

Abstract
Neuroendocrine prostate cancer (NEPC) is an aggressive subtype of prostate cancer that most commonly evolves from preexisting prostate adenocarcinoma (PCA). Using Next Generation RNA-sequencing and oligonucleotide arrays, we profiled 7 NEPC, 30 PCA, and 5 benign prostate tissue (BEN), and validated findings on tumors from a large cohort of patients (37 NEPC, 169 PCA, 22 BEN) using IHC and FISH. We discovered significant overexpression and gene amplification of AURKA and MYCN in 40% of NEPC and 5% of PCA, respectively, and evidence that that they cooperate to induce a neuroendocrine phenotype in prostate cells. There was dramatic and enhanced sensitivity of NEPC (and MYCN overexpressing PCA) to Aurora kinase inhibitor therapy both in vitro and in vivo, with complete suppression of neuroendocrine marker expression following treatment. We propose that alterations in Aurora kinase A and N-myc are involved in the development of NEPC, and future clinical trials will help determine from the efficacy of Aurora kinase inhibitor therapy.
Keywords: neuroendocrine prostate cancer, aurora kinase A, n-myc, drug targets
Introduction
Neuroendocrine prostate cancer (NEPC) is an aggressive subtype of prostate cancer that can arise de novo, but much more commonly arises after hormonal therapy for prostate adenocarcinoma (PCA) (1). NEPC differs histologically from PCA, and is characterized by the presence of small round blue neuroendocrine cells, which do not express androgen receptor (AR) or secrete prostate specific antigen (PSA), but usually express neureondocrine markers such as chromogranin A, synaptophysin, and neuron specific enolase (NSE)(2). The prostate cancer specific TMPRSS2-ERG gene rearrangement (3) has been reported in approximately 50% of NEPC (4), similar to the frequency in PCA(5). This suggests clonal origin of NEPC from PCA and distinguishes NEPC from small carcinomas of other primary sites (4, 6–7). NEPC frequently metastasizes to visceral organs, responds only transiently to chemotherapy, and most patients survive less than one year (1). The poor molecular characterization of NEPC accounts in part for the lack of disease specific therapeutics. In this study, we sought to better understand the molecular transformation of NEPC and to identify new drug targets.
Results
We evaluated forty-five NEPC tumors, and observed a spectrum ranging from pure small cell carcinoma to tumors with mixed features of both PCA and NEPC (Supplementary Fig. 1). The TMPRSS2-ERG gene fusion was detected by Fluorescence In Situ Hybridization (FISH) break-apart assay in 44% of NEPC. Importantly, in tumors demonstrating both PCA and NEPC foci, there was perfect concordance with regards to the TMPRSS2-ERG status (Fig. 1A). NEPC foci lacked ERG protein expression by immunohistochemistry (IHC), even in tumors harboring the TMPRSS2-ERG rearrangement, with a sharp margin separating NEPC and PCA components in mixed tumors. This margin also corresponded directly to presence or absence of androgen receptor (AR) expression (in PCA and NEPC, respectively), consistent with ERG protein expression being driven by androgen and requiring AR signaling.
Figure 1.
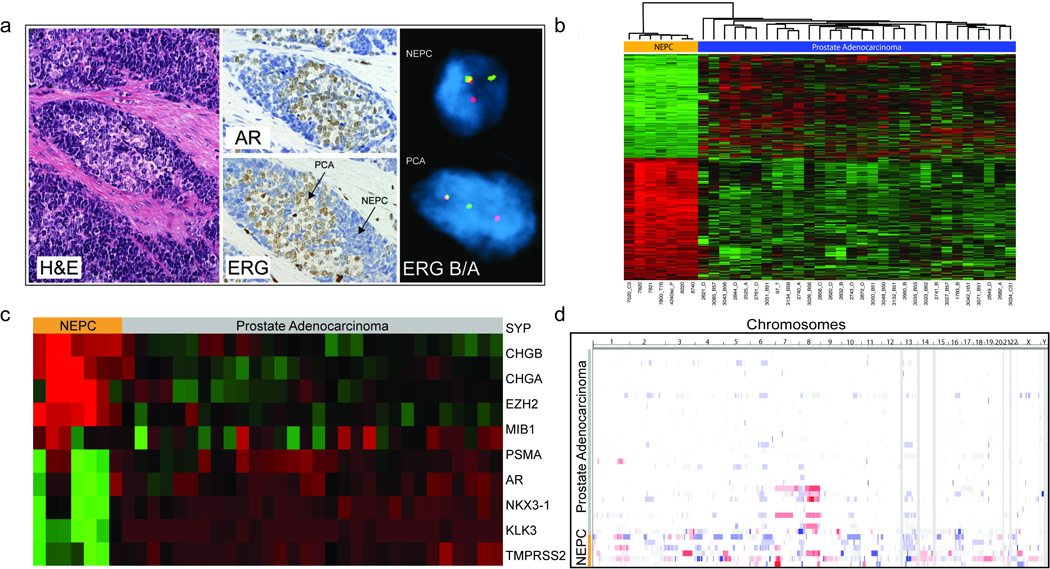
Characterization of NEPC: (A) Tumor with mixed features of NEPC and PCA. Hematoxylin and eosin (H&E) staining, immunohistochemical analysis for androgen receptor (AR) and ERG, and FISH for ERG breakapart (indicating gene fusion) (B) Gene expression of the 936 of 25932 genes showing differential expression between 7 NEPC cases and 30 PCAs, after a Benjamini-Hochberg correction for multiple hypothesis testing of <0.001 (Red =High Expression, Green = Low Expression). (C) Gene Expression of select genes comparing NEPC and PCA, including neuroendocrine associated genes (SYP, CHGB, CHGA), EZH2, MIB1 (Ki67), PSMA, AR, and androgen regulated genes (NKX3-1, KLK3 (PSA), TMPRSS2). (D) Graphical representation of the genomic landscape of PCA (in order of increasing Gleason Score) and NEPC, as determined by Affymetrix 6.0 oligonucleotide array (Red= Copy Number Gain, Blue= Copy Number Loss, White= No Change).
Using Next Generation RNA sequencing (RNA-Seq) and oligonucleotide arrays, we sequenced seven NEPC and thirty localized PCA tumors. Clinical characteristics are summarized in Supplementary Tables 1–2. There were significant gene expression differences between NEPC and PCA, with 936 of 25,932 evaluated genes showing differential expression after correction for multiple hypothesis (Benjamini-Hochberg P<0.001) (Fig. 1B, Supplementary Table 3). There were no global gene expression differences between primary and secondary NEPC. As expected, NEPC demonstrated low expression of known androgen-regulated genes (e.g., KLK3 (PSA), TMPRSS2, NXK3.1) and high expression of neuroendocrine-associated genes (e.g., CGA and SYP), though there were some tumors with mixed molecular features (Fig. 1C). EZH2, a polycomb gene shown to be associated with aggressive behavior in a number of cancer types including prostate (8), was significantly overexpressed in NEPC compared to PCA (P=0.0001). Somatic copy number alteration assessment revealed discrete, statistically significant differences in the number of genomic amplifications and deletions in NEPC as compared to PCA (Fig. 1D).
After integration of gene expression and copy number data, we evaluated for targetable lesions and discovered significant overexpression and gene amplification of AURKA (Aurora kinase A) in NEPC compared to PCA (P=1.46×10−5) (Fig. 2A, Supplementary Fig. 2). AURKA mRNA was overexpressed in all 7 cases, but amplified in 4/7 NEPC. AURKA amplification was associated with overexpression (P=0.0006), but AURKA overexpression also occurred without amplification (potentially through other mechanisms). Within NEPC, the level of AURKA overexpression was not differential based on amplification (Supplemental Fig. 3A). AURKA is a serine/threonine kinase involved in mitotic spindle formation, centrosome separation, and G2- M transition during the cell cycle (9), although it also has oncogenic properties (9).
Figure 2.
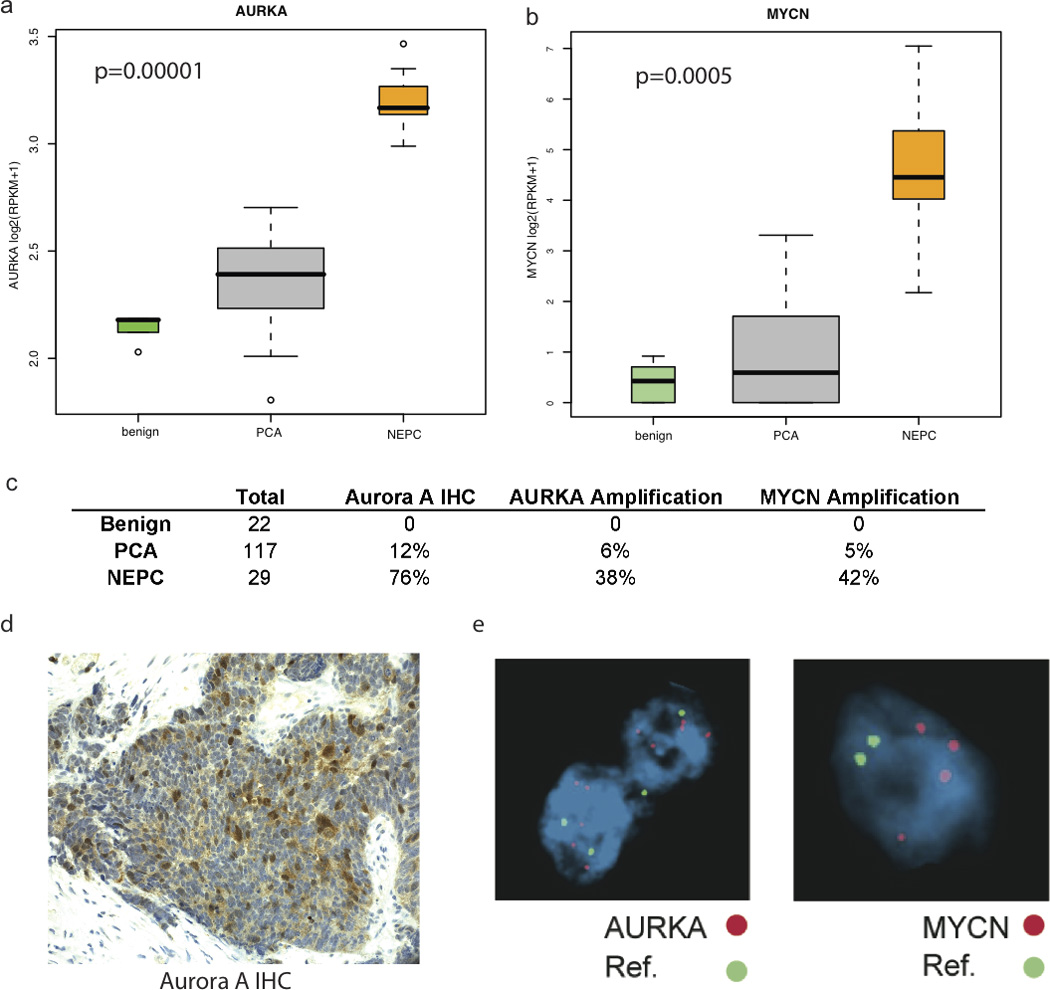
Evaluation of Aurora Kinase and N-Myc: (A) Gene expression of AURKA in Benign Prostate Tissue, PCA, and NEPC, as measured by RNA-Seq. RPKM= Reads per kilobase of exon per million mapped reads. (B) Gene expression of AURKA in Benign Prostate Tissue, PCA, and NEPC, as measured by RNA-Seq. RPKM= Reads per kilobase of exon per million mapped reads (C) Table summarizing IHC and FISH data from tumors from large cohort of PCA, NEPC, and benign prostate (D) Representative example of positive Aurora kinase A overexpression by IHC, and MYCN and AURKA amplification by FISH in human NEPC. Green= Centromeric Control Probes, Red = AURKA and MYCN loci as labeled in NEPC.
In human neuroblastoma models, AURKA has also been shown to interact with and stabilize the oncogene N-myc (MYCN) (10), independent of its role in mitosis. As neuroblastoma is another highly aggressive neuroendocrine tumor, we queried the dataset for MYCN gene expression and discovered significant overexpression in NEPC compared to PCA (P=0.0005) (Fig. 2B). N-myc is a transcription factor in the MYC family, involved in nervous system development and not normally expressed in the prostate (11) or previously linked to prostate cancer. The Affymetrix 6.0 array does not have adequate coverage of the MYCN locus on 2p24 with the nearest markers 2.5 Kb from the 3’ and 5’ end of the gene, and is therefore suboptimal in evaluating for MYCN copy number gain.
We screened benign prostate and prostate tumors from a larger cohort of patients (22 benign, 169 primary PCA, 37 NEPC) (Fig. 2C), and found AURKA was overexpressed by immunohistochemistry (IHC) in none of the benign prostate cases, 12% of PCA, and 76% of NEPC. AURKB was also over-expressed in NEPC although to a lesser degree, and AURKC was minimally expressed in either NEPC or PCA (Supplementary Fig. 3B). In NEPC, there was strong cytoplasmic expression of Aurora kinase A in the majority of tumor cells (>50%), but in PCA represented <5% of tumor cells and expression was weaker and demonstrated a speckled pattern (Fig. 2D). AURKA was amplified by FISH in none of the benign prostate, 5% of PCA, and 40% of NEPC. Neither AURKB or AURKC were amplified. MYCN was amplified by FISH in none of the benign prostate, 4% of PCA, and 40% of NEPC. In nearly all positive cases (>90%), amplification of AURKA or MYCN was concurrent (Supplementary Fig. 4). Notably, one patient who progressed from PCA to NEPC after three years demonstrated amplification of AURKA and MYCN in his primary PCA, suggesting that these genomic aberrations can arise early.
Transfection of MYCN in either benign prostate RWPE-1 or LNCaP (PCA cells) induced expression of AURKA and phosphorylated histone 3 (a downstream marker of Aurora kinase activity (12)) (Fig. 3A, B). Chromatin immunoprecipation (ChIP) of LNCaP cells stably transfected with MYCN revealed that N-myc does not bind the promoter of AURKA but did bind to E-box binding elements associated with the N-myc-responsive promoter of telomerase reverse transcriptase (hTERT) as previously described (13) (Supplementary Fig. 5). Instead, we found that the N-myc protein physically interacts with Aurora kinase A (as seen by coimmunoprecipitation) and enhances Aurora kinase A protein stability (Fig. 3C).
Figure 3.
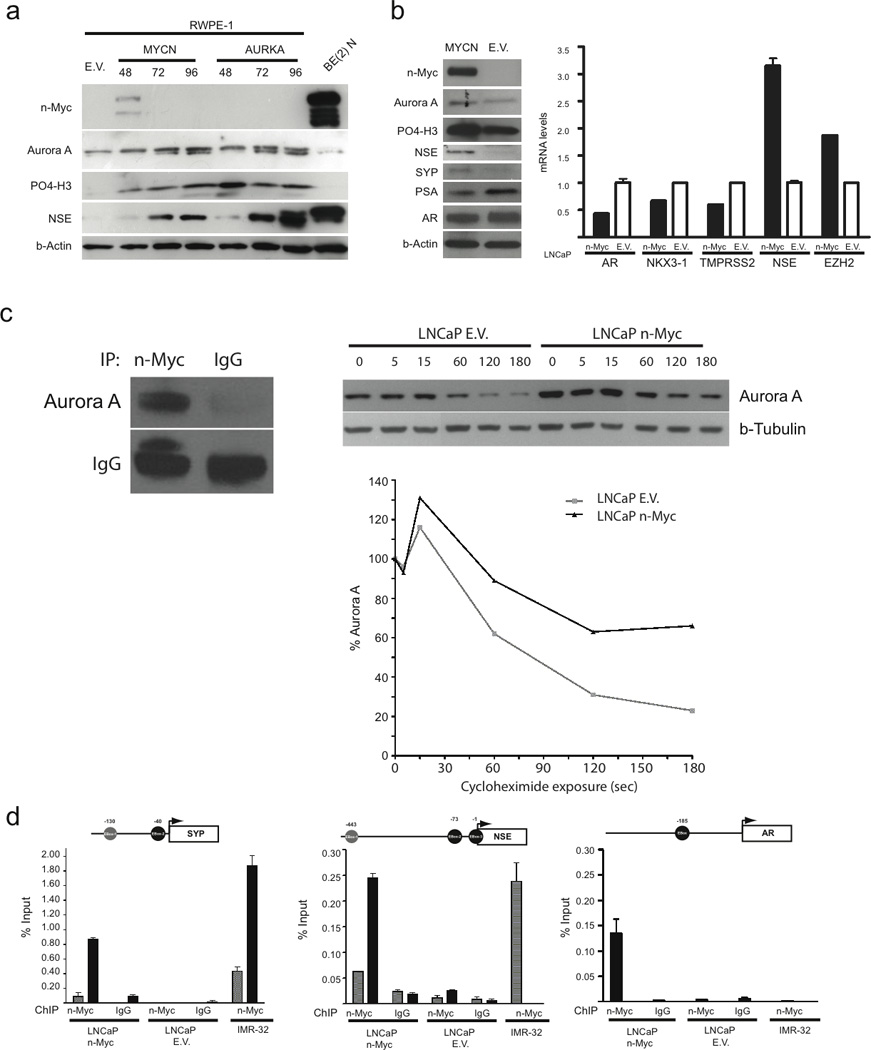
(A) Immunoblot analysis for protein expression of Aurora kinase A, Phosphorylated histone 3 (P04-H3), neuron specific enolase (NSE) and synaptophysin (SYP) after transient transfection of MYCN, AURKA, or Empty Vector (EV) in RWPE-1 cells. BE(2)N is a neuroblastoma cell line as positive control for NSE. (B) Stable LNCaP cell line over-expressing N-myc compared to empty vector (EV) : Immunoblot analysis for protein expression of N-myc, Aurora kinase A, P04-H3, NSE, SYP, PSA, AR, beta actin. qRT-PCR and microarray (MA) data showing induction of NSE (qRT-PCR) and EZH2 (MA) gene expression and suppression of AR (qRT-PCR) and androgen regulated genes (NKX3-1, TMPRSS2 (MA)) (C) Left: Immunoprecipitation of LNCaP-n-Myc cell lysates using antibodies directed against N-Myc or control IgG antibodies and Western blot using antibodies directed against Aurora kinase A (Aurora A) or control IgG antibodies. Right: LNCaP control (LNCaP E.V.) and LNCaP-n-Myc cells were treated with cycloheximide (CHX) for the indicated time (in minutes) and Aurora kinase A or beta-tubulin levels were assessed by immunoblotting. The normalized percent of Aurora kinase A relative to beta-tubulin and to time point 0 for LNCaP E.V. (gray line) or LNCaP-n-Myc (black line). (D) N-Myc directly binds to the SYP, NSE, and AR promoters in LNCaP-n-Myc cells and not LNCaP-EV. Not-to-scale schematic representation of SYP, NSE, and AR promoters showing the E-box sites (grey and black circles) indicated for each. The transcription start site for each gene is indicated with an arrow. Below each schematic are bar graphs showing the amount of enriched DNA (relative to input chromatin preparation) for each E-box site in the indicated cell lines following ChIP using either anti-N-Myc (right) or anti-IgG (left) antibodies. IMR-32 is a MYCN amplified neuroblastoma cell line. In IMR-32 cells, Nmyc binds promoters of SYP and NSE, but not AR.
Overexpression of either AURKA or MYCN into RWPE-1 cells induced expression of the neuroendocrine markers, NSE and SYP, which are not normally expressed in benign prostate (Fig. 3A), suggesting that AURKA and MYCN may be involved in neuroendocrine differentiation. Knockdown of AURKA with siRNA suppressed NSE expression in the NEPC cell line, NCI-H660 (Supplementary Fig. 6). Furthermore, LNCaP cells stably transfected with MYCN (LNCaP-n-Myc) phenotypically resembled NEPC, with upregulation of the neuroendocrine marker NSE, downregulation of AR and androgen regulated genes (TMPRSS2, NKX3-1), and upregulation of EZH2 compared to control LNCaP cells (Fig. 3B). Chromatin immunoprecipitation revealed that N-myc binds the promoters of NSE, SYP, and AR, suggesting direct modulation of the neuroendocrine phenotype by transcription factor binding (Fig. 3D). In MYCN-amplified neuroblastoma cells (IMR-32), N-myc also bound NSE and SYP promoters, but not AR (suggesting N-myc binding of AR promoter may be prostate-specific).
Based on these findings, we posited that treatment with an Aurora kinase inhibitor would have a preferential effect on NEPC compared to PCA. To test this hypothesis in vitro, we used two experimental models: LNCaP cells stably transfected with MYCN (which phenotypically resemble NEPC), and the NCI-H660 cell line. NCI-H660 was originally derived at time of autopsy from a patient with small cell carcinoma initially thought to be lung cancer but later classified as prostate (14–15). RNA sequencing revealed that NCI-H660 has a similar molecular signature as our NEPC tumors (including overexpression of AURKA and MYCN), and FISH also demonstrated AURKA and MYCN copy number gain (Supplementary Fig. 7A, B) as well as over-expression of phosphorylated Aurora A (compared to phosphorylated forms of Aurora B and C, Supplementary Fig. 7C), as another measure of kinase activity status.
LNCaP-n-Myc cells demonstrated enhanced in vitro sensitivity to the Aurora kinase inhibitor PHA-739358 (Nerviano Medical Sciences, Milan, Italy) compared to control LNCaP cells (LNCaP-EV) (Fig. 4A). Similarly, NCI-H660 also demonstrated enhanced sensitivity to PHA- 739358 compared to two PCA cell lines (DU145, VCaP) and benign RWPE-1 cells (Fig. 4b). Knockdown of Aurora A in NCI-H660 with multiple shRNAs showed similar results (Supplementary Fig 8). Given the role of Aurora kinase A in the cell cycle, we performed FACS analysis of cells following PHA-739358 treatment confirming a dose dependent G2/M arrest with PHA-739358 treatment in LNCaP and LNCaP-MYCN cells. Polyploidy was induced by PHA-739358 in all cells (i.e., LNCaP, LNCaP-MYCN, and NCI-H660). However, there was no significant G2/M arrest in NCI-H660 cells, supporting our hypothesis that Aurora A may have alternative mechanisms of action in NEPC (Supplementary Fig. 9).
Figure 4.
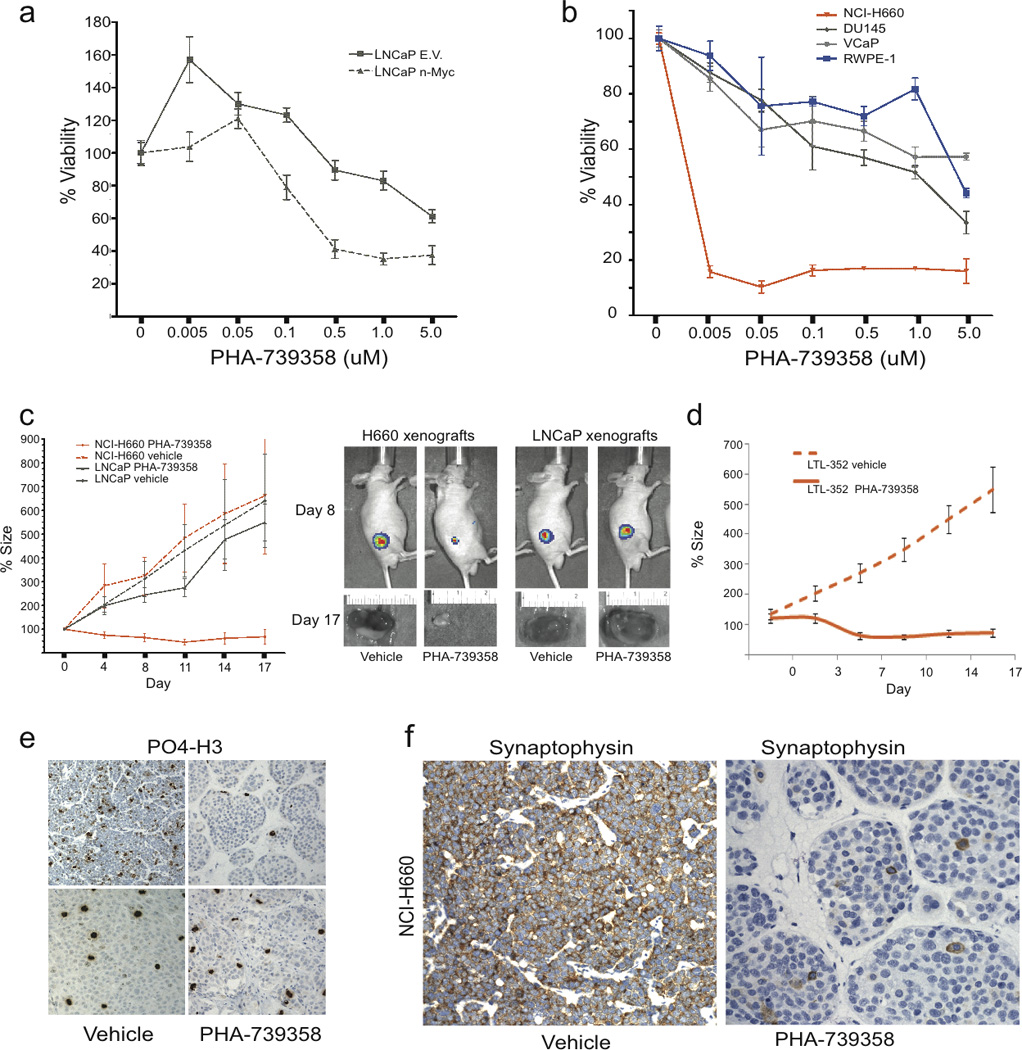
NEPC demonstrates enhanced sensitivity to Aurora Kinase Inhibitor therapy compared to PCA (A) Viability assay of LNCaP cells transfected with MYCN or Empty Vector (EV) at 72 hours after treatment with vehicle or indicated doses of the pan-Aurora kinase inhibitor PHA-739358. (B) Viability assay of RWPE (blue circles), VCaP (gray diamonds), DU145 (gray triangles), and NCI-H660 (orange triangles) at 72 hours after treatment with vehicle or indicated doses of PHA-739358. (C) Percent tumor size after treatment of LNCaP (gray) and NCI-H660 (red) xenografts with vehicle (dotted lines) or PHA-739358 30 mg/kg IP BID (solid lines) twice a day for 5 days relative to day 0. Luciferase imaging at day 8 and tumor photographs at day 17 of representative tumors following treatment with either vehicle or PHA-73935. (D) Percent tumor size after treatment of LTL-362 xenografts with vehicle (dotted lines) or PHA-739358 30 mg/kg IP BID (solid lines) twice a day for 5 days relative to day 0. (E) Immunohistochemical staining for phosphorylated histone 3 (PO4-H3) in NCI-H660 or LNCaP tumors at day 4 of treatment with either vehicle or PHA-739358. (F) Immunohistochemistry for the neuroendocrine marker, synaptophysin, in NCI-H660 xenografts treated with vehicle (positive) and PHA-739358 (negative).
We then tested PHA-739358 in xenografts using two NEPC models: 1) NCI –H660 xenografts, which appear histologically similar to NEPC, are positive for neuroendocrine markers by IHC, and though harboring the TMPRSS2-ERG gene fusion (16), are negative for ERG protein (Fig. 4F and Supplementary Fig. 10–11), and 2) the LTL-352 xenograft, derived from patient with a history of metastatic PCA that progressed to NEPC after forty months of androgen deprivation therapy (17). When treated with PHA-739358, there was average tumor shrinkage of 50–87%, in both NCI-H660 and LTL-352 xenografts (P<0.001), compared to no effect in LNCaP xenografts (Fig. 4C, D,) and a cystostatic effect in VCaP xenografts (Supplementary Fig. 12) and was without significant toxicity (Supplementary Fig. 13). Phosphorylated histone 3 expression was significantly inhibited in the treated NCI-H660 xenografts (indicating on-target drug effect) and not in the LNCaP xenografts (Fig. 4E). Notably, SYP expression was also completely suppressed in the treated NCI-H660 xenografts (Fig. 4F), again supporting a role of Aurora kinase in modulating the neuroendocrine phenotype.
Discussion
Although de novo NEPC are considered uncommon and represents only 0.5–2% of all prostate cancers, focal neuroendocrine differentiation is present 10–100% of localized prostate adenocarcinomas and increases with disease progression (18–19). Autopsy studies suggest that a subset of patients with prostate cancer die from pure AR- negative NEPC(20–21) but this incidence may be under-recognized. Patients are not typically biopsied late in the stages of PCA to evaluate for NEPC progression, but can be suspected in patients with progressive disease despite a normal or modestly elevated PSA and elevated serum markers of neuroendocrine differentiation (i.e., chromogranin A or NSE)(1). Serum markers rise and the amount of neuroendocrine differentiation in tumors increases with long-term androgen deprivation (18, 22). Depletion of androgen in cell culture also promotes neuroendocrine differentiation of LNCaP adenocarcinoma cells (23). These data suggest that with the recent introduction of new highly potent androgen deprivation therapies into the clinical arena, the incidence of NEPC may escalate.
Although histologically similar, NEPC differs from small cell carcinomas of other primary sites in that there is evolution from PCA, as suggested by concordance of the TMPRSS2-ERG gene rearrangement and other molecular abnormalities (such TP53 mutations) in mixed tumors (24). Although PCA and NEPC can co-exist within the same tumor foci, we found dramatic gene expression and copy number differences between them. It is possible that a few key mutations occur that drive and select for the neuroendocrine phenotype and a number of passenger alterations follow as a result of the phenotype. The molecular events associated with this transformation have not been well defined.
We found AURKA and MYCN to be overexpressed and amplified in NEPC, and provide evidence that they functionally cooperate and can induce a neuroendocrine phenotype in prostate cells. AURKA is best known for its role in mitosis, but its interaction with the oncogene N-myc has been described in neuroblastoma (10) and now NEPC, and may play a role in other neuroendocrine carcinomas. MYCN is most commonly amplified in neuroblastoma, although has been reported in other central nervous system tumors as well as up to 40% of small cell lung cancers (25). We show for the first time that MYCN is abnormally expressed and amplified in NEPC and in 5% of PCA, and is involved in neuroendocrine differentiation and in stabilizing AURKA.
Based on these results, it remains unclear why MYCN and AURKA are co-amplified in nearly all PCA and NEPC, despite their location on separate chromosomes (2p24 and 20q13, respectively). In neuroblastoma, a positive feedback loop has been described in which AURKA also induces MYCN(10), and perhaps it is cooperation of these two lesions that drives the phenotype. Notably, AURKA and MYCN are also overexpressed and amplified in approximately 5% of PCA, which may represent a high-risk population that could benefit from early intervention. We did not see any association with standard clinical parameters (Supplementary Fig. 14). Larger studies are needed to determine the prognostic role of AURKA and MYCN amplification in PCA.
Currently, there is no standard treatment for patients with NEPC, accounted for in part by its poor molecular characterization. Based on the biologic role of AURKA in NEPC, its interaction with MYCN, and the dramatic and preferential sensitivity of NEPC preclinical models to Aurora kinase inhibition, we propose that Aurora kinase inhibitors may potentially benefit patients with similar molecular alterations. Their potential therapeutic role should be further explored in this subset of patients as well as other tumors with AURKA and MYCN alterations, either alone or in combination with cytotoxic chemotherapy. Although a recent Phase II trial evaluating PHA- 739358 for patients with castration resistant prostate cancer failed to achieve its primary endpoint of PSA-response (26), we propose that it was an unselected population, and that PSA may not be an ideal trial endpoint (since NEPC does not secrete PSA). As new targeted agents are being introduced into the clinical arena, patient selection based on molecular subtyping is essential as we transition into an era of personalized oncology.
Methods
Description of the clinical cohort
All tissue samples were collected as part of an Institutional Review Board (IRB) approved protocol at Weill Cornell Medical College (WCMC). Deidentified frozen NEPCs were obtained from WCMC (tumor metastases obtained from lung, soft tissue, and spinal cord metastases), University of Michigan rapid autopsy program (metastases), Henri Mondor Hospital (prostatectomy case), University of Pittsburgh (biobank program), and University of British Columbia (metastatic tumor passaged as second generation xenograft). Additional formalin fixed- paraffin embedded (FFPE) NEPC tumors for validation studies were obtained from WCMC, University of Tubingen, and University of Pittsburgh. Frozen and FFPE localized PCA and benign prostate tissues were collected at time of radical prostatectomy at WCMC. All cases were reviewed by the study’s pathologist (M.A.R.) and high density tumor foci with less than 10% stroma were selected for RNA and DNA extraction. Benign prostate samples were selected from blocks containing no tumor tissue, in order to minimize contamination. Clinical features of the frozen NEPC and PCA tumor cases are summarized in Supplementary Methods.
RNA-Sequencing (RNA-Seq) and Copy Number Assessment
The complete transcriptomes of seven NEPC tumors, 30 prostate adenocarcinomas, 6 benign prostatic epithelial samples, and 6 prostate cell lines were sequenced on an Illumina GA II Sequencer. Paired- end sequencing was performed, reads were mapped to human genome (hg18) using ELAND alignment software. Gene expression was quantified using RSEQtools (27). A full description of the PCA cases was reported in Pflueger et al(28). Tumor DNA together with paired blood DNA was extracted from high density foci and areas of genomic gain and loss were assessed using the Affymetrix 6.0 SNP array platform. For complete description of methodology, see Supplementary Methods.
Immunohistochemistry (IHC) and Fluorescent In Situ Hybridization (FISH)
Validation studies were performed on 169 primary PCA, 37 NEPC, 22 benign formalin fixed, paraffin embedded prostate tissue samples using IHC for protein expression and FISH for gene amplification or ERG gene rearrangement. For reagents used and detailed methods, see Supplementary Information. IHC was considered positive if >1% of tumor cells displayed immunoreactivity in cell cytoplasm (Aurora kinase A) or nucleus (Aurora kinase B, ERG). ERG rearrangement was assessed using dual-color break-apart interphase FISH assay as described previously3,17. In cases where FISH did not work, the TMPRSS2-ERG gene fusion was evaluated using reverse transcriptase PCR to screen for fusion transcript expression (as described in Tomlins et al. 2005)(3).
Statistical Analysis
Wilcoxon test was applied for mRNA differential analysis, followed by Benjamini-Hochberg correction for multiple hypothesis testing. Pearson correlation and Fisher Exact test were implemented for gene-gene expression correlation and genomic aberration association analysis, respectively. T test method was used to determine differences in tumor volumes in xenograft studies, with criteria for significance <0.05.
Transfection, Quantitiative PCR, Immunoblot Analysis
Functional studies were performed using NCI-H660 cell line as a model of NEPC, and VCaP and LNCaP as models of PCA. All cell lines were purchased from ATCC (Manassas, VA), and the 293FT cell line was purchased from Invitrogen (Carlsbad, CA) and maintained according to the manufacturers’ protocols. A complete description of the materials and methods is provided in the Supplementary Methods.
Drug Treatment
PHA-739358 was obtained from Nerviano Medical Sciences (Milan, Italy) in powder form (MW 414.36). Prostate cell lines for this study were obtained from the American Type Culture Collection (Manassas, VA). RWPE (20 ×103 per well), NCI-H660 (20 × 103), DU145 (5 ×103 per well), VCaP (20 ×103 per well) and LNCaP (1.5 ×104 per well) cells were seeded on 96-well-tissue culture plates. At 24 hours, cell lines were treated with vehicle (0.5% DMSO) or escalating doses of PHA-739358 (5nM, 50 nM, 100nM, 500 nM, 1uM, 5uM). At 48hr, 72h, 96h, and 120h, viability was assessed by performing WST-1 assay (Roche, Indianapolis, IN) reading absorbance at 450 nm according to the manufacturer’s instructions. Xenografts were prepared by injection of 1 million NCI-H660 cells, VCaP cells, or LNCaP cells into NU/J mice (Jackson Laboratories, Bar Harbor, Maine). L T L-362 xenografts were established at BC Cancer Center Living Tumor Laboratory as previously described(17). 3×3×2mm2 tissue fragments were subcutaneously engrafted into fourteen 6–8 week old NOD/SCID mice. All xenograft tumors were allowed to grow to an average tumor weight of 100 mm3. 20 LNCaP, 20 VCaP, 40 NCI-H660, 14 LTL-362 mice were randomized to treatment with PHA-739358 at 30 mg/kg intraperitoneal dosing on days 1–5 or vehicle. Body weight, tumor volume based on caliper measurements (0.5236 × length × width) and luciferase imaging (Supplementary Methods) were performed every 3 or 4 days after treatment. Mice were sacrificed on day 17, and tumors were evaluated for weight, gross pathology, histology, and IHC. Three tumors were processed during treatment (on day 4) to evaluate for phosphorylated histone 3 expression by IHC.
Additional Methods
Detailed methodology is described in the Supplementary Methods.
Supplementary Material
Acknowledgements
We thank Naoki Kitabayashi, Juan-Miguel Mosquera, Isabelita Vengco, Derek Oldridge, and Terry Vuong for their technical assistance and Nerviano Medical Sciences for providing PHA-739358.
Grant Support: This study was supported by the Ann and William Bresnan Foundation, Prostate Cancer Foundation (PCF), PCF Young Investigator Award (H.B.), Department of Defense New Investigator Award (D.R.), RO1-CA116337 (M.A.R.), RO1-125612 (M.A.R.), 2 P50 CA69568 (K.P.).
Footnotes
Conflicts of Interest: None
Significance: We report the largest in depth molecular analysis of NEPC and new insight into molecular events involved in prostate cancer progression.
References
- 1.Palmgren JS, Karavadia SS, Wakefield MR. Unusual and underappreciated: small cell carcinoma of the prostate. Semin Oncol. 2007;34:22–29. doi: 10.1053/j.seminoncol.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 2.Wang W, Epstein JI. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am J Surg Pathol. 2008;32:65–71. doi: 10.1097/PAS.0b013e318058a96b. [DOI] [PubMed] [Google Scholar]
- 3.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 4.Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, Chaux A, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol. 2011 doi: 10.1038/modpathol.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mosquera JM, Mehra R, Regan MM, Perner S, Genega EM, Bueti G, et al. Prevalence of TMPRSS2-ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clin Cancer Res. 2009;15:4706–4711. doi: 10.1158/1078-0432.CCR-08-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheble VJ, Braun M, Beroukhim R, Mermel CH, Ruiz C, Wilbertz T, et al. ERG rearrangement is specific to prostate cancer and does not occur in any other common tumor. Mod Pathol. 2010;23:1061–1067. doi: 10.1038/modpathol.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williamson SR, Zhang S, Yao JL, Huang J, Lopez-Beltran A, Shen S, et al. ERG-TMPRSS2 rearrangement is shared by concurrent prostatic adenocarcinoma and prostatic small cell carcinoma and absent in small cell carcinoma of the urinary bladder: evidence supporting monoclonal origin. Mod Pathol. 2011 doi: 10.1038/modpathol.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 9.Zhou H, Kuang J, Zhong L, Kuo WL, Gray JW, Sahin A, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 10.Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 11.Strieder V, Lutz W. Regulation of N-myc expression in development and disease. Cancer Lett. 2002;180:107–119. doi: 10.1016/s0304-3835(02)00020-4. [DOI] [PubMed] [Google Scholar]
- 12.Hsu JY, Sun ZW, Li X, Reuben M, Tatchell K, Bishop DK, et al. Mitotic phosphorylation of histone H3 is governed by Ipl1/aurora kinase and Glc7/PP1 phosphatase in budding yeast and nematodes. Cell. 2000;102:279–291. doi: 10.1016/s0092-8674(00)00034-9. [DOI] [PubMed] [Google Scholar]
- 13.Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A, et al. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proc Natl Acad Sci U S A. 2005;102:731–736. doi: 10.1073/pnas.0405495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carney DN, Gazdar AF, Bepler G, Guccion JG, Marangos PJ, Moody TW, et al. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res. 1985;45:2913–2923. [PubMed] [Google Scholar]
- 15.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, et al. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57:205–225. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 16.Setlur SR, Mertz KD, Hoshida Y, Demichelis F, Lupien M, Perner S, et al. Estrogen-dependent signaling in a molecularly distinct subclass of aggressive prostate cancer. J Natl Cancer Inst. 2008;100:815–825. doi: 10.1093/jnci/djn150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tung WL, Wang Y, Gout PW, Liu DM, Gleave M. Use of irinotecan for treatment of small cell carcinoma of the prostate. Prostate. 2010 doi: 10.1002/pros.21283. [DOI] [PubMed] [Google Scholar]
- 18.Hirano D, Okada Y, Minei S, Takimoto Y, Nemoto N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol. 2004;45:586–592. doi: 10.1016/j.eururo.2003.11.032. discussion 92. [DOI] [PubMed] [Google Scholar]
- 19.Berruti A, Mosca A, Porpiglia F, Bollito E, Tucci M, Vana F, et al. Chromogranin A expression in patients with hormone naive prostate cancer predicts the development of hormone refractory disease. J Urol. 2007;178:838–843. doi: 10.1016/j.juro.2007.05.018. quiz 1129. [DOI] [PubMed] [Google Scholar]
- 20.Brawn PN, Speights VO. The dedifferentiation of metastatic prostate carcinoma. Br J Cancer. 1989;59:85–88. doi: 10.1038/bjc.1989.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vashchenko N, Abrahamsson PA. Neuroendocrine differentiation in prostate cancer: implications for new treatment modalities. Eur Urol. 2005;47:147–155. doi: 10.1016/j.eururo.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Yuan TC, Veeramani S, Lin MF. Neuroendocrine-like prostate cancer cells: neuroendocrine transdifferentiation of prostate adenocarcinoma cells. Endocr Relat Cancer. 2007;14:531–547. doi: 10.1677/ERC-07-0061. [DOI] [PubMed] [Google Scholar]
- 24.Hansel DE, Nakayama M, Luo J, Abukhdeir AM, Park BH, Bieberich CJ, et al. Shared TP53 gene mutation in morphologically and phenotypically distinct concurrent primary small cell neuroendocrine carcinoma and adenocarcinoma of the prostate. Prostate. 2009;69:603–609. doi: 10.1002/pros.20910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meulenbeld HJ, Bleuse JP, Vinci EM, Raymond E, Vitali G, Santoro A, et al. Randomized Phase II study of danusertib in 2nd line metastatic castration -resistant prostate cancer. ASCO. 2011;2011 doi: 10.1111/j.1464-410X.2012.11404.x. [DOI] [PubMed] [Google Scholar]
- 27.Gerstein M. RSEQtools. [cited; Available from: http://rseqtools.gersteinlab.org.
- 28.Pflueger D, Terry S, Sboner A, Habegger L, Esgueva R, Lin PC, et al. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2011;21:56–67. doi: 10.1101/gr.110684.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.