968 [DOI] [PubMed] [Google Scholar]
- 8. Hoogewerf A. J., Kuschert G. S., Proudfoot A. E., Borlat F., Clark-Lewis I., Power C. A., Wells T. N. (1997) Glycosaminoglycans mediate cell surface oligomerization of chemokines. Biochemistry 36, 13570–13578 [DOI] [PubMed] [Google Scholar]
- 9. Frevert C. W., Goodman R. B., Kinsella M. G., Kajikawa O., Ballman K., Clark-Lewis I., Proudfoot A. E., Wells T. N., Martin T. R. (2002) Tissue-specific mechanisms control the retention of IL-8 in lungs and skin. J. Immunol. 168, 3550–3556 [DOI] [PubMed] [Google Scholar]
- 10. Frevert C. W., Kinsella M. G., Vathanaprida C., Goodman R. B., Baskin D. G., Proudfoot A., Wells T. N., Wight T. N., Martin T. R. (2003) Binding of interleukin-8 to heparan sulfate and chondroitin sulfate in lung tissue. Am. J. Respir. Cell Mol. Biol. 28, 464–472 [DOI] [PubMed] [Google Scholar]
- 11. Tanino Y., Coombe D. R., Gill S. E., Kett W. C., Kajikawa O., Proudfoot A. E., Wells T. N., Parks W. C., Wight T. N., Martin T. R., Frevert C. W. (2010) Kinetics of chemokine-glycosaminoglycan interactions control neutrophil migration into the airspaces of the lungs. J. Immunol. 184, 2677–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rajarathnam K., Clark-Lewis I., Sykes B. D. (1995) 1H NMR solution structure of an active monomeric interleukin-8. Biochemistry 34, 12983–12990 [DOI] [PubMed] [Google Scholar]
- 13. Clore G. M., Appella E., Yamada M., Matsushima K., Gronenborn A. M. (1990) Three-dimensional structure of interleukin 8 in solution. Biochemistry 29, 1689–1696 [DOI] [PubMed] [Google Scholar]
- 14. Kuschert G. S., Hoogewerf A. J., Proudfoot A. E., Chung C. W., Cooke R. M., Hubbard R. E., Wells T. N., Sanderson P. N. (1998) Identification of a glycosaminoglycan binding surface on human interleukin-8. Biochemistry 37, 11193–11201 [DOI] [PubMed] [Google Scholar]
- 15. Handel T. M., Johnson Z., Crown S. E., Lau E. K., Proudfoot A. E. (2005) Regulation of protein function by glycosaminoglycans—as exemplified by chemokines. Annu. Rev. Biochem. 74, 385–410 [DOI] [PubMed] [Google Scholar]
- 16. Lortat-Jacob H., Grosdidier A., Imberty A. (2002) Structural diversity of heparan sulfate binding domains in chemokines. Proc. Natl. Acad. Sci. USA 99, 1229–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goger B., Halden Y., Rek A., Mosl R., Pye D., Gallagher J., Kungl A. J. (2002) Different affinities of glycosaminoglycan oligosaccharides for monomeric and dimeric interleukin-8: a model for chemokine regulation at inflammatory sites. Biochemistry 41, 1640–1646 [DOI] [PubMed] [Google Scholar]
- 18. Das S. T., Rajagopalan L., Guerrero-Plata A., Sai J., Richmond A., Garofalo R. P., Rajarathnam K. (2010) Monomeric and dimeric CXCL8 are both essential for in vivo neutrophil recruitment. PLoS One 5, e11754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nasser M. W., Raghuwanshi S. K., Grant D. J., Jala V. R., Rajarathnam K., Richardson R. M. (2009) Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 183, 3425–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rajarathnam K., Sykes B. D., Kay C. M., Dewald B., Geiser T., Baggiolini M., Clark-Lewis I. (1994) Neutrophil activation by monomeric interleukin-8. Science 264, 90–92 [DOI] [PubMed] [Google Scholar]
- 21. Rajarathnam K., Prado G. N., Fernando H., Clark-Lewis I., Navarro J. (2006) Probing receptor binding activity of interleukin-8 dimer using a disulfide trap. Biochemistry 45, 7882–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rajagopalan L., Rajarathnam K. (2004) Ligand selectivity and affinity of chemokine receptor CXCR1. Role of N-terminal domain. J. Biol. Chem. 279, 30000–30008 [DOI] [PubMed] [Google Scholar]
- 23. Lee J., Cacalano G., Camerato T., Toy K., Moore M. W., Wood W. I. (1995) Chemokine binding and activities mediated by the mouse IL-8 receptor. J. Immunol. 155, 2158–2164 [PubMed] [Google Scholar]
- 24. Fan X., Patera A. C., Pong-Kennedy A., Deno G., Gonsiorek W., Manfra D. J., Vassileva G., Zeng M., Jackson C., Sullivan L., Sharif-Rodriguez W., Opdenakker G., Van Damme J., Hedrick J. A., Lundell D., Lira S. A., Hipkin R. W. (2007) Murine CXCR1 is a functional receptor for GCP-2/CXCL6 and interleukin-8/CXCL8. J. Biol. Chem. 282, 11658–11666 [DOI] [PubMed] [Google Scholar]
- 25. Yung S., Chan T. M. (2009) Intrinsic cells: mesothelial cells—central players in regulating inflammation and resolution. Perit. Dial. Int. 29 (Suppl. 2), S21–S27 [PubMed] [Google Scholar]
- 26. Yung S., Thomas G. J., Stylianou E., Williams J. D., Coles G. A., Davies M. (1995) Source of peritoneal proteoglycans. Human peritoneal mesothelial cells synthesize and secrete mainly small dermatan sulfate proteoglycans. Am. J. Pathol. 146, 520–529 [PMC free article] [PubMed] [Google Scholar]
- 27. Gill S., Wight T. N., Frevert C. W. (2010) Proteoglycans: key regulators of pulmonary inflammation and the innate immune response to lung infection. Anat. Rec. (Hoboken) 293, 968–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rittirsch D., Huber-Lang M. S., Flierl M. A., Ward P. A. (2009) Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 4, 31–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grommes J., Soehnlein O. (2011) Contribution of neutrophils to acute lung injury. Mol. Med. 17, 293–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.