

Abstract
Aims
This study aimed to determine the role of the renin-angiotensin system (RAS) in high salt (HS) diet-induced left ventricular hypertrophy (LVH).
Methods and Results
Swiss mice were subjected to regular salt (RS) diet (0.6% NaCl), HS diet (4% NaCl) and HS plus irbesartan (50 mg/kg/day) or ramipril (1 mg/kg/day). After 8 weeks, arterial pressure was similar in all groups and similar to baseline, whereas LV/body weight ratio was higher in HS mice than in RS mice (p<0.005). There were also significant increases in collagen density, ACE activity, AT1 receptor density and extracellular signal-regulated kinase (ERK1/2) phosphorylation in the left ventricle. Interestingly, increases in wall thickness and ERK1 phosphorylation were more marked in the septum than in the rest of the left ventricle. Irbesartan or ramipril treatment prevented LVH and the increase in ERK phosphorylation and reduced collagen content and AT1 upregulation but upregulated AT2 receptors.
Conclusions
In normal mice, HS diet induces septum-predominant LVH and fibrosis through activation of the cardiac RAS-ERK pathway, which can be blocked by irbesartan or ramipril, indicating a key role of the cardiac RAS in HS diet-induced LVH.
Keywords: Angiotensin II Type 1 Receptor Blockers; metabolism; pharmacology; Angiotensin-Converting Enzyme Inhibitors; pharmacology; Animals; Autoradiography; Biphenyl Compounds; pharmacology; Blotting, Western; Female; Fibrosis; Heart Ventricles; pathology; ultrasonography; Hypertrophy, Left Ventricular; physiopathology; Immunohistochemistry; JNK Mitogen-Activated Protein Kinases; metabolism; Mice; Mitogen-Activated Protein Kinases; pharmacology; Phosphorylation; Ramipril; pharmacology; Renin; metabolism; Renin-Angiotensin System; drug effects; physiology; Sodium, Dietary; administration & dosage; Tetrazoles; pharmacology; p38 Mitogen-Activated Protein Kinases; metabolism
Keywords: High salt diet, Left ventricular hypertrophy, Blood pressure, Renin angiotensin system, Mitogen-activated protein kinases, Mice
Introduction
Left ventricular hypertrophy (LVH) contributes to cardiovascular morbidity and mortality. High salt (HS) intake affects not only arterial pressure but also left ventricular mass and cardiac fibrosis in hypertensive rats [1–3] and may participate in LVH in hypertensive patients [5]. Interestingly, in normal rats, HS intake has been shown to induce LVH [1, 3, 5–7] and interstitial fibrosis [3, 6] with an increased blood pressure [3] or without changes in blood pressure [1, 5–7] or sympathetic activity [1, 5]. However, the mechanisms involved in HS intake-induced LVH in normal animals, especially the role of the renin-angiotensin system (RAS) need to be clarified. It has been shown that during HS loading, a depletion of circulating RAS occurs [7–10] while myocardial angiotensin II increased in normal or Dahl salt-sensitive rats [8, 9]. Interestingly, during HS loading, angiotensin AT1 receptor mRNA and/or protein increased in the heart of normal rats [7, 8]. Inhibition of the RAS by losartan or perindopril reduced HS intake-induced LVH in Dahl salt-sensitive rats or partial renal ablation-induced hypertensive rats, independently of blood pressure change [2, 10]. These segmental data suggest that the cardiac RAS may be activated during HS intake. Therefore, to clarify the role of the cardiac RAS in HS diet-induced LVH and to evaluate the cellular signalling pathways involved, normal mice were subjected to different salt diets in the absence and presence of irbesartan (AT1 blocker) or ramipril (ACE inhibitor). LV posterior and septal wall thicknesses were measured using echocardiography. Blood pressure was followed in conscious mice and the cardiac RAS was assessed by measuring ACE activity and angiotensin II receptor binding capacity in the left ventricle. Since mitogen-activated protein kinases, including extracellular signal-regulated kinases (ERK), c-Jun NH(2)-terminal kinases (JNK) and p38, mediate angiotensin II-induced LVH [11], the activation or not of these signalling pathways in the heart may help to determine whether the cardiac RAS is activated during salt loading. For this purpose, the expression and activation (phosphorylation) of ERK1/2, JNK and p38 in the left ventricle were determined.
Methods
All animals were handled according to the Guidelines for the care and use of laboratory animals published by the US NIH (NIH publication No. 86-23, revised 1985) and to the animal protection law of France. Swiss mice (male; 8 weeks of age; n=12 per group; Charles River Laboratories France) were subjected to either regular salt diet (0.6% NaCl, RS), high salt diet (4% NaCl, HS), HS diet plus ramipril (HS+Ramipril), or HS diet plus irbesartan (HS+Irbesartan) for 8 weeks. Salt was incorporated into the chow by the manufacturer. Based on previous studies in mice [12, 13], doses of 1 mg/kg/day of ramipril and 50 mg/kg/day of irbesartan were used. Drugs were added to the drinking water. Water was changed 3 times per week. Ramipril and irbesartan concentrations were adjusted to achieve an intake of ~1 mg/kg/day and ~50 mg/kg/day respectively, based on the measured water intakes of animals in each group. After completing in-vivo experiments, hearts were excised from the animals. In six mice from each group, the heart was frozen in liquid nitrogen cooled-isopentane and stored at −80°C for subsequent histological and receptor binding studies. In the six other mice from each group, the atria and right ventricle were removed and the left ventricle (LV) was dissected into the septum and free wall according to the natural limit of the right ventricle. Left ventricular, right ventricular and total atrial weights were measured and tissues rapidly frozen in liquid nitrogen for tissue assays.
Blood pressure measurements
Systolic blood pressure (SBP) was monitored by the tail cuff technique in all mice in the conscious state at baseline, 4 and 8 weeks with the aid of a computerized system. At each time point, 10–15 values were averaged for each mouse.
Echocardiography
Echocardiographic studies were performed at baseline, and at 4 and 8 weeks in mice anaesthetized with a mixture of ketamine (65 μg/g), acepromazine (2 μg/g), and xylazine (13 μg/g). Mice were positioned prone on a warmed bag containing physiological solution and a 9-Mhz transducer (VINGMED CFM750 echocardiograph) was placed under the bag. The parasternal short-axis view at the level of two papillary muscles was used to position the M-mode cursor perpendicular to the LV posterior and septal walls. For each study, 3–5 consecutive cardiac cycles were analyzed. Measured parameters were: heart rate; LV diameter at the end of diastole and systole; LV fractional shortening defined as (LV end-diastolic diameter − LV end-systolic diameter)/LV end-diastolic diameter ×100; LV posterior wall thickness and septal wall thickness at the end of diastole and systole
Left ventricular fibrosis analysis
Hearts were cut into 4-μm sections which were mounted onto slides and stained with picosirius red F3BA (0.1% solution in saturated aqueous picric acid) to colour collagen. Interstitial fibrosis in the LV free wall and the septum was measured using Image-Pro Plus 5.0 (Micromécanique, Evry, France) and results were expressed as a percentage of the total area. In each heart, 6 to 8 images were analyzed.
Cardiac angiotensin converting enzyme activity
Cardiac ACE activity was measured as previously described [14]. Briefly, LV samples were homogenized in Tris · HCl (pH 7.4) and centrifuged at 1000g. The collected supernatant was then centrifuged at 15000g. The resulting pellet was dissolved in 1 ml Tris · HCl with 8.5 mM 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate and incubated at 37°C in the presence of z-Phe-His-Leu-OH (Bachem) and Na-Tris · HCl (50 mM HCl, 1% NaCl, pH 8). A negative control was included for each sample by addition of enalaprilat (10−5 M). The reaction was stopped by the addition of 10% trichloroacetic acid and centrifugation. The supernatant was then incubated for 10 min at 37°C after addition of NaOH (0.28N) and o-phthaldialdehyde (Sigma) and the reaction was stopped by HCl (2N). Fluorometric measurements were done at 365 nm excitation and 500 nm emission (F-2000 Fluorescence Spectrophotometer, Hitachi). ACE activity was normalized to total protein in the supernatant (Bio-Rad protein assay).
In-situ autoradiographic quantitative receptor binding assay
Angiotensin II binding studies were performed in transverse heart cryosections (10 μm) as previously described [15]. Consecutive sections were incubated for 15 min at room temperature in a buffer (pH 7.4) containing 0.3 mM bacitracin, 10 mM sodium phosphate, 150 mM NaCl, 1 M EDTA and 0.2% proteinase-free bovine serum albumin to remove endogenous bound ligand. Thereafter, the sections were transferred and incubated for 30 minutes in the same mixture without the bacitracin, in the presence or absence, of losartan (AT1 blocker, 0.5 μM, Merck Sharp & Dohme-Chibret), PD-123319 (AT2 blocker, 0.5 μM, Sigma) or angiotensin II (0.5 μM, Sigma). Finally, the sections were incubated for 90 min in the presence of 32 pM (3-[125I]iodotyrosyl4,Sar1-Ile8)Angiotensin II (2000 Ci/mmol; Amersham) with or without losartan, PD-123319, or angiotensin II (same concentrations as above) to identify AT2, AT1, and angiotensin II specific binding sites, respectively. After being washed and dried, the sections were exposed to imaging plates with a range of known amounts of (3-[125I]iodotyrosyl4,Sar1-Ile8)Angiotensin II and were quantified using the Bio-Imaging Analysis system (Fuji MacBAS 1000, Fuji Medical Systems). Pixels accumulated within the LV section were normalized for surface area and converted to fmol/mm2 by direct density comparison with the (3-[125I]iodotyrosyl4,Sar1-Ile8)Angiotensin II calibration curve.
Cardiac ERK, JNK and p38 expressions by Western blot analysis
The frozen LV free wall and septum were separately pulverized, homogenized and centrifuged. Sample proteins (40 μg/lane) contained in the supernatant were size-fractionated by SDS-polyacrylamide gels (10 %, 3h) and transferred to PVDF membranes. A prestained protein-weight marker and a control sample (used for normalization) were loaded onto each gel. After blocking non specific binding sites, membranes were probed with specific antibodies to detect total and phosphorylation levels of ERKs, JNKs and p38 (Santa Cruz Biotechnology, 1:5000 for all total forms, 1:2500, 1:1000 and 1:1000 respectively for ERK, JNK and p38 phosphorylated forms, diluted in 2% milk TTBS, 1.5 h). Subsequently, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit IgG (1:5000, 1h). Specific immunoreactive proteins were detected by enhanced chemiluminescence (Amersham) and quantified densitometrically. ERKs, JNKs or p38 phosphorylation level was expressed as the ratio of the phosphorylated form to the total form.
Statistical analysis
Statistical analyses were performed using StatView software (v5, Abacus Concepts Inc). Results are expressed as mean±SEM. One-way analysis of variance for repeated measures was used for intra-group studies. Two-way analysis of variance was performed for comparisons between groups at the same stage. If there was a significant difference, Student-Newman-Keuls multiple comparison test was used to identify differences between means. When only two means were compared, an appropriate Student’s t-test was used. A p< 0.05 was considered significant.
Results
At baseline and at 8 weeks, body weight was similar between groups. No difference was detected among groups for food intake (Table 1). In contrast, water intake was nearly doubled in HS, HS/Irbesartan and HS/Ramipril mice compared with LS mice (Table 1). In the preliminary study, we verified the effect of HS diet on blood sodium, creatinine and protein concentrations and did not observe any significant difference between HS mice (serum sodium: 151±2 mmol/l; serum creatinine: 31±2 mmol/l; serum protein: 42±2 g/l) and LS mice (serum sodium: 149±3 mmol/l; serum creatinine: 32±3 mmol/l; serum protein: 42±2 g/l).
Table 1.
Arterial pressure, body weight, heart weight, heart rate, LV diameter and fractional shortening measured by echocardiography, plasma renin activity and cardiac ACE activity
| RS (n=12) | HS (n=12) | HS+Irbesartan (n=12) | HS+Ramipril (n=12) | |
|---|---|---|---|---|
| Systolic blood pressure, mmHg | ||||
| Baseline | 125±3 | 123±4 | 124±2 | 124±2 |
| 8 weeks | 127±2 | 126±3 | 125±2 | 122±1 |
| Body weight, g | ||||
| Baseline | 33.9±0.8 | 34.8±0.6 | 33.4±0.6 | 33.2±0.6 |
| 8 weeks | 42.6±1.3‡ | 42.5±1.0‡ | 42.1±1.0‡ | 41.9±0.8‡ |
| Food intake, g/24h | 6.4±0.2 | 6.7±0.3 | 6.3±0.2 | 6.2±0.3 |
| Water intake, ml/24h | 6.0±0.8 | 10.9±0.5 * | 11.2±0.4 * | 12.5±1.2 * |
| Heart weight, mg | 188±6 | 218±5 * | 178±3† | 184±5† |
| Heart/body weight ratio | 4.3±0.1 | 5.3±0.1 * | 4.2±0.1† | 4.4±0.2† |
| LV weight #, mg | 135±4 | 158±5 * | 128±2† | 132±6† |
| LV/body weight ratio # | 2.9±0.1 | 3.6±0.2 * | 3.1±0.1† | 3.1±0.2† |
| Heart rate, beats/min | ||||
| Baseline | 284±16 | 273±15 | 294±12 | 291±12 |
| 8 weeks | 257±13 | 270±13 | 267±13 | 270±10 |
| LVEDD, mm | ||||
| Baseline | 4.4±0.1 | 4.4±0.1 | 4.4±0.1 | 4.4±0.1 |
| 8 weeks | 4.8±0.1‡ | 4.8±0.1‡ | 4.7±0.1‡ | 4.7±0.1‡ |
| Fractional shortening, % | ||||
| Baseline | 34±1 | 33±1 | 30±3 | 33±2 |
| 8 weeks | 35±6 | 34±2 | 34±1 | 32±2 |
| Cardiac ACE activity #, nmol/mg protein | 3.4±0.3 | 5.6±0.3* | 5.5±0.2* | 3.9±0.2† |
RS: regular salt diet (0.6% NaCL); HS: high salt diet (4% NaCl); HS+Irbesartan: HS diet plus irbesartan (50 mg/kg/day); HS+Ramipril: HS diet plus ramipril (1 mg/kg/day); LV: left ventricle; LVEDD: LV end-diastolic diameter.
LV weight and LV/Body weight ratio were measured in 6 mice of each group after 8 weeks of salt diets and treatments.
cardiac ACE activity were measured in 5 mice in each group. There was no difference in systolic blood pressure between groups at baseline and after 8 weeks of salt diet and treatment.
p<0.05 versus RS mice,
p<0.05 versus HS and
p<0.05 versus corresponding baseline.
Blood pressure and cardiac pathology
At baseline, there was no difference between groups with regard to SBP. Eight weeks of HS diet alone or with irbesartan or ramipril did not affect SBP (Table 1). Similar results were also observed after 4 weeks of salt diet and treatment (data not shown).
Compared with RS mice, heart weight and heart/body weight ratio were significantly increased in HS mice. This was essentially due to increases in LV mass (+17±4%, p<0.001) and LV/body weight ratio (+23±6%, p<0.005). These increases were absent in HS+Irbesartan and HS+Ramipril mice (Table 1).
HS diet significantly increased interstitial fibrosis in the LV free wall and in the septum (Fig. 1). Treatment with irbesartan or ramipril markedly reduced the collagen deposition.
Fig. 1.
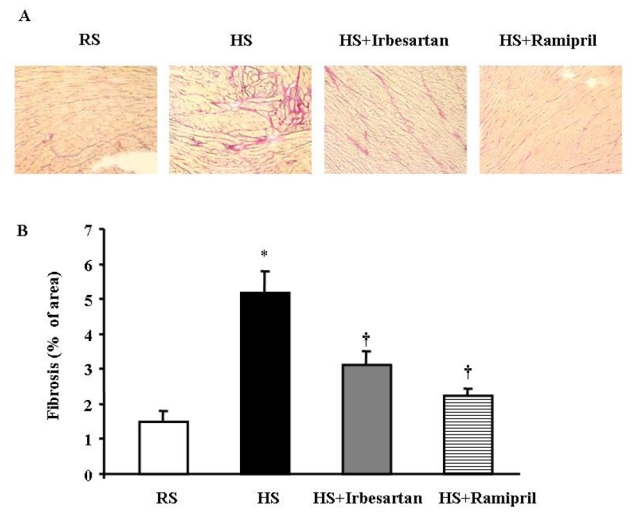
Eight weeks of high salt diet increased interstitial fibrosis while irbesartan or ramipril treatment limited interstitial fibrosis. (A) Representative left ventricular (LV) tissues stained with Picrosirius red (presented at a magnification of X 10). (B) Quantitative morphometric analysis of interstitial fibrosis. (n=5 in each group). * p<0.05 versus RS mice and † p<0.05 versus HS mice.
Left ventricular dimensional parameters and left ventricular function
Heart rate was similar for all groups at baseline and after 8 weeks (Table 1). LV dimensional and functional parameters were similar at baseline for all groups. After 8 weeks of salt diet, LV end-diastolic diameter and fractional shortening were similar in all groups (Table 1). In contrast to RS mice, HS mice showed a slight increase in LV posterior end-diastolic wall thickness but a marked and significant increase in septal end-diastolic wall thickness (Fig. 2).
Fig. 2.
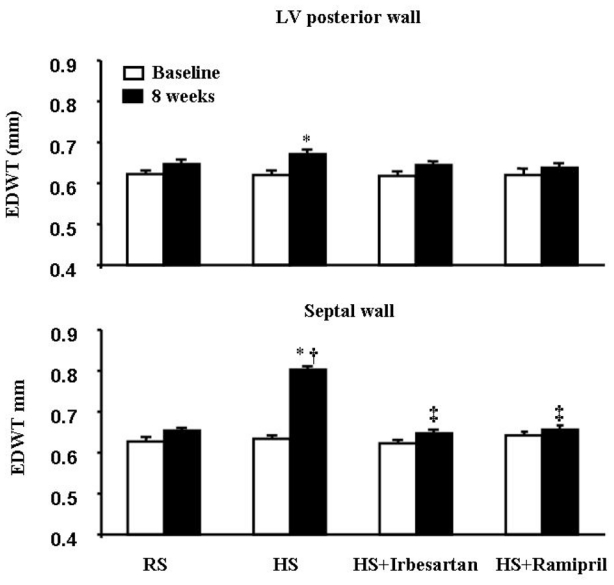
High salt (HS) diet slightly increased LV end-diastolic posterior wall thickness but markedly increased end-diastolic ventricular septal wall thickness. These changes were completely prevented by either irbesartan or ramipril treatment. n=12 in each group. * p<0.05 versus baseline, † p<0.05 versus RS mice and ‡ p<0.05 versus HS mice after 8 weeks.
Treatment with irbesartan or ramipril prevented the increases in LV posterior end-diastolic wall thickness and septal end-diastolic wall thickness (Fig. 2)
At 8 weeks, calculated LV mass using echocardiography showed that LV end-diastolic diameter, LV posterior end-diastolic wall thickness and septal end-diastolic wall thickness was closely correlated with LV mass at autopsy (y= 0.94 x+0.50, r=0.852, p<0.0001).
Cardiac angiotensin converting enzyme activity
ACE activity in the left ventricle was significantly higher in HS and HS+Irbesartan mice than in RS mice, indicating an activated cardiac ACE during HS diet, which was not affected by irbesartan. LV ACE activity in HS+Ramipril mice was similar to that in RS mice, indicating an effective ACE inhibition by ramipril (Table 1).
Angiotensin II receptors in the left ventricle
In-situ binding assay showed that LV density of AT1 receptors in HS mice was 2-fold higher than in RS mice (Fig. 3). In HS+Irbesartan mice, LV AT1 density was significantly lower than that of HS mice. In HS+Ramipril mice, LV AT1 density was significantly lower than that of HS mice and HS+irbesartan mice.
Fig. 3.
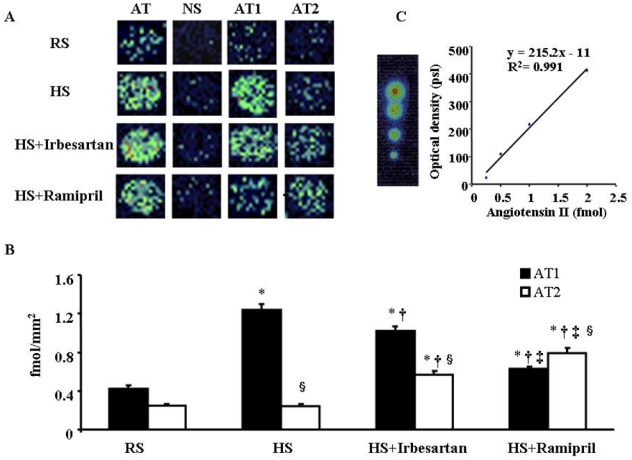
(3-[125I]iodotyrosyl4,Sar1-Ile8)Angiotensin II binding in LV sections. A: Pseudocolour images of LV sections from mice subjected to regular salt (RS) or high salt (HS) diet and HS mice treated with irbesartan (HS+Irbesartan) or ramipril (HS+Ramipril) for 8 weeks. LV sections were incubated with (3-[125I]iodotyrosyl4,Sar1-Ile8)Angiotensin II alone (AT: total angiotensin II binding) or in the presence of unlabelled angiotensin II, PD-123319, or losartan which showed the nonspecific (NS), AT1 and AT2 binding, respectively. B: Mean values of AT1 and AT2 densities in the left ventricle (n=5 in each group). C: Typical calibration curve and corresponding quantification. * p<0.05 versus RS mice, † p<0.05 versus HS mice, ‡ p<0.05 versus HS+Irbesartan and § p<0.05 versus corresponding AT1 value.
In RS mice, LV density of AT2 receptors was lower than AT1 density (Fig. 3). HS did not change AT2 density while treatment with irbesartan or ramipril significantly increased AT2 density in the left ventricle, which was more pronounced in ramipril-treated than in irbesartan-treated mice.
ERK, JNK and p38 expressions
ERK1/2 phosphorylation in the LV tissue increased significantly after 8 weeks of HS diet, as indicated by > 50% increases in phosphorylated ERK1 and ERK2 (Fig. 4). The increase in phosphorylated ERK1 was more marked in the septum than in the rest of the LV wall. Treatment with irbesartan or ramipril completely prevented ERK1/2 activation. The total ERK1/2 protein levels were similar whatever the animal group.
Fig. 4.
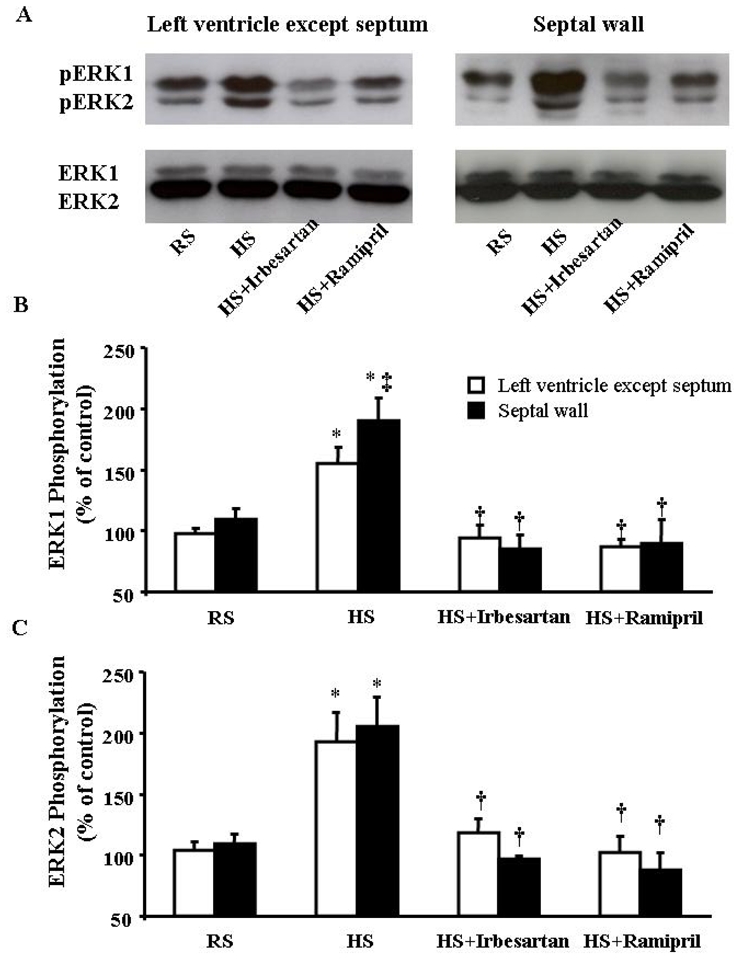
A. Typical examples illustrate the expression of phosphorylated ERK1/2 (pERK1 and pERK2) and total ERKs (ERK1 and ERK2) proteins in the left ventricle (except septum) and septal wall in response to regular salt (RS), high salt (HS) diet and treatment with irbesartan (HS+Irbesartan) or ramipril (HS+Ramipril). B and C. ERK1/2 phosphorylation levels were calculated as % of total ERK1/2. HS diet for 8 weeks significantly increased the phosphorylation of ERK1/2. The increase in pERK1 was greater in the septum than in the left ventricle excluding the septum. n=6 in each group. * p<0.05 versus RS mice; † p<0.05 versus HS mice and ‡ p<0.05 versus LV wall.
HS diet did not modify the expression and phosphorylation of JNK or p38 (Table 2). Neither irbesartan nor ramipril modified the expression and phosphorylation of these proteins.
Table 2.
Phosphorylation of JNKs and p38 in different regions of the left ventricle
| RS (n=6) | HS (n=6) | HS+Irbesartan (n=6) | HS+Ramipril (n=6) | |
|---|---|---|---|---|
| JNK1 phosphorylation (% ) | ||||
| LV except septum | 105±4 | 108±8 | 111±6 | 105±7 |
| Septum | 98±6 | 105±7 | 108±6 | 100±6 |
| JNK2 phosphorylation (% ) | ||||
| LV except septum | 98±3 | 105±8 | 98±6 | 89±7 |
| Septum | 103±7 | 108±5 | 116±9 | 102±8 |
| p38 phosphorylation (% ) | ||||
| LV except septum | 96±2 | 98±5 | 99±3 | 99±3 |
| Septum | 102±5 | 96±5 | 107±4 | 107±3 |
Phosphorylation was calculated as the ratio of phosphorylated form to total form x100.
Discussion
The present study confirms that HS diet in normal mice induces LV hypertrophy and fibrosis, independently of arterial pressure. However, two novel findings were observed: 1) HS diet-induced LVH in normal mice mainly occurs at the interventricular septum and 2) this septum-predominant LVH is related to the activation of cardiac RAS, which can be prevented by an ACE inhibitor or AT1 receptor blocker.
In normal rodents, most studies have shown no effect of HS intake on blood pressure [1, 5–7]. Similarly, we did not observe any significant effect of 4% salt diet on blood pressure. This can be explained by a physiological regulation of water-electrolyte homeostasis through increasing diuresis in response to HS loading. In accordance with a previous study [12], we did not observe any effect of ramipril and irbesartan on blood pressure. This is in line with a depressed circulating RAS during salt loading [7–10].
Our data showing that HS diet-induced ventricular hypertrophy predominantly occurred in the ventricular septum is particularly interesting because previous studies in humans have shown a high prevalence of disproportionate ventricular septal hypertrophy in different pathologies (aortic valvular disease, pulmonary or essential hypertension) [16–18]. The reasons why the interventricular septum is more susceptible to developing hypertrophy remain unclear. This may be related to its embryonic development during which three components participate in its formation [19] or to its relatively flatter contour compared with the LV free wall, which results in a greater systolic stress being applied to it [20].
Previous studies have given seemingly contradictory messages about the RAS in HS intake-induced LVH. In hypertensive rats fed with DOCA+salt, inhibition of the RAS did not affect LVH [21, 22] while in rats fed with HS alone, losartan or perindopril reduced LVH [2, 10]. Our data showing that both irbesartan and ramipril prevented HS diet-induced LVH indicate a role of the RAS in normal mice. Accordingly, we found an increase in cardiac ACE activity in HS mice, which persisted during irbesartan treatment but was inhibited by ramipril. We also found a 2-fold increased AT1 receptor binding capacity in the LV tissue of HS mice, which was partially attenuated by irbesartan or ramipril. In addition, the activation of ERK1/2 in the LV tissue of HS mice also supports an activated cardiac RAS because ERK1/2 mediates angiotensin II-induced LVH [11]. Overall, these data indicate that a HS diet activates the cardiac RAS to stimulate LVH and fibrosis.
Generally, AT2 receptors oppose the action of AT1 receptors and participate in the inhibitory effect of AT1 antagonism or ACE inhibition on hypertrophy and fibrosis through activation of kinins and intracellular cGMP signalling [23, 24]. Our study showed that irbesartan or ramipril treatment augmented AT2 density in the LV tissue. This might allow accumulated angiotensin during irbesartan treatment or angiotensin I during ramipril treatment to stimulate AT2 receptors to generate nitric oxide. However, since we did not examine the role of AT2 receptors, the significance of the AT2 receptor upregulation induced by irbesartan and ramipril needs further investigation.
This study did not examine the role of aldosterone in HS diet-induced LVH and fibrosis. However, a potential role of aldosterone should not be ignored because a HS diet can increase expression and activity of aldosterone synthase and increase aldosterone concentrations in rat hearts [7, 9] and aldosterone antagonism prevents HS loading-induced LVH and fibrosis [25]. Furthermore, angiotensin II can stimulate the adrenal gland to produce aldosterone while aldosterone combined with HS intake increases cardiac angiotensin AT1 receptor density [26]. All of these data indicate a complicated interaction between angiotensin and aldosterone in the renin-angiotensin-aldosterone system. Also, our data showing that irbesartan or ramipril completely prevented LVH but only partially prevented fibrosis may suggest a potential role of aldosterone in this setting.
The mechanism by which a HS diet induces the activation of the RAS has not been completely elucidated. It is known that HS intake depletes circulating angiotensin II, which upregulates angiotensin AT1 receptors. Unlike circulating angiotensin II, cardiac angiotensin II concentration is increased [8, 9] due to augmented cardiac ACE activity during HS loading, which stimulates AT1 receptors to initiate hypertrophic signalling pathways. Increased AT1 receptors can also be activated by mechanical stress [27] due to increased arterial pressure during nocturnal activity in animals. Another possible mechanism involved in the activation of the RAS by high salt may be related to the increased cardiac aldosterone concentration during HS loading [7, 8], which upregulates cardiac AT1 receptors [25].
Previous studies have demonstrated different roles for the three branches of mitogen-activated protein kinases. In transgenic mice, specific activation of JNK in the heart did not induce cardiac hypertrophy but caused lethal cardiomyopathy in juvenile animals [28] whereas specific activation of p38 in the heart induced heart failure with a thinned ventricular wall [29]. In contrast, specific activation of ERK1/2 in the heart caused a prominently hypertrophic response [30]. Accordingly, we found that a HS diet selectively activated ERKs, with a predominant ERK1 activation in the septum, but not JNK or p38. The absence of ERK1/2 activation and septum-predominant ventricular hypertrophy in irbesartan or ramipril-treated mice further supports a role of the AT1 receptor-ERK1/2 pathway in HS induced ventricular hypertrophy.
One limitation of our study is that cardiac angiotensin II levels were not measured. In addition, due to the inability of the technique used to finely distinguish regional changes in angiotensin AT1 receptors in small hearts, we could not determine LV regional changes in AT1 receptors.
In summary, this study shows that in normal mice, the first impact of HS intake on the cardiovascular system is a cardiac remodelling effect. This effect is related to selective upregulation of the cardiac RAS, which initiates activation of the mitogen-activated protein kinases cascade, especially the phosphorylation of ERK1/2. Blockade of the RAS at the ACE or AT1 receptor level inhibited ERK1/2 activation, and thereby septum-predominant ventricular hypertrophy and interstitial fibrosis. Since LVH in humans is associated with cardiovascular mortality and morbidity and septal hypertrophy is highly prevalent in different cardiovascular diseases, our data may be relevant in the clinical setting.
Acknowledgments
We thank Jane-Lise Samuel for her scientific support and helpful comments and Monique Philippe for her technical assistance. Irbesartan was a kind gift of Bristol-Myers Squibb and Sanofi-Synthelabo laboratories.
Funding
This work was supported by INSERM, the Université Paris-Est and the Société Française d’Hypertension Artérielle. This project was also supported by a grant from Bristol-Myers Squibb and Sanofi-Synthelabo laboratories.
Footnotes
Conflict of Interest
None declared
References
- 1.Frohlich ED, Chien Y, Sesoko S, Pegram BL. Relationship between dietary sodium intake, hemodynamics and cardiac mass in SHR and WKY rats. Am J Physiol. 1993;264:R30–R34. doi: 10.1152/ajpregu.1993.264.1.R30. [DOI] [PubMed] [Google Scholar]
- 2.De Simone G, Devereux RB, Camargo MJF, Wallerson DC, Sealey JE, Laragh JH. Reduction of development of left ventricular hypertrophy in salt-loaded Dahl salt-sensitive rats by angiotensin II receptor inhibition. Am J Hypertens. 1996;9:216–222. doi: 10.1016/0895-7061(95)00338-x. [DOI] [PubMed] [Google Scholar]
- 3.Yu HC, Burrell LM, Black MJ, Wu LL, Dilley RJ, Cooper ME, Johnston CI. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation. 1998;98:2621–2628. doi: 10.1161/01.cir.98.23.2621. [DOI] [PubMed] [Google Scholar]
- 4.Schmieder RE, Langenfeld MRW, Friedrich A, Schobel HP, Gatzka CD, Weihprecht H. Angiotensin II related to sodium excretion modulates left ventricular structure in human essential hypertension. Circulation. 1996;94:1304–1309. doi: 10.1161/01.cir.94.6.1304. [DOI] [PubMed] [Google Scholar]
- 5.Yuan BX, Leenen FH. Dietary sodium intake and left ventricular hypertrophy in normotensive rats. Am J Physiol. 1991;261:H1397–H1401. doi: 10.1152/ajpheart.1991.261.5.H1397. [DOI] [PubMed] [Google Scholar]
- 6.Bourraindeloup M, Adamy C, Candiani G, Cailleret M, Bourin MC, Badoual T, Su JB, Adubeiro S, Roudot-Thoraval F, Dubois-Rande JL, Hittinger L, Pecker F. N-acetylcysteine treatment normalizes serum tumor necrosis factor-alpha level and hinders the progression of cardiac injury in hypertensive rats. Circulation. 2004;110:2003–2009. doi: 10.1161/01.CIR.0000143630.14515.7C. [DOI] [PubMed] [Google Scholar]
- 7.Takeda Y, Yoneda T, Demura M, Miyamori I, Mabuchi H. Sodium-induced cardiac aldosterone synthesis causes cardiac hypertrophy. Endocrinology. 2000;141:1901–1904. doi: 10.1210/endo.141.5.7529. [DOI] [PubMed] [Google Scholar]
- 8.Ding Y, Lv J, Mao C, Zhang H, Wang A, Zhu L, Zhu H, Xu Z. High-salt diet during pregnancy and angiotensin-related cardiac changes. J Hypertens. doi: 10.1097/HJH.0b013e328337da8f. Published online ahead of print 3 March 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayorh MA, Ganafa AA, Emmett N, Socci RR, Eatman D, Fridie IL. Alterations in aldosterone and angiotensin II levels in salt-induced hypertension. Clin Exp Hypertens. 2005;27:355–367. [PubMed] [Google Scholar]
- 10.Nakamura F, Nagano M, Higaki J, Higashimori K, Morishita R, Mikami H, Ogihara T. The angiotensin-converting enzyme inhibitor, perindopril, prevents cardiac hypertrophy in low-renin hypertensive rats. Clin Exp Pharmacol Physiol. 1993;20:135–140. doi: 10.1111/j.1440-1681.1993.tb01660.x. [DOI] [PubMed] [Google Scholar]
- 11.Pellieux C, Sauthier T, Aubert JF, Brunner HR, Pedrazzini T. Angiotensin II-induced cardiac hypertrophy is associated with different mitogen-activated protein kinase activation in normotensive and hypertensive mice. J Hypertens. 2000;18:1307–1317. doi: 10.1097/00004872-200018090-00017. [DOI] [PubMed] [Google Scholar]
- 12.Yang XP, Liu YH, Mehta D, Cavasin MA, Shesely E, Xu J, Liu F, Carretero OA. Diminished cardioprotective response to inhibition of angiotensin-converting enzyme and angiotensin II type 1 receptor in B(2) kinin receptor gene knockout mice. Circ Res. 2001;88:1072–1079. doi: 10.1161/hh1001.090759. [DOI] [PubMed] [Google Scholar]
- 13.Wassmann S, Czech T, van Eickels M, Fleming I, Bohm M, Nickenig G. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice. Circulation. 2004;110:3062–3067. doi: 10.1161/01.CIR.0000137970.47771.AF. [DOI] [PubMed] [Google Scholar]
- 14.Gaertner R, Prunier F, Philippe M, Louedec L, Mercadier JJ, Michel JB. Scar and pulmonary expression and shedding of ACE in rat myocardial infarction. Am J Physiol Heart Circ Physiol. 2002;283:H156–H164. doi: 10.1152/ajpheart.00848.2001. [DOI] [PubMed] [Google Scholar]
- 15.Adamy C, Oliviero P, Eddahibi S, Rappaport L, Samuel J-L, Teiger E, Chassagne Cardiac modulations of ANG II receptor expression in rats with hypoxic pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2002;283:H733–H740. doi: 10.1152/ajpheart.01088.2001. [DOI] [PubMed] [Google Scholar]
- 16.Goodman DJ, Harrison DC, Popp RL. Echocardiographic features of primary pulmonary hypertension. Am J Cardiol. 1974;33:438–443. doi: 10.1016/0002-9149(74)90329-4. [DOI] [PubMed] [Google Scholar]
- 17.Nunez BD, Lavie CJ, Messerli FH, Schmieder RE, Garavaglia GE, Nunez M. Comparison of diastolic left ventricular filling and cardiac dysrhythmias in hypertensive patients with and without isolated septal hypertrophy. Am J Cardiol. 1994;74:585–589. doi: 10.1016/0002-9149(94)90748-x. [DOI] [PubMed] [Google Scholar]
- 18.Dunn RB. Regional blood flow and metabolic levels in the left ventricular free wall and septum during aortic insufficiency: implications for the development of asymmetric septal hypertrophy. J Am Coll Cardiol. 1986;8:1182–1188. doi: 10.1016/s0735-1097(86)80399-0. [DOI] [PubMed] [Google Scholar]
- 19.Lamers WH, Moorman AF. Cardiac septation: a late contribution of the embryonic primary myocardium to heart morphogenesis. Circ Res. 2002;91:93–103. doi: 10.1161/01.res.0000027135.63141.89. [DOI] [PubMed] [Google Scholar]
- 20.Heng MK, Janz RF, Jobin J. Estimation of regional stress in the left ventricular septum and free wall: An echocardiographic study suggesting a mechanism for asymmetric septal hypertrophy. Am Heart J. 1985;110:84–90. doi: 10.1016/0002-8703(85)90519-8. [DOI] [PubMed] [Google Scholar]
- 21.Haddad F, Bodell PW, Baldwin KM. Pressure-induced regulation of myosin expression in rodent heart. J Appl Physiol. 1995;78:1489–1495. doi: 10.1152/jappl.1995.78.4.1489. [DOI] [PubMed] [Google Scholar]
- 22.Young MJ, Funder JW. The renin-angiotensin-aldosterone system in experimental mineralocorticoid-salt-induced cardiac fibrosis. Am J Physiol Endocrinol Metab. 1996;271:E883–E888. doi: 10.1152/ajpendo.1996.271.5.E883. [DOI] [PubMed] [Google Scholar]
- 23.Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Inves. 1997;99:1926–1935. doi: 10.1172/JCI119360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartunek J, Weinberg EO, Tajima M, Rohrbach S, Lorell BH. Angiotensin II type 2 receptor blockade amplifies the early signals of cardiac growth response to angiotensin II in hypertrophied hearts. Circulation. 1999;99:22–25. doi: 10.1161/01.cir.99.1.22. [DOI] [PubMed] [Google Scholar]
- 25.Lal A, Veinot JP, Leenen FHH. Prevention of high salt diet-induced cardiac hypertrophy and fibrosis by spironolactone. Am J Hypertens. 2003;16:319–323. doi: 10.1016/s0895-7061(02)03268-5. [DOI] [PubMed] [Google Scholar]
- 26.Robert V, Heymes C, Silvestre JS, Sabri A, Swynghedauw B, Delcayre C. Angiotensin AT1 receptor subtype as a cardiac target of aldosterone: role in aldosterone-salt-induced fibrosis. Hypertension. 1999;33:981–986. doi: 10.1161/01.hyp.33.4.981. [DOI] [PubMed] [Google Scholar]
- 27.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol. 2004;6:499–506. doi: 10.1038/ncb1137. [DOI] [PubMed] [Google Scholar]
- 28.Petrich BG, Eloff BC, Lerner DL, Kovacs A, Saffitz JE, Rosenbaum DS, Wang Y. Targeted activation of c-Jun N-terminal kinase in vivo induces restrictive cardiomyopathy and conduction defects. J Biol Chem. 2004;279:15330–15338. doi: 10.1074/jbc.M314142200. [DOI] [PubMed] [Google Scholar]
- 29.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao RP, Kass DA, Wang Y. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci USA. 2001;98:12283–12288. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bueno OF, De Windt LJ, Tymitz KM, Witt SA, Kimball TR, Klevitsky R, Hewett TE, Jones SP, Lefer DJ, Peng CF, Kitsis RN, Molkentin JD. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]