

The size and copy number of an organelle depend on an equilibrium of membrane fusion and fission. In vitro reconstitution of yeast vacuole fission and fusion shows that TORC1 selectively stimulates fission but does not change fusion activity. This explains the morphological transitions of yeast vacuoles in response to nutrient availability.
Abstract
Size and copy number of organelles are influenced by an equilibrium of membrane fusion and fission. We studied this equilibrium on vacuoles—the lysosomes of yeast. Vacuole fusion can readily be reconstituted and quantified in vitro, but it had not been possible to study fission of the organelle in a similar way. Here we present a cell-free system that reconstitutes fragmentation of purified yeast vacuoles (lysosomes) into smaller vesicles. Fragmentation in vitro reproduces physiological aspects. It requires the dynamin-like GTPase Vps1p, V-ATPase pump activity, cytosolic proteins, and ATP and GTP hydrolysis. We used the in vitro system to show that the vacuole-associated TOR complex 1 (TORC1) stimulates vacuole fragmentation but not the opposing reaction of vacuole fusion. Under nutrient restriction, TORC1 is inactivated, and the continuing fusion activity then dominates the fusion/fission equilibrium, decreasing the copy number and increasing the volume of the vacuolar compartment. This result can explain why nutrient restriction not only induces autophagy and a massive buildup of vacuolar/lysosomal hydrolases, but also leads to a concomitant increase in volume of the vacuolar compartment by coalescence of the organelles into a single large compartment.
INTRODUCTION
Numerous organelles fragment into smaller vesicles during cell division, vesicular traffic, or in response to changes in environmental conditions, changing the size, copy number, and morphology of these compartments. Examples include mitochondria (Yoon and McNiven, 2001; Shaw and Nunnari, 2002; Chan, 2006), plastids (Osteryoung and McAndrew, 2001), peroxisomes (Yan et al., 2005; Schrader and Fahimi, 2006), endosomes (van der Goot and Gruenberg, 2006), the nuclear envelope (Newport and Forbes, 1987; Prunuske and Ullman, 2006), Golgi (Corda et al., 2002; Shorter and Warren, 2002; Colanzi et al., 2003; Rabouille and Jokitalo, 2003), lysosomes (Storrie and Desjardins, 1996), and vacuoles (Weisman, 2003). Whereas membrane fission of coated vesicles has been studied in considerable detail (Conner and Schmid, 2003; Mancias and Goldberg, 2005; McMahon and Gallop, 2005; Antonny, 2006), organelle fragmentation is less well understood. Genetic and biochemical approaches have identified a number of proteins and lipids involved in such reactions, but it is unclear how they cooperate to split organelles into smaller vesicles. Few organelle fragmentation reactions have been reconstituted in vitro: disassembly of the Golgi in mammalian cell extracts (Misteli and Warren, 1994; Rabouille et al., 1995; Shorter and Warren, 1999) or in semi-intact cells (Acharya et al., 1998; Kano et al., 2000); the fission of endosomal/lysosomal hybrid organelles (Luzio et al., 2003); and nuclear fragmentation (Newport and Forbes, 1987; Higa et al., 2006) in Xenopus egg extracts. The cell-free reconstitution of these organelle fragmentation reactions has considerably supported the study of their mechanism.
Yeast vacuoles are relatively large (≤5 μm), so that their shape, size, and number during fragmentation are clearly visible in the light microscope. Size and copy number of yeast vacuoles change depending on the cell cycle stage and on environmental conditions (Weisman, 2003). Hypertonic shock induces rapid fragmentation of vacuoles into multiple small vesicles, whereas hypotonic treatment has the opposite effect. Nutrient restriction leads to a strong induction of vacuolar hydrolases and to a coalescence of all vacuoles of a cell into a single organelle, which provides increased volume (Baba et al., 1994). Vacuole coalescence and fragmentation depend on the equilibrium of the activities of the vacuolar fusion and fission machineries, which thus determine vacuole morphology (Baars et al., 2007). To study this equilibrium and the factors influencing it, it is desirable to be able to study fusion and fission separately.
For the study of vacuole fusion a powerful in vitro system has been established that makes it possible to measure this reaction with purified organelles in the test tube (Conradt et al., 1994). The assay is based a mixture of two separately prepared vacuole populations—one containing an enzymatically inactive pro–alkaline phosphatase and the other one the necessary maturation enzyme (Haas et al., 1994). Fusion will produce mature and active alkaline phosphatase, which serves as a tracer for fusion. This system has made it possible to identify many proteins and lipids implicated in vacuole fusion and to characterize multiple reaction intermediates (Ostrowicz et al., 2008; Wickner, 2010). It has revealed that vacuole fusion shares many fundamental characteristics with other fusion reactions in cells, such as the requirement for soluble N-ethylmaleimide–sensitive factor attachment protein receptors, Rab-GTPases, Rab-effector proteins (homotypic fusion and vacuole protein sorting complex), and their control factors.
An in vitro system to study vacuole fission had not been available. However, because vacuoles fragment during the cell cycle or in hypertonic media, optical screening of mutants could identify mutants that show enlarged vacuoles and are defective in the salt-inducible fragmentation of vacuoles (Weisman, 2003; Efe et al., 2005; LaGrassa and Ungermann, 2005). Crucial factors for vacuole fragmentation, such as the phosphatidylinositol-3-phosphate-5-kinase Fab1 and its effectors and regulators Vac7p, Vac14p, Fig4p, and Atg18p (Cooke et al., 1998; Gary et al., 1998, 2002; Bonangelino et al., 2002; Dove et al., 2002, 2004; Duex et al., 2006a, 2006b; Efe et al., 2007) are conserved between different eukaryotes, acting also on endosomal/lysosomal compartments in plants and mammals. In addition, the class C core vacuole/endosome tethering complex subunit Vps3p is required for fragmentation, and the casein kinase Yck3p is necessary to maintain the fragmented state under hypertonic conditions (LaGrassa and Ungermann, 2005; Peplowska et al., 2007). Vacuole fragmentation also requires a dynamin-related GTPase, Vps1p (Peters et al., 2004). This indicates a potential mechanistic similarity of vacuole fragmentation to the fragmentation of other organelles, in which dynamin-related GTPases have been implicated as well. Examples include mitochondria, peroxisomes, and chloroplasts (Bleazard et al., 1999; Hoepfner et al., 2001; Gao et al., 2003; Li and Gould, 2003; Okamoto and Shaw, 2005; Kuravi et al., 2006).
Here we report the cell-free reconstitution of the fragmentation of purified Saccharomyces cerevisiae vacuoles. We validated the authenticity of the cell-free reaction by studying the effects of mutations that generate defects of vacuole fragmentation in vivo. We demonstrate the utility of the in vitro approach by using it to identify a regulation of vacuole fragmentation by TOR kinase, confirm its relevance in vivo, and explore its role for regulating vacuole size and number.
RESULTS
Vacuolar membrane fission can be induced on isolated vacuoles
In living cells, vacuoles fragment during the cell cycle or in response to hypertonic shock (Weisman et al., 1987; Catlett and Weisman, 2000). We attempted to reproduce vacuole fragmentation in a test tube with purified vacuoles. We incubated the organelles with ATP as an energy source and a cytosolic extract as a source of soluble proteins. After 1 h of incubation at 27°C, the membranes were stained with the lipophilic dye BODIPY FL-DHPE, and vacuolar morphology was visualized by confocal fluorescence microscopy. During the 60 min after the start of an in vitro fragmentation reaction, ≤90% of the large vacuoles that had been visible at the beginning fragmented into smaller vesicles (Figure 1, A and B) that often remained clustered together. We analyzed the size of the small fragmentation products by electron microscopy (Figure 1C). Fragmentation reactions incubated in the absence of ATP contained many intact vacuoles (d = 2.1 ± 0.6 μm), visible as large circular structures with an electron-translucent, white lumen. After incubation with ATP, these large structures were absent, and numerous small vesicles (d = 0.45 ± 0.27 μm) appeared. For routine analyses, vacuole fragmentation was analyzed by fluorescence microscopy. The organelles were quantified on confocal images taken from seven to 10 random fields and grouped into three classes according to their diameter: small (d ≤ 0.6 μm), medium (0.6 < d <1.5 μm), and large (d ≥ 1.5 μm). Because clusters of the small vesicles are hard to resolve in the light microscope, their number could not be reliably determined by direct counting. Because their average diameter was known from electron microscopy, however, their number could be deduced from the surface they occupy in the image. Therefore we used this approach to assay the number of small vesicles. Electron microscopy (Figure 1C) showed that large vacuoles had an average surface of 12.5 μm2 and the small vesicles of 0.64 μm2. This suggests that a large vacuole could at most produce ∼20 vacuolar fragments. In reasonable agreement with this, analysis of a fragmentation reaction (Figure 1B) from fluorescence micrographs according to the described procedure suggests that a large vacuole gave rise to ∼17 fragments during the reaction. To facilitate comparisons, we defined a fragmentation index representing the degree of fragmentation. The proportion (R) of large vacuoles in the total vesicle pool of small (Ns), medium (Nm), and large (Nl) vacuoles is R = Nl/(Ns + Nm + Nl). For the fragmentation index (FI) this proportion of large vacuoles is related to that in a positive control (Rpos), which is set to 100%: FI = Rpos/R × 100 (%).
FIGURE 1:

Vacuole fragmentation in vivo and in vitro. (A) Vacuoles were isolated from BJ3505 cells and subjected to in vitro fragmentation reactions. The membranes were stained with BODIPY-FL DHPE, fixed in cavity slides with 0.2% LMP-agarose, and visualized by confocal fluorescence microscopy. Examples for small (arrow) and medium-sized (arrowhead) vesicles are indicated. (B) Values for the numbers of vesicles of different diameter d: small (Ns; d ≤ 0.6 μm), medium (Nm; 0.6 <d < 1.5 μm), and large (Nl; d ≥ 1.5 μm). Vesicles were quantified and FI calculated as described in Materials and Methods. Of 121 large vacuoles, 113 were consumed during the reaction. The small-vesicle fraction increased from 951 to 2816, indicating a gain of 1865 small vesicles. This suggests that one large vacuole produced on average 1865/113 = 17 small vesicles. (C) Vacuoles were incubated in an in vitro fragmentation reaction in the presence or absence of ATP, fixed with glutaraldehyde, and analyzed by electron microscopy. Bars, 2 μm.
In vitro fragmentation activity increased with salt concentration, with optimal values obtained between 100 and 150 mM KAc (Figure 2A). Higher salt concentrations inhibited fragmentation. Activity was influenced by the type of salt. NaCl and KCl did not induce fragmentation, but NaAc supported fragmentation as well as KAc (Figure 2B). Sorbitol, used as a control for a nonionic but osmotically active substance, did not support fragmentation either. We decided to use KAc because K+ ions are the predominant intracellular cation species. The salt dependence of vacuole fragmentation could have different reasons: Fragmentation into small vesicles increases the surface-to-volume ratio of the membranes, requiring extrusion of water. Exogenously added KAc might osmotically extract water and thus allow the vesicles to adjust their volume to the decreasing radius. Yet this cannot be the only factor because we could not replace KAc by sorbitol and because KAc concentrations >150 mM decreased fragmentation activity. It was also insufficient to provide just a certain ionic strength because the chloride salts could not substitute for the acetate salts. Thus a specific ionic milieu is required.
FIGURE 2:

Parameters relevant to fragmentation in vitro. (A) Salt concentration. Fragmentation reactions were performed with 0, 50, 100, 150, and 250 mM of KAc. For each concentration, parallel reactions were set up with (positive control) and without ATP (negative control). p < 0.03 (−ATP vs. +ATP) at 100 and 150 mM KAc. (B) Ionic composition. Fragmentation reactions were performed with 100 mM of four different salts—KAc, KCl, NaCl, or NaAc—or with 300 mM sorbitol. Each solution was adjusted to a pH of 6.8. Values for KAc/+ATP were normalized to 100% before averaging. p < 0.01 for the differences between KAc and all other salts except NaAc. (C) Temperature. Fragmentation reactions were run for 60 min at 0, 20, 25, 27, 30, and 35°C with and without ATP. Values for 27°C/+ATP were normalized to 100% before averaging. p < 0.01 (−ATP vs. +ATP) for 25, 27, and 30°C. (D) Gdi1p suppresses fusion activity that can be measured under conditions of in vitro fragmentation. Reactions were run in the presence or absence of Gdi1p (10 μM) and an ATP regenerating system. After 70 min, fusion activity was determined. p < 0.001 for all differences relative to the 27°C/−GDI values. n = 3. (E) Time. Fragmentation reactions with and without ATP and Gdi1p (0.3 mg/ml) were run at 27°C for the indicated periods of time. Values for 60 min/+ATP were normalized to 100% before averaging. p < 0.01 (−ATP vs. +ATP) for all time points ≥20 min.
Vacuole fragmentation depends on the incubation temperature and on time. On ice, virtually no fragmentation was observed, whereas the efficiency of the reaction was maximal at 27°C (Figure 2C). Higher temperatures decreased apparent fragmentation. They even produced a fraction of vacuoles that were bigger than at the onset of the incubation (data not shown), suggesting that some vacuoles might have fused. To test whether competing fusion activity might distort our results on fragmentation, time-course experiments were performed in the absence or presence of GDP dissociation inhibitor (GDI), a protein inhibiting fusion by inactivating the vacuolar Rab-GTPase Ypt7p (Figure 2D; Haas et al., 1995). Vacuole fragmentation was determined 0, 20, 40, 60, and 90 min after starting a reaction (Figure 2E). After 60 min, most vacuoles had been converted into small vesicles, showing that in this phase of the incubation, fragmentation activity prevails. The time courses in the absence and presence of GDI overlapped up to the 60-min time point, indicating that the apparent fragmentation activity is not significantly diminished by fusion in this reaction phase. Later, vacuoles reappeared that were larger than the starting material, suggesting that fusion activity dominates the reaction after prolonged incubation times. This fusion activity can be demonstrated by an enzymatic fusion assay (see discussion and Figure 8 later in the paper). In sum, the in vitro system lets the organelles go through a cycle of initial fragmentation and subsequent refusion, and it may allow the study of events that shift the balance from preferential fragmentation to preferential fusion.
FIGURE 8:

Vacuole fragmentation in mutants related to TORC1 signaling. (A) Vacuole fragmentation in Δsit4 mutants in vivo. Deletion mutants for SIT4 and isogenic wild-type (BY4742) cells were grown to logarithmic phase (OD600 = 1) and stained with FM4-64. The culture medium was supplemented with 0.4 M NaCl. After 10 min at 25°C, cells were centrifuged and immediately analyzed by fluorescence microscopy. The cells (>100 per condition and experiment) were evaluated and classified according to the number of vacuoles visible per cell. n = 3. (B, C) Vacuole fragmentation in tap42 mutants. Cells carrying the temperature-sensitive alleles tap42-106 or tap42-109 (Duvel et al., 2003), tap42-11 (Di Como and Arndt, 1996), or the corresponding wild-type TAP42 allele were grown to logarithmic phase (OD600 = 1) and stained with FM4-64 at 25°C in YPD. After a shift to 37°C (40 min for tap42-11, 60 min for tap42-106 and tap42-109), the medium was supplemented with 0.4 M NaCl. After 10 min of further incubation at 37°C, cells were analyzed for vacuole fragmentation as in A. n = 3.
In vitro fragmentation shares numerous properties with vacuole fragmentation in vivo
We tested whether the reconstituted reaction reflected properties of vacuole fragmentation in an intact cell. Complex cell biological reactions typically require an energy source, the presence of specific proteins, and a physiological temperature. Temperature dependence has been shown (Figure 2C). We next tested the ATP and cytosol dependence of the process. Whereas fragmentation proceeded efficiently in the presence of cytosolic proteins. it was reduced by >80% if cytosol was omitted from the reactions (Figure 3A). Omitting ATP or using EDTA to chelate the Mg2+ that is essential for hydrolysis of ATP reduced fragmentation by >90% (Figure 3B). Thus vacuole fragmentation in vitro is temperature-dependent and ATP-dependent and requires cytosolic factors.
FIGURE 3:
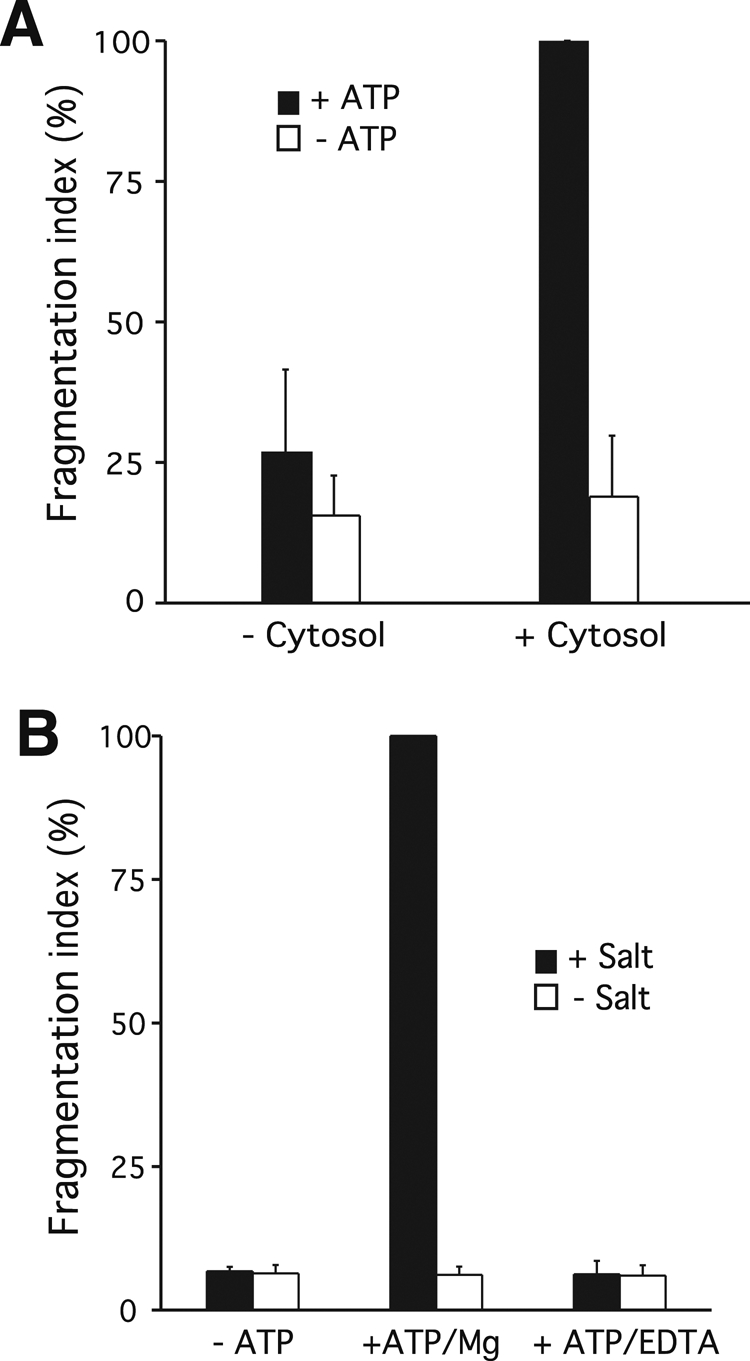
ATP and cytosol dependence of fragmentation in vitro. (A) Cytosolic factors. Fragmentation reactions were run in the presence or absence of cytosolic proteins (1 mg/ml). Values +cytosol/+ATP were normalized to 100% before averaging. p < 001 for the difference between +ATP values. (B) Energy source. In vitro fragmentation was performed in the presence or absence of ATP. Alternatively, hydrolysis of the ATP present in the sample was prevented by chelating Mg2+ with EDTA (7 mM). p < 001 for all differences relative to the +Mg/−ATP value.
Vacuole fragmentation in vivo depends on the dynamin-related GTPase Vps1p (Peters et al., 2004) and on the production of phosphatidylinositol-3,5-bisphosphate (PI-3,5-P2; Bonangelino et al., 1997, 2002; Cooke et al., 1998; Gary et al., 1998; Jin et al., 2008). We tested whether fragmentation in vitro reproduces these requirements. We purified vacuoles from a strain deleted for VPS1 (Δvps1) or from a strain carrying a thermosensitive vps1ts allele as the sole source of Vps1p (Ekena et al., 1993). By use of a moderate salt shock, which induces rapid and synchronous vacuole fragmentation (Weisman, 2003), it was shown that Δvps1 cells have defects in vacuole fragmentation in vivo (Peters et al., 2004). In the same assay, vps1ts cells fragmented their vacuoles as in wild type at 25°C but not after brief incubation at 37°C, when the vps1ts protein becomes nonfunctional (Figure 4A). In the in vitro assays vacuoles isolated from Δvps1 cells showed <15% of the fragmentation activity of wild-type vacuoles (Figure 4B). For testing the vps1ts vacuoles in vitro the cells were grown at 25°C, the temperature at which vps1ts remains functional (Ekena et al., 1993). Before vacuole extraction, the cells were shifted to 37°C for 20 min. This treatment reduced the fragmentation activity of vps1ts vacuoles by >80% (Figure 4C) compared with the wild-type control.
FIGURE 4:

Fragmentation of vacuoles from vps1 and fab1 mutants. (A) Fragmentation in vivo. vps1ts mutants or isogenic wild-type cells were grown to logarithmic phase at 25°C and stained with FM4-64. Then the cells were split and either shifted to 37°C for 20 min or left at 25°C. The medium was supplemented with 0.4 M NaCl. After further 10 min at 25 or 37°C, cells were centrifuged and immediately analyzed by fluorescence microscopy. The cells (>100 per condition and experiment) were evaluated and classified according to the number of vacuoles visible per cell. (B) Vacuoles were prepared from BJ3505 wild-type or isogenic Δvps1-knockout strains. Fission activity of the organelles in vitro was assayed in presence or absence of ATP. Values for WT/+ATP were normalized to 100% before averaging. p < 0.001 for all differences relative to the WT+ATP value. n = 3. (C) Temperature-sensitive Vps1p. BJ3505 Δvps1 was complemented with centromeric plasmids expressing wild-type VPS1 or a thermosensitive allele, vps1ts (Ekena et al., 1993). The cells were grown overnight at permissive temperature (25°C) and subjected to a 20-min temperature shock (37°C) during the spheroplasting step of vacuole preparation. Vacuoles isolated from these cells were compared in in vitro fragmentation assays in the presence and absence of ATP. Values for WT/+ATP were normalized to 100% before averaging. p < 0.001 for all differences relative to the WT/+ATP value. n = 3. (D) Inhibition of fragmentation by antibodies to Vps1p. Vacuoles were isolated from wild-type BJ3505 cells. The organelles were analyzed in in vitro fragmentation reactions containing affinity-purified antibodies to Vps1p (0.5 μM) or, as a negative control, the same concentration of antibodies to the vacuolar membrane protein Vph1p. p < 0.01 for all differences relative to the buffer/+ATP value. (E) Δfab1 mutants and corresponding wild-type cells, both in Δpep4 background (Mayer et al., 2000), were grown to logarithmic phase (OD600 = 1) and stained with FM4-64. The culture medium was supplemented with 0.4 M NaCl. After 10 min at 25°C, cells were centrifuged and immediately analyzed by fluorescence microscopy. The cells (>100 per condition and experiment) were evaluated and classified according to the number of vacuoles visible per cell. n = 3. (F) Vacuoles were prepared from Δfab1 mutants and corresponding wild-type cells. The in vitro fission activity of the organelles was assayed as in B.
To inactivate Vps1p more acutely, we used affinity-purified antibodies to this protein. They allow us to rapidly interfere with Vps1p function on the surface of isolated wild-type vacuoles (Peters et al., 2004). Incubation of the vacuoles with 0.5 μM anti-Vps1p suppressed fragmentation activity as efficiently as the omission of ATP (Figure 4D). Control antibodies, which bind the abundant vacuolar V-ATPase subunit Vph1p but do not inhibit its proton pump function (Bayer et al., 2003), did not significantly inhibit fragmentation. These results suggest that the dynamin-like GTPase Vps1p is directly involved in vacuole fragmentation in vitro. The requirement for the PI-3,5-P2 producing Fab1 complex was tested using a mutant deleted for the gene of its catalytic subunit Fab1p. Δfab1 cells do not fragment their vacuoles upon salt shock (Figure 4E; Bonangelino et al., 2002). Vacuoles prepared from this mutant were also defective for fragmentation in vitro (Figure 4F). These observations confirm that in vitro fragmentation reproduces important aspects of the reaction in living cells.
In vitro vacuole fragmentation depends on the electrochemical potential and on GTP hydrolysis
The accessibility of the membranes in the in vitro system allows us to identify low–molecular weight inhibitors of fragmentation. We tested whether GTP hydrolysis is required to support vacuole fragmentation, by using GTPγS. This poorly hydrolysable analogue of GTP can arrest GTPases in the GTP-bound state and prevent their enzymatic cycling. GTPγS inhibited fragmentation in vitro by >90% (Figure 5A). The IC50 was 0.3 mM and complete inhibition occurred at 1 mM GTPγS (Figure 5B). GTPγS might inhibit fragmentation because it disassembles high–molecular weight forms of Vps1p and weakens their interaction with vacuoles (Peters et al., 2004). A multimeric form of Vps1p is expected to support vacuole fragmentation, in analogy to the fission step in endocytosis, which depends on multimeric dynamin I (Muhlberg et al., 1997; Sweitzer and Hinshaw, 1998). Using the in vitro system we could identify several other low–molecular weight inhibitors of vacuole fragmentation (Figure 5C). Among these were concanamycin A (2.5 μM), valinomycin (3 μM), carbonyl cyanide p-trifluoromethoxyphenyl hydrazone (FCCP; 10 μM), microcystin LR (2.5 μM), and rapamycin (7 μM). Valinomycin is a K+ ionophore that interferes with the membrane potential. Concanamycin A inhibits vacuole acidification by blocking the proton pump activity of the vacuolar H+-ATPase. FCCP is a protonophore that collapses the pH gradient across the vacuole membrane. We also tested the microtubule- and actin-directed drugs colchicine and latrunculin A, which had no significant influence. The effects of valinomycin, concanamycin, and FCCP suggest that fragmentation depends on the electrochemical potential across the membranes. Furthermore, the in vitro effects of these inhibitors are consistent with in vivo observations indicating that vacuoles in V-ATPase mutants cannot fragment and that concanamycin A suppresses vacuole fragmentation in wild-type cells (Baars et al., 2007). This further supports the physiological authenticity of the reconstituted reaction.
FIGURE 5:

Sensitivity of fragmentation to low–molecular weight inhibitors. (A) Poorly hydrolysable GTP analogue. Vacuoles from BJ3505 wild-type cells were subjected to fragmentation reactions with and without ATP in the presence or absence of 1 mM GTPγS. p < 0.001 for all differences relative to the −GTPγS/+ATP value. (B) As in A, but the concentration of GTPγS was varied as indicated. p < 0.001 (−ATP vs. +ATP) for concentrations of <1 mM. (C) Inhibitors of TOR or of the electrochemical potential. Fragmentation reactions were run as in A, but with concanamycin A (2.5 μM), rapamycin (7 μM), valinomycin (3 μM), FCCP (10 μM), microcystin LR (25 μM), latrunculin A (10 μM), colchicine (1 mM), or without inhibitor (DMSO solvent only). p < 0.01 for all differences relative to the −inhibitor/+ATP value. Values −inhibitor/+ATP were normalized to 100% before averaging for A–C.
Rapamycin-sensitive TOR complex 1 activity controls vacuole fragmentation
Vacuole fragmentation in vitro was also blocked by rapamycin (Figure 5C), a drug inhibiting TOR kinase activity in the TOR complex 1 (TORC1; Heitman et al., 1991; Loewith et al., 2002), and by microcystin LR, an inhibitor of protein phosphatases of types 1 and 2A (MacKintosh et al., 1990). TOR is a regulator of cell growth, whose activity is down-regulated in starving cells, and PP2A activity mediates many TOR outputs (De Virgilio and Loewith, 2006). Rapamycin mimics this down-regulation and thus induces starvation responses, such as autophagy (Wullschleger et al., 2006). Upon starvation or rapamycin treatment, the vacuoles in living yeast cells coalesce into a single big organelle, leading to a increase in vacuole size (Cardenas and Heitman, 1995; Catlett and Weisman, 2000; Crespo et al., 2005; Dubouloz et al., 2005).
Because vacuoles exist in an equilibrium of fusion and fragmentation in vivo (Baars et al., 2007), we investigated whether these two processes are controlled by TOR. We used tor1-1/tor2-1 double mutants, which express rapamycin-resistant alleles of the two TOR genes in yeast, TOR1 and TOR2. Many TOR-dependent processes—at least those essential for growth—are less sensitive to the drug in this strain (Wullschleger et al., 2006). We isolated vacuoles from tor1-1/tor2-1 double mutants and wild-type strains and compared their fragmentation activities in the presence of rapamycin (Figure 6A). Rapamycin inhibited the fragmentation of wild-type vacuoles to the background levels observed in the absence of ATP, whereas vacuoles from the rapamycin-resistant tor1-1/tor2-1 cells retained their activity. Note that all fragmentation reactions were performed with cytosol from wild-type TOR1/TOR2 cells. That vacuoles isolated from tor1-1/tor2-1 cells retain their rapamycin resistance indicates that the relevant pool of TOR is associated with the vacuoles and does not readily diffuse into the cytosol. This is consistent with the fact that in living cells one of the two TOR complexes is bound to vacuoles (Sturgill et al., 2008).
FIGURE 6:

TOR and rapamycin dependence of vacuole fragmentation. (A) Vacuoles were isolated from tor1-1/tor2-1 double mutants or from isogenic wild-type cells. Fragmentation reactions were run in vitro in the presence or absence of rapamycin (7 μM) and ATP. p < 0.01 for the difference of the +ATP/+rapamycin values. (B) Fragmentation in vivo. tor1-1/tor2-1 double mutants or isogenic wild-type cells were grown to logarithmic phase and stained with FM4-64. Rapamycin (2 μM from a 100× stock in DMSO) or DMSO was added to the cultures. Cells were incubated for 40 min at 25°C with shaking. The culture medium was supplemented with 0.4 M NaCl. After 10 min at 25°C, cells were centrifuged and immediately analyzed by fluorescence microscopy. The cells (>100 per condition and experiment) were evaluated and classified according to the number of vacuoles visible per cell. n = 3. (C) Representative confocal images of the vacuoles in groups of cells analyzed in B.
We tested the authenticity of this in vitro result in the in vivo situation, using the induction of vacuole fragmentation by hypertonic shock (Bonangelino et al., 2002). Vacuole membranes in living yeast cells were stained with the fluorescent vital dye FM4-64 (Vida and Emr, 1995). Upon addition of 0.4 M NaCl to the medium, wild-type cells rapidly (<10 min) fragmented their large vacuoles into multiple small vesicles (Figure 6, B and C). Cells preincubated with rapamycin for 40 min did not show this reaction. In contrast, tor1-1/tor2-1 cells fragmented their vacuoles equally well in the presence or absence of rapamycin. Because TOR functions via two large protein complexes, TORC1 and TORC2, we investigated which complex is responsible for vacuole fragmentation. The fact that TORC1 is localized on vacuoles (Sturgill et al., 2008) and that only TORC1 but not TORC2 is sensitive to rapamycin (Loewith et al., 2002) points to TORC1. We tested this further by deleting the gene for the nonessential TORC1 subunit Tco89p (Reinke et al., 2004) and assaying vacuole structure of the cells before and after NaCl treatment (Figure 7A). Already in normal medium, Δtco89 cells showed fewer and slightly larger vacuoles than the wild type. Upon addition of NaCl, the Δtco89 cells did not show the pronounced increase in vacuole number that is observable in wild-type cells. Vacuoles prepared from Δtco89 cells showed a corresponding deficiency for ATP-dependent fragmentation in vitro (Figure 7B). Collectively these data suggest that fragmentation of the vacuolar compartment is positively regulated by TORC1 in vivo and in vitro and that TORC1 activity is necessary for executing the fragmentation signal provided by hypertonic shock.
FIGURE 7:
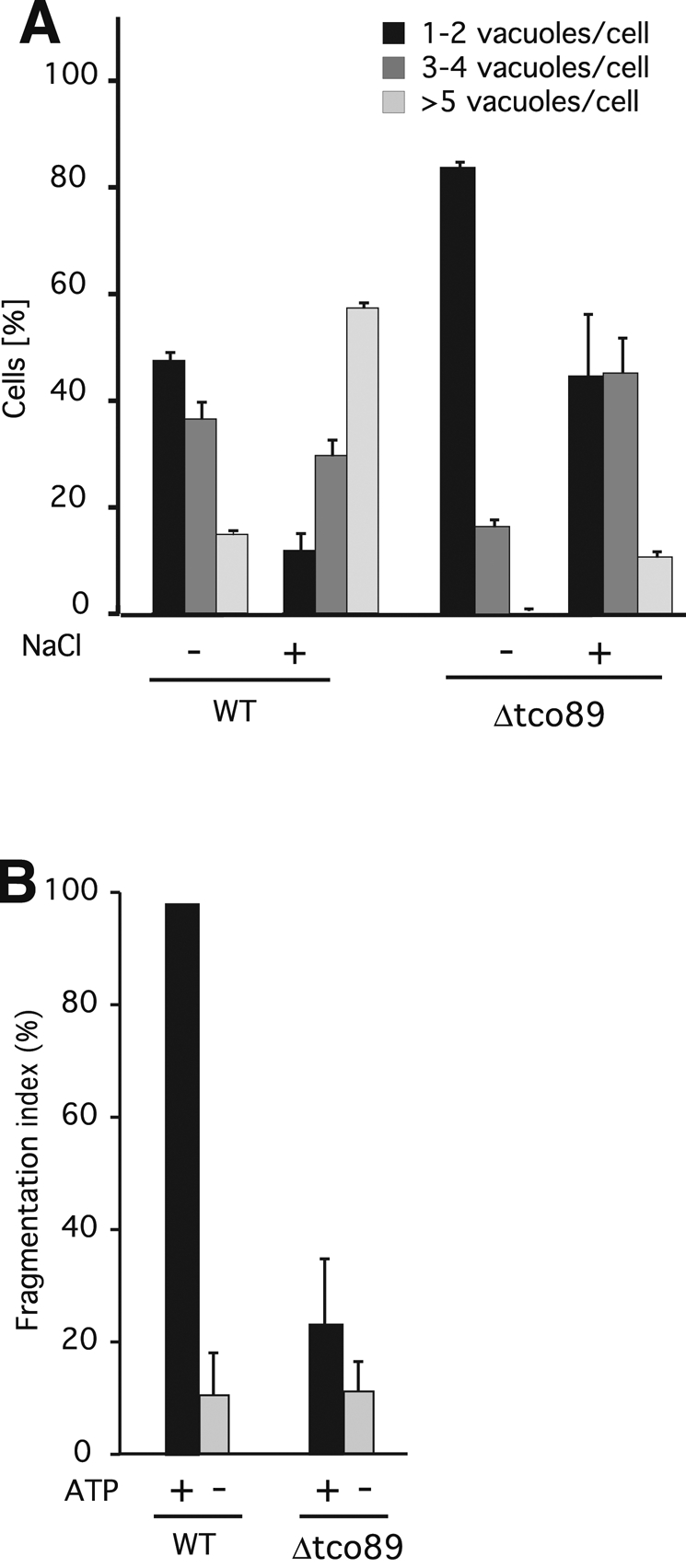
Fragmentation activity of Δtco89 mutants. (A) Δtco89 mutants and corresponding wild-type cells were grown to logarithmic phase (OD600 = 1) and stained with FM4-64. The culture medium was supplemented with 0.4 M NaCl. After 10 min at 25°C, cells were centrifuged and immediately analyzed by fluorescence microscopy. The cells (>100 per condition and experiment) were evaluated and classified according to the number of vacuoles visible per cell. n = 3. (B) Vacuoles were prepared from Δtco89 mutants and corresponding wild-type cells. The in vitro fission activity of the organelles was assayed as in Figure 4B.
Vacuole fragmentation depends on the PP2A-like phosphatase Sit4p
Few direct target proteins for TOR have been identified in yeast. Among these are the TORC1 targets Sch9p and Sfp1p, as well as Ypk1p/2p, Slm1p, and Slm2p, which act in TORC2 signaling (Audhya et al., 2004; Kamada et al., 2005; Urban et al., 2007; Lempiainen et al., 2009). Conditional ATP analogue–sensitive alleles of SCH9 did not interfere with vacuole fragmentation, and the constitutively active allele SCH92D3E (Urban et al., 2007) did not render vacuole fragmentation rapamycin resistant. Equivalent mutants for YPK1 and deletion mutants for SLM1, SLM2, and SFP1 had no effects on vacuole fragmentation either (data not shown). TORC1 signaling often implicates the PP2A-like phosphatase Sit4p and the PP2A Pph21p/Pph22p. Deletion of the PP2A-like SIT4 inhibited fragmentation (Figure 8A). Sit4p binds to Tap42p, a cofactor that can be phosphorylated by TORC1 and that controls Sit4p and Pph21/Pph22p activities in many essential TOR-dependent processes (Di Como and Arndt, 1996; Gorner et al., 1999; Jacinto et al., 2001; Duvel and Broach, 2004). We tested the role of Tap42p and of its major downstream targets for vacuole fragmentation. Strains expressing the temperature-sensitive alleles tap42-106, tap42-109 (Duvel et al., 2003), or tap42-11 (Di Como and Arndt, 1996) as the sole source of Tap42p fragmented their vacuoles as efficiently as those carrying a wild-type allele, even at restrictive temperature (Figure 8, B and C). This suggests that vacuole fragmentation might be regulated by a signaling branch that depends on Sit4p and TORC1 but is insensitive to the conditional tap42 alleles used here.
Vacuole fusion is largely rapamycin insensitive
Both fragmentation and fusion activities determine vacuole structure in vivo (Baars et al., 2007). An apparent inhibition of vacuole fragmentation by rapamycin could thus have three reasons: A genuine inhibition of fragmentation activity by rapamycin, a stimulation of fusion activity, or a combination of both effects. Because our microscopic observations (see prior discussion) suggested that fusion was possible and prevailed upon prolonged incubation of the vacuoles in our fragmentation assay, we tested a potential influence of TORC1 on fusion using an in vitro assay system for vacuole fusion. This assay reproduces numerous physiological aspects of vacuole fusion. It is based on the proteolytic conversion of pro–alkaline phosphatase in one fusion partner by the appropriate maturation enzymes provided by the other fusion partner (Haas et al., 1994). The assay was used to score fusion activity after 60 min of incubation under the conditions that support in vitro fragmentation of vacuoles (“fragmentation conditions”) or under conditions that had been optimized to study vacuole fusion only (“fusion conditions”; Reese et al., 2005). Even under conditions optimized to reconstitute in vitro vacuole fragmentation, the vacuoles showed significant fusion activities, reaching more than half of the signal obtainable under conditions optimized for vacuole fusion. Thus both fusion and fragmentation can occur during the in vitro fragmentation assay. However, in contrast to fragmentation, which is sensitive to rapamycin, fusion activity was only weakly affected (Figure 9). Of importance, it did not show the increase that would have been required to explain a potential interference of the fusion machinery with the apparent vacuole fragmentation activity. This suggests that rapamycin-sensitive TOR activity regulates vacuole fragmentation rather than fusion.
FIGURE 9:
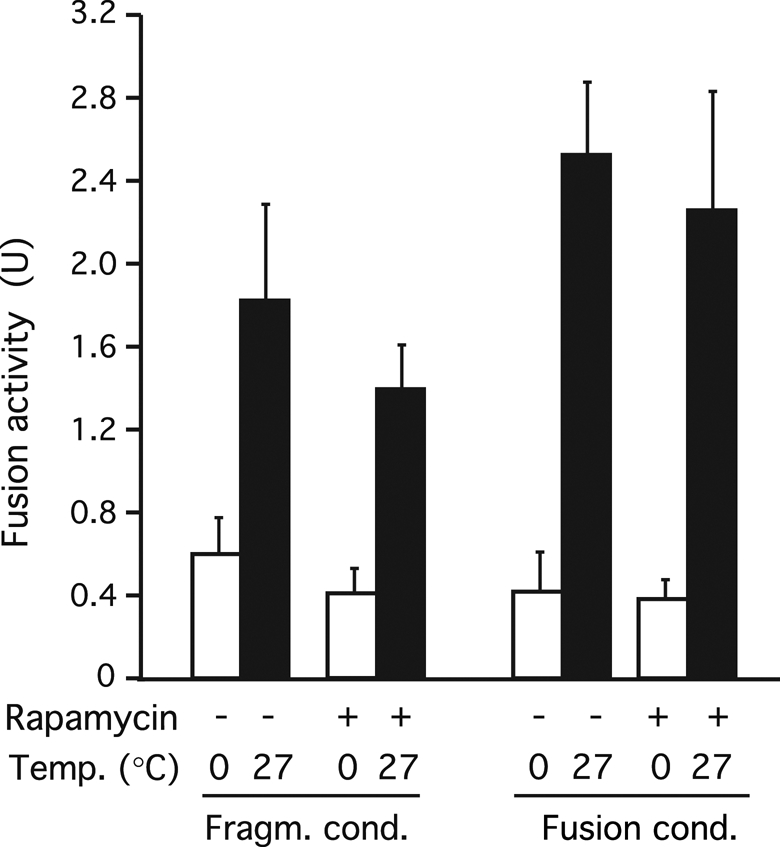
Effect of rapamycin on vacuole fusion. Vacuoles were isolated from BJ3505 and DKY6281 strains and used in in vitro reactions optimized for fragmentation or for fusion. The vacuoles were mixed and incubated in the presence or absence of rapamycin. After 90 min at 27°C or on ice, fusion activity was assayed in both sets of samples via the activity of the alkaline phosphatase reporter enzyme. p = 0.35 (+rapamycin vs. −rapamycin for fragmentation conditions).
DISCUSSION
In response to nutrient restriction the vacuoles of yeast cells coalesce into a single organelle of strongly increased volume, probably reflecting the need for increased hydrolytic capacity for autophagy (Baba et al., 1994). TORC1 is a key factor in nutrient signaling (Wullschleger et al., 2006). Our study demonstrates that TORC1 stimulates vacuole fragmentation, one of the two elementary processes controlling rapid adjustments of vacuole size and copy number. Because vacuole structure is defined by an equilibrium of fusion and fragmentation, starvation-induced down-regulation of TORC1 can suffice to induce vacuole coalescence by decreasing the ratio of the rates of fragmentation and fusion (Figure 10). Vacuole coalescence could result from a decrease in fragmentation activity, an increase in fusion activity, or both. Our data indicate that TORC1 activity can be limiting for vacuole fragmentation but not for vacuole fusion. This suggests that coalescence of the vacuolar compartment under starvation conditions is determined by down-regulation of fragmentation in the face of a constitutive opposing fusion activity, resulting in the formation of a single large organelle.
FIGURE 10:

(A) Components involved in vacuole fusion and fragmentation and (B) their influence on the fusion–fission equilibrium under different conditions. The scheme shows only components relevant to this study and not all components involved in fusion and fission.
Few organelle fragmentation reactions have been reconstituted in vitro, such as mitotic fragmentation of the Golgi and the nuclear envelope or the separation of endosomal/lysosomal hybrid organelles (Pfaller and Newport, 1995; Pryor et al., 2000; Shorter and Warren, 2002; Colanzi et al., 2003). They are all based on higher eukaryotic cells. In yeast cells, which are genetically more tractable, organelle fragmentation has so far only been studied in vivo (Hoppins et al., 2007; Nagotu et al., 2010). Yeast vacuoles offer advantages for studying organelle fragmentation. They have a diameter of ≤5 μm and a simple structure, that is, their limiting membrane can be clearly visualized by fluorescence microscopy. Furthermore, hypertonic shock induces rapid and quantitative vacuole fragmentation in vivo (Bonangelino et al., 2002), which facilitates mutant screening. Using this criterion, we screened the 4881 nonessential yeast genes for defects in vacuole fragmentation (Michaillat and Mayer, unpublished results). Dissection of this complex cell-biological reaction is facilitated by combining this in vivo approach with an in vitro system that reconstitutes vacuole fragmentation in a physiologically relevant form. The fragmentation reaction reconstituted here shares with the in vivo process its sensitivities to low–molecular weight inhibitors and numerous requirements, such as dependence on Vps1p, V-ATPase pump activity, the electrochemical potential, and TORC1 and Fab1. It did not show discrepancies from any known requirements for vacuole fragmentation. This indicates that the in vitro system reiterates important aspects of the physiological process. The system is read out by confocal microscopy, followed by counting the numbers of large and small vacuoles, which limits throughput. Despite these inconveniences, the cell-free system is a major advance, allowing analyses of the fragmentation process that are more difficult or even impossible in vivo. This includes the addition or withdrawal of individual proteins, the use of antibodies to selectively inactivate individual proteins on the organelle surface, the use of cell-impermeable inhibitors, and the dissection of the reaction into intermediates. Therefore the in vitro system provides additional tools that are not available in microscopic in vivo analyses.
Vacuole fragmentation and fusion are in an equilibrium that determines vacuole structure (Weisman, 2003; Baars et al., 2007). Both reactions also occurred in our in vitro reactions. Whether net fragmentation or fusion of the organelle becomes apparent then depends on the ratio of the rates of these two reactions. This necessitates some precautions since a stimulation of fusion activity could shift the equilibrium and hence be misinterpreted as an inhibition of fragmentation. We addressed this problem in two ways. First, we used only inhibitors and mutants that are known not to stimulate fusion: GTPγS (Haas et al., 1994; Reese and Mayer, 2005), concanamycin A (Bayer et al., 2003; Muller et al., 2003), rapamycin, valinomycin, and FCCP (Mayer et al., 1996; Peters et al., 2001), antibodies to the V-ATPase (Bayer et al., 2003), withdrawal of ATP or prevention of its hydrolysis by EDTA (Conradt et al., 1994; Mayer et al., 1996), Δvps1 and vps1ts (Peters et al., 2004), withdrawal of cytosol (Conradt et al., 1992), and GDI (Haas et al., 1995). We verified and confirmed the lack of stimulation of fusion by these reagents under our assay conditions (data not shown). Second, fragmentation activity strongly predominates during the initial 60 min of the reaction. This predominance is sufficiently pronounced so that even addition of GDI, which reduces fusion activity by >90% (Figure 2E), does not significantly accelerate the kinetics of apparent fragmentation. Therefore, it appears that the changes in vacuole size and number are mainly defined via fragmentation activity during the first 60 min of incubation. The reappearance of large vacuoles, which occurs upon prolonged incubations, suggests that fusion can dominate the equilibrium at later time points. This is an interesting and potentially useful phenomenon. Whether it reproduces a physiological switch that lets the organelles go through an obligatory cycle of fragmentation and subsequent coalescence in the test tube must be carefully tested in future studies. Such a regulated switch might shift the fusion/fragmentation equilibrium to control organelle size and number, for example, in response to osmo- or nutrient-sensing signaling networks. The TOR requirement identified here could be part of such a mechanism.
Why do vacuoles fragment? Vacuoles fragment and fuse as part of the mechanism for their transmission to daughter cells (Weisman, 2003). They fulfill important functions in hydrolysis, osmoregulation, and storage of amino acids and ions (Klionsky et al., 1990) and modify their shape, number, and size in response to environmental changes. During logarithmic growth, cells of most yeast strains contain two to five vacuoles of intermediate size. Upon hypertonic shock, vacuoles fragment. They rapidly (<1 min) lose water and volume, but the membrane surface seems to remain constant. Transfer into hypotonic media promotes the opposite reaction—vacuole coalescence. Vacuoles may respond in this way because fragmentation and coalescence allow them to rapidly readjust their surface-to-volume ratio. This ratio determines membrane tension on the organelle, which in turn might influence central vacuolar functions, such as the activity of vacuolar transporters and ion channels. Vacuoles also coalesce into a single big organelle upon nutrient limitation and entry into stationary phase. Because starvation strongly induces the expression of vacuolar hydrolases (Knop et al., 1993), the resulting increase in hydrolytic capacity probably necessitates an expansion of the vacuolar volume in order to facilitate the degradation of large amounts of cytoplasmic material, which autophagy transfers into vacuoles under these conditions (Baba et al., 1994). Control over fragmentation and fusion of vacuoles could thus adjust vacuole size and number to physiological changes. It might, in addition, adjust vacuole volume and number to growth of the other cell constituents, that is, to integrate vacuole growth with the growth of the rest of the cell. Testing this hypothesis and separating these two aspects will require the identification of the relevant TORC1 targets on vacuoles.
The precise analysis of signaling events downstream of TORC1 was not the purpose of our study, but we nevertheless obtained information on a few candidate pathways. In contrast to Cdc28p, for which many targets are known, few direct target proteins for TOR have been identified. Our experiments did not reveal a role for any of them in vacuolar fragmentation. In addition, knockouts of key genes involved in TORC1-controlled pathways, such as the stress response pathway (MSN2, MSN4) and the nitrogen discrimination pathway (GLN3, URE2, GAT1), did not show effects on vacuole fragmentation. Many known outputs of TORC1 activity are regulated via PP2A- and PP2A-like protein phosphatases. Astonishingly, we identified a strong fragmentation defect for Δsit4 mutants but not for tap42-106, tap42-109, or tap42-11 mutants, which affect TORC1 control over SIT4 in other PP2A-regulated pathways (Di Como and Arndt, 1996; Jacinto et al., 2001; Duvel et al., 2003; De Virgilio and Loewith, 2006). This TAP42-dependent branch of TORC1 signaling has mainly been characterized using processes essential for survival. Vacuole fragmentation, which is nonessential under rich growth conditions (Weisman, 2003), provides an example for a TORC1 output that may be controlled via PP2A in a way that is independent of TAP42, or at least insensitive to the three well-characterized conditional alleles we used. It might implicate other associations of PP2As, such as those with Cdc55p, Rts1p, and Tpd3p, or, for Sit4p, with the SAP proteins (Jablonowski et al., 2009).
Development of cells depends on the coordination of proliferation and cell growth. These aspects are controlled by two central kinases—the cyclin-dependent kinase Cdc28p, which governs proliferation via the cell cycle, and TOR kinase, which regulates growth (Wullschleger et al., 2006). Whereas there are examples of organelle fragmentation being linked to the cell cycle and to control by Cdc28p, for example for the Golgi (Shorter and Warren, 2002; Colanzi et al., 2003) or for the nuclear envelope (Newport and Forbes, 1987), TOR has remained largely unexplored in these respects. A recent observation links mTOR activity to the behavior of mammalian lysosomes. Upon starvation and induction of autophagy, lysosomes fuse with many newly formed autophagosomes, thereby forming enlarged hybrid organelles—autolysosomes. During exit from the starved state, which reactivates mTOR, autolysosomes shed tubular and vesicular structures, which are assumed to be substrates for re-forming lysosomes (Yu et al., 2010; Rong et al., 2011). This shedding of large vesicular and tubular structures is a membrane fission reaction and in this sense equivalent to the vacuolar fragmentation that we studied. Apparent fragmentation of autolysosomes could result from an inhibition of fusion or from a stimulation of fission activity by mTOR. In analogy to our observations, we propose that mTOR activity selectively stimulates the (auto-) lysosomal fission machinery in order to trigger regeneration of lysosomes.
MATERIALS AND METHODS
Strains
See Table 1.
TABLE 1:
Strains used in this work.
| Strain | Genotype | Source or reference |
|---|---|---|
| BJ3505 | Mat a pep4::HIS3 prb1-D1.6R lys2-208 trp1-D101 ura3-52 gal2 can | Pieren et al. (2010) |
| DKY6281 | Mat a leu2-3, -112 ura3-52 his3-∆200 trp1-∆901 lys2-801 suc2-∆9 pho8::TRP1 | Pieren et al. (2010) |
| Δvps1 | BJ3505 Δvps1 | Peters et al. (2004) |
| vps1wt | BJ3505 Δvps1 + p416GPD vps1wt | Ekena et al. (1993) |
| vps1ts | BJ3505 Δvps1 + p416GPD vps1ts | Ekena et al. (1993) |
| BY4741 | MATα his3Δ1 met15Δ0 ura3Δ0 | Brachmann et al. (1998) |
| BY4741/2 | MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 | Brachmann et al. (1998) |
| ∆tco89 | BY4741/2 Δtco89 | R. Loewith |
| ∆ure2 | BY4741 Δure2 | This study |
| ∆gln3 | BY4741 Δgln3 | EUROSCARF |
| ∆gat1 | BY4741 Δgat1 | EUROSCARF |
| ∆sit4 | BY4741 Δsit4 | This study |
| W303-1A | MAT a ade2-1 can1-100 his3-11 15leu2-3 112trp1-1 ura3-1 GAL | Duvel et al. (2003) |
| Y3033 | W303-1A tap42::HIS3 + pRS414-TAP42 wt | Duvel et al. (2003) |
| Y3034 | W303-1A tap42::HIS3 + pRS414-tap42-106 ts | Duvel et al. (2003) |
| Y3035 | W303-1A tap42::HIS3 + pRS414-tap42-109 ts | Duvel et al. (2003) |
| TB50a | Mat a; leu2-3, ura3-52; trp1; rme1;his3D GAL+ HMLa | R. Loewith; Heitman et al. (1991) |
| RL206-4d | TB50a TOR1-1 TOR2-1 | R. Loewith |
| tap42-11 | TB50a sch9::kanMX tap42::HphMX4 + pRS414::SCH9wt + pRS415::tap42-11ts | R. Loewith |
| tap42 wt | TB50a sch9::kanMX tap42::HphMX4 + pRS414::SCH9wt + pRS415::tap42wt | R. Loewith |
| sch9 2D3E | TB50a sch9::kanMX tap42::HphMX4 + pRS414::sch9D3E + pRS415::TAP42wt | R. Loewith |
| sch9 as | TB50a sch9::kanMX + pRS414::SCH9as (Sch9T492G) | R. Loewith |
| ypk1 as | TB50a ypk1::kanMX ypk2::HIS3MX + pRS461::YPK1as | R. Loewith |
| ypk1 wt | TB50a ypk1::kanMX ypk2::HIS3MX + pRS461::YPK1wt | R. Loewith |
| ∆tor1 | BY4741 Δtor1 | EUROSCARF |
| ∆sfp1 | BY4741 Δsfp1 | EUROSCARF |
| ∆msn2 | BY4741 Δmsn2 | EUROSCARF |
| ∆msn4 | BY4741 Δmsn4 | EUROSCARF |
| ∆rtg1 | BY4741 Δrtg1 | EUROSCARF |
| ∆rtg3 | BY4741 Δrtg3 | EUROSCARF |
| ∆mpk1 | BY4741 Δmpk1 | EUROSCARF |
| ∆npr1 | BY4741 Δnpr1 | EUROSCARF |
| ∆mks1 | BY4741 Δmks1 | EUROSCARF |
EUROSCARF, European Saccharomyces cerevisiae Archive for Functional Analysis, Institute for Molecular Biosciences, Johann Wolfgang Goethe-University Frankfurt, Frankfurt, Germany. R. Loewith, University of Geneva, Geneva, Switzerland.
Plasmids introduced into strains are denoted by a +.
Reagents
Reagents included BODIPY FL-DHPE (Molecular Probes, Invitrogen, Carlsbad, CA), FM4-64 (Molecular Probes), concanamycin A (Alexis Biochemicals, San Diego, CA), rapamycin (Alexis), valinomycin (Sigma-Aldrich, St. Louis, MO), FCCP (Sigma-Aldrich), GTPγS (Roche, Indianapolis, IN), and microcystin LR (Alexis). Polyclonal antibodies to Vps1p and Vph1p were produced by injecting the soluble proteins subcutaneously into rabbit (Vps1p) or goat (Vph1p). Antibodies were affinity purified from the serum. Gdi1p was recombinantly expressed and purified from Escherichia coli (Mayer et al., 1996).
Cytosol preparation
Ten l of S. cerevisiae strain K91-1A were grown to early logarithmic phase in yeast extract/peptone/dextrose (YPD; 150 rpm, 30°C). Cells were harvested (5 min, 4°C, 4200 × g) and resuspended in one pellet volume of chilled cytosol buffer (200 mM sorbitol, 40 mM 1,4-piperazinediethanesulfonic acid [PIPES]/KOH, pH 6.8, 0.5 mM MgCl2, 1 mM dithiothreitol [DTT], 1 mM phenylmethylsufonylfluoride, and 1× protease inhibitor cocktail: 0.1 mM Pefablock SC [Roche], 0.2 μM leupeptin, 0.1 mM o-phenanthrolin, and 0.75 μM pepstatin A). Cells were pooled, centrifuged (5 min, 4°C, 4200 × g), and resuspended in cytosol buffer as a thick slurry. A thin stream of the slurry was poured into liquid nitrogen, yielding nuggets of frozen cells. They were cracked for 5–10 min in a Waring blender filled with liquid nitrogen. The homogenized powder was thawed and centrifuged (20 min, 4°C, 4000 × g). The supernatant was centrifuged again (Beckman Ti45 rotor, 30 min, 4°C, 120,000 × g). Lipid was aspirated from the top, and the supernatant was centrifuged as before. The cytosol was filtered (0.2 μm), adjusted to 30 mg/ml protein concentration with cytosol buffer, frozen in 100-μl aliquots in liquid nitrogen, and stored at −80°C.
Lyticase preparation
β-1,3-Glucanase from Oerskovia xanthineolytica was expressed in E. coli strain RSB 805. The protein was purified from the periplasmic space. A 10-l amount of RSB 805 was grown in Luria Bertani (LB) medium (5 g/l yeast extract, 10 g/l tryptone, 5 g/l NaCl) with 100 μg/ml ampicillin to OD600 = 0.6. Isopropyl-β-d-thiogalactoside 0.4 mM was added, and, after another 5 h at 30°C, cells were harvested (10 min, room temperature, 4200 × g), washed with 50 ml of washing buffer (25 mM Tris/Cl, pH 7.4), and centrifuged as before. Pellets were resuspended in 200 ml of washing buffer, supplemented with 2 mM EDTA and an equal volume of 40% (wt/vol) sucrose in washing buffer, and gently shaken at room temperature for 20 min. Cells were harvested (10 min, room temperature, 4200 × g), and the supernatant was completely removed. Flasks were chilled on ice. The cells from 1 l of culture were resuspended in 20 ml of ice-cold 0.5 mM MgSO4, gently shaken (4°C, 20 min), and centrifuged (10 min, 4°C, 4200 × g). Protein concentration of the supernatant varied around 1 mg/ml.
Vacuole isolation
For vacuole preparation, cells were precultured in YPD medium overnight (30°C, 150 rpm). Logarithmic cultures were inoculated (30°C, 14–16 h, 150 rpm) in baffled 2-l Erlenmeyer flasks with 1 l of YPD. At OD600 = 1.6–1.8, cells were centrifuged (3 min, 4000 × g, 4°C, JLA-10.500 rotor), resuspended in 50 ml of 0.1 M Tris/HCl (pH 8.9) with 10 mM DTT, incubated (7 min, 30°C), centrifuged (2 min, 4000 × g, 4°C), resuspended in 15 ml of SB (50 mM KPi, pH 7.5, 600 mM sorbitol in YPD with 0.2% glucose and 8 mg/ml lyticase), and transferred into 30-ml Corex tubes. Cells were incubated (25 min, 30°C), reisolated (2 min, 4000 × g, 4°C, JA20 rotor), and gently resuspended in 2.5 ml of 15% Ficoll 400 in PS buffer (10 mM PIPES/KOH, pH 6.8, 200 mM sorbitol). DEAE-dextrane (300 μl) was added from a fresh stock (0.4 mg/ml) in 15% Ficoll in PS. The cells were incubated (2 min, 30°C), chilled again, transferred to a SW41 tube, and overlaid with 3 ml of 8% Ficoll, 3 ml of 4% Ficoll, and 1.5 ml of 0% Ficoll (all in PS). After centrifugation (75 min, 150,000 × g, 2°C), vacuoles were harvested from the 0–4% interphase. Water purity is a critical issue, which can influence the obtainable absolute fragmentation activities. In the course of these studies we noted that purification of the water used for the media over a charcoal cartridge enhances the absolute activities of the vacuoles for some strain backgrounds, such as TB50.
Vacuole fragmentation in vitro
Reactions contained 3 μg of vacuoles in 30 μl of reaction buffer (20 mM PIPES/KOH, pH 6.8, 200 mM sorbitol, 5 mM MgCl2, 0.75 mM ATP, 1 mg/ml K91-1A cytosol extract, 0.2× protease inhibitor cocktail [final concentration 40 μM Pefablock SC, 2.1 μM leupeptin, 80 μM o-phenanthrolin, 1.5 μM pepstatin A], 30 mM creatine phosphate, 52.5 U/ml creatine kinase, and 100 mM or 150 mM KAc). The optimal KAc concentration differed between experiments due to batch differences in the culture media. The experiments were hence performed with two concentrations of KAc—100 and 150 mM—and the one resulting in the higher control activities was chosen to evaluate the experiment. To suppress the competing fusion activity, 0.3 mg/ml GDI (Reese et al., 2005) can be added (this was used, e.g., for experiments involving tor1-1/tor2-1 mutants). The ATP regenerating system, containing MgCl2, ATP, creatine phosphate, and creatine kinase, was added from a frozen stock. Creatine kinase was stored at –20°C in 10 mM PIPES/KOH, pH 6.8, and 50% glycerol. Negative controls without ATP contained creatine phosphate and PS buffer but no ATP and creatine kinase. Fragmentation reactions were incubated for 60 min at 27°C. Then vacuoles in the 30-μl reaction were stained with 2 μl of BODIPY FL-DHPE (10 μg/ml in 70% ethanol). A 3-μl amount was withdrawn and mixed with 6 μl of 0.2% LMP-agarose held at 37°C. A 1-μl amount of this suspension was transferred to cavity slides and analyzed by fluorescence confocal microscopy (Zeiss [Thornwood, NY] LSM 510; excitation 488 nm, 100×/1.4 numerical aperture PlanNeofluoar, electronic zoom 2, pinhole size 1.00 Airy unit). Laser intensity was 32.5, detector gain was 880, amplifier offset was −0.287, and amplifier gain was 1.
As an alternative, reactions could also be performed in poly-l-lysine–coated chambers. Coverglass chambers (Lab-Tek 155411; Nalge Nunc International, Rochester, NY) were washed once with ethanol 70% and twice with sterile water. After drying, a solution of poly-l-lysine (0.1% wt/vol) in water was added in the chamber and incubated at room temperature for 10 min. Then the solution was removed and the chamber was closed and stored at room temperature overnight. Coated chambers were used for fragmentation assays the next day. An 800-μl amount of vacuoles with a concentration of 0.3–0.5 mg/ml in PS buffer was stained with 10 μM rhodamine 123 for 3 min at 27°C. Then 500 μl of 15% Ficoll 400 in PS was added and mixed gently. The suspension was transferred into siliconized 2-ml Eppendorf tubes and overlaid with 300 μl of 4% Ficoll in PS and 400 μl of PS. The tubes were centrifuged (10 min, 8900 × g, 2°C), and the vacuoles were recovered from the 0–4% interphase. Vacuoles were incubated 30 min at room temperature in the coated chamber. Then the chambers were washed twice with PS, making sure to always keep some liquid in the chamber to avoid drying of the vesicles. Finally, the buffer was supplemented to a complete reaction mixture, and the chambers were incubated at 27°C for 1 h and directly analyzed by confocal fluorescence microscopy.
Quantification of the reaction in vitro
Pictures were taken close to the coverslip, where vacuoles sediment, or at the bottom of the chamber. Vacuoles were counted and grouped into classes according to their diameter d: small, d ≤ 0.6 μm; medium, 0.6 μm < d < 1.5 μm; large, d ≥ 1.5 μm. Vacuolar fragments in clusters were often too small and numerous to permit direct counting. Therefore their number was approximated via the area they occupied in the pictures (using the software ImageJ [National Institutes of Health, Bethesda, MD]). The number of small vacuolar fragments in the section was calculated by dividing this area by the area of an average small vacuole that has a diameter of 0.4 μm. The numbers of large, medium, and small vacuoles (Nl, Nm, and Ns, respectively) were used to calculate the ratio R = Ns/(Ns + Nm + Nl). To simplify representation and comparisons of fragmentation activity, we defined a fragmentation index FI: FI = Rpos/R×100 (%), where Rpos is the ratio for the positive control and R the ratio for the sample to be evaluated. Pictures of seven random fields per sample, containing the membrane equivalent to 10–50 vacuoles, were taken. The minimal number of vacuole equivalents analyzed was 200 per condition and experiment. FI of the positive controls (+ATP) ranged between 100 and 400 for all experiments evaluated.
Electron microscopy
Vacuoles were subjected to 10× fragmentation reactions. At the end of the reaction period the samples were supplemented with 2% glutaraldehyde, incubated for 30 min on ice, and supplemented with 50 μM glycine. After centrifugation (15 min, 100,000 × g, 4°C) the supernatant was withdrawn. The pellet was resuspended in 500 μl of buffer F (0.1 M Na-phosphate, pH 7.3, 200 mM sorbitol), centrifuged as before. This wash was repeated, and the final pellet was resuspended in a small volume of 1% agarose in buffer F. After solidification, we added 2% OsO4 in buffer F, incubated at room temperature (1 h), washed again with buffer F, and dehydrated in a series of 50, 70, 90, and 100% ethanol. Finally, the pellet was incubated with an ethanol/epon (1:1) mixture (30 min) and then with pure epon overnight at ambient temperature. After polymerization at 60°C (3 d), the samples were cut and analyzed by electron microscopy.
Vacuole fusion assay
Fusion reactions were performed either under fragmentation conditions as described earlier or under conditions promoting only fusion as described in Peters et al. (2004): 3 μg of each type of purified vacuole (DKY6281 and BJ3505) was incubated in a total volume of 30 μl of reaction buffer (20 mM PIPES/KOH, pH6.8, 200 mM sorbitol, 120 mM KCl, 0.5 mM MnCl2, 0.5 mM MgCl2, 0.5 mM ATP, 20 μM Pefablock SC, 1 μM leupeptin, 40 μM o-phenanthrolin, 0.75 μM pepstatin A, 20 mM creatine phosphate, and 35 U/ml creatine kinase). After 60 min at 27°C, alkaline phosphatase activity was determined by conversion of p-nitrophenol phosphate, which can be measured as change in OD400.
In vivo vacuole fragmentation
Yeast was precultured in YPD or selective medium (25°C). Overnight cultures were grown (14–16 h, 25°C, 150 rpm) in logarithmic phase (OD600 < 1). To visualize vacuolar membranes, in vivo cells were stained with FM4-64 (Vida and Emr, 1995). cultures were adjusted to OD600 = 0.4, and 10 μM FM4–64 was added from a 100 mM stock in dimethyl sulfoxide (DMSO). Cells were incubated for 1 h at 25°C, harvested (1 min, 3000 × g), washed twice in fresh medium, resuspended in YPD at OD600 = 0.4, and shaken for 1–2 h at 25°C. Fragmentation was induced by adding 0.4 M NaCl. After 10 min at 25°C, cells were centrifuged (15 s, 8000 × g), resuspended in 0.05 volume of the supernatant, and immediately analyzed by spinning disk confocal fluorescence microscopy (QLC100; Visitron, Puchheim, Germany) at minimal intensity of excitation light (488 nm). To quantify vacuole morphology, photos of at least 10 random fields were taken. The number of vacuolar vesicles in 100 cells per experiment and condition was determined, and cells were grouped into three categories: 1–2, 3–4, or >5 vacuoles per cell. Data from at least three independent experiments were averaged and the standard deviations calculated.
Statistics
Data from three to five independent experiments were averaged. Error bars represent the SD. Statistical significance was tested using Student's t test.
Acknowledgments
We thank Robbie Loewith, Claudio de Virgilio, Jim Broach, and Tom Stevens for strains; Monique Reinhardt, Véronique Comte, and Andrea Schmidt for assistance; and the Cellular Imaging Facility for support with microscopy. This work was supported by the Swiss National Science Foundation, the European Research Council, and the Human Frontier and Science Program.
Abbreviations used:
- GDI
GDP dissociation inhibitor
- PI-3,5-P2
phosphatidylinositol-3,5-bisphosphate
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-08-0703) on January 11, 2012.
REFERENCES
- Acharya U, Mallabiabarrena A, Acharya JK, Malhotra V. Signaling via mitogen-activated protein kinase kinase (MEK1) is required for Golgi fragmentation during mitosis. Cell. 1998;92:183–192. doi: 10.1016/s0092-8674(00)80913-7. [DOI] [PubMed] [Google Scholar]
- Antonny B. Membrane deformation by protein coats. Curr Opin Cell Biol. 2006;18:386–394. doi: 10.1016/j.ceb.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Audhya A, Loewith R, Parsons AB, Gao L, Tabuchi M, Zhou H, Boone C, Hall MN, Emr SD. Genome-wide lethality screen identifies new PI4,5P2 effectors that regulate the actin cytoskeleton. EMBO J. 2004;23:3747–3757. doi: 10.1038/sj.emboj.7600384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baars TL, Petri S, Peters C, Mayer A. Role of the V-ATPase in regulation of the vacuolar fission-fusion equilibrium. Mol Biol Cell. 2007;18:3873–3882. doi: 10.1091/mbc.E07-03-0205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Takeshige K, Baba N, Ohsumi Y. Ultrastructural analysis of the autophagic process in yeast: detection of autophagosomes and their characterization. J Cell Biol. 1994;124:903–913. doi: 10.1083/jcb.124.6.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer MJ, Reese C, Buhler S, Peters C, Mayer A. Vacuole membrane fusion: V0 functions after trans-SNARE pairing and is coupled to the Ca2+-releasing channel. J Cell Biol. 2003;162:211–222. doi: 10.1083/jcb.200212004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleazard W, McCaffery JM, King EJ, Bale S, Mozdy A, Tieu Q, Nunnari J, Shaw JM. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonangelino CJ, Catlett NL, Weisman LS. Vac7p, a novel vacuolar protein, is required for normal vacuole inheritance and morphology. Mol Cell Biol. 1997;17:6847–6858. doi: 10.1128/mcb.17.12.6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonangelino CJ, Nau JJ, Duex JE, Brinkman M, Wurmser AE, Gary JD, Emr SD, Weisman LS. Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p. J Cell Biol. 2002;156:1015–1028. doi: 10.1083/jcb.200201002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998;14:115–132. doi: 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Cardenas ME, Heitman J. FKBP12-rapamycin target TOR2 is a vacuolar protein with an associated phosphatidylinositol-4 kinase activity. EMBO J. 1995;14:5892–5907. doi: 10.1002/j.1460-2075.1995.tb00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catlett NL, Weisman LS. Divide and multiply: organelle partitioning in yeast. Curr Opin Cell Biol. 2000;12:509–516. doi: 10.1016/s0955-0674(00)00124-1. [DOI] [PubMed] [Google Scholar]
- Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- Colanzi A, Suetterlin C, Malhotra V. Cell-cycle-specific Golgi fragmentation: how and why? Curr Opin Cell Biol. 2003;15:462–467. doi: 10.1016/s0955-0674(03)00067-x. [DOI] [PubMed] [Google Scholar]
- Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- Conradt B, Haas A, Wickner W. Determination of four biochemically distinct, sequential stages during vacuole inheritance in vitro. J Cell Biol. 1994;126:99–110. doi: 10.1083/jcb.126.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt B, Shaw J, Vida T, Emr S, Wickner W. In vitro reactions of vacuole inheritance in Saccharomyces cerevisiae. J Cell Biol. 1992;119:1469–1479. doi: 10.1083/jcb.119.6.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke FT, Dove SK, McEwen RK, Painter G, Holmes AB, Hall MN, Michell RH, Parker PJ. The stress-activated phosphatidylinositol 3-phosphate 5-kinase Fab1p is essential for vacuole function in S. cerevisiae. Curr Biol. 1998;8:1219–1222. doi: 10.1016/s0960-9822(07)00513-1. [DOI] [PubMed] [Google Scholar]
- Corda D, Hidalgo Carcedo C, Bonazzi M, Luini A, Spano S. Molecular aspects of membrane fission in the secretory pathway. Cell Mol Life Sci. 2002;59:1819–1832. doi: 10.1007/PL00012508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo JL, Diaz-Troya S, Florencio FJ. Inhibition of target of rapamycin signaling by rapamycin in the unicellular green alga Chlamydomonas reinhardtii. Plant Physiol. 2005;139:1736–1749. doi: 10.1104/pp.105.070847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Virgilio C, Loewith R. Cell growth control: little eukaryotes make big contributions. Oncogene. 2006;25:6392–6415. doi: 10.1038/sj.onc.1209884. [DOI] [PubMed] [Google Scholar]
- Di Como CJ, Arndt KT. Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases. Genes Dev. 1996;10:1904–1916. doi: 10.1101/gad.10.15.1904. [DOI] [PubMed] [Google Scholar]
- Dove SK, McEwen RK, Mayes A, Hughes DC, Beggs JD, Michell RH. Vac14 controls PtdIns(3,5)P(2) synthesis and Fab1-dependent protein trafficking to the multivesicular body. Curr Biol. 2002;12:885–893. doi: 10.1016/s0960-9822(02)00891-6. [DOI] [PubMed] [Google Scholar]
- Dove SK, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. EMBO J. 2004;23:1922–1933. doi: 10.1038/sj.emboj.7600203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19:15–26. doi: 10.1016/j.molcel.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Duex JE, Nau JJ, Kauffman EJ, Weisman LS. Phosphoinositide 5-phosphatase Fig 4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryot Cell. 2006a;5:723–731. doi: 10.1128/EC.5.4.723-731.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duex JE, Tang F, Weisman LS. The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol. 2006b;172:693–704. doi: 10.1083/jcb.200512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvel K, Broach JR. The role of phosphatases in TOR signaling in yeast. Curr Top Microbiol Immunol. 2004;279:19–38. doi: 10.1007/978-3-642-18930-2_2. [DOI] [PubMed] [Google Scholar]
- Duvel K, Santhanam A, Garrett S, Schneper L, Broach JR. Multiple roles of Tap42 in mediating rapamycin-induced transcriptional changes in yeast. Mol Cell. 2003;11:1467–1478. doi: 10.1016/s1097-2765(03)00228-4. [DOI] [PubMed] [Google Scholar]
- Efe JA, Botelho RJ, Emr SD. The Fab1 phosphatidylinositol kinase pathway in the regulation of vacuole morphology. Curr Opin Cell Biol. 2005;17:402–408. doi: 10.1016/j.ceb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Efe JA, Botelho RJ, Emr SD. Atg18 regulates organelle morphology and Fab1 kinase activity independent of its membrane recruitment by phosphatidylinositol 3,5-bisphosphate. Mol Biol Cell. 2007;18:4232–4244. doi: 10.1091/mbc.E07-04-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekena K, Vater CA, Raymond CK, Stevens TH. The VPS1 protein is a dynamin-like GTPase required for sorting proteins to the yeast vacuole. Ciba Found Symp. 1993;176:198–211. doi: 10.1002/9780470514450.ch13. discussion, 211–214. [DOI] [PubMed] [Google Scholar]
- Gao H, Kadirjan-Kalbach D, Froehlich JE, Osteryoung KW. ARC5, a cytosolic dynamin-like protein from plants, is part of the chloroplast division machinery. Proc Natl Acad Sci USA. 2003;100:4328–4333. doi: 10.1073/pnas.0530206100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary JD, Sato TK, Stefan CJ, Bonangelino CJ, Weisman LS, Emr SD. Regulation of Fab1 phosphatidylinositol 3-phosphate 5-kinase pathway by Vac7 protein and Fig4, a polyphosphoinositide phosphatase family member. Mol Biol Cell. 2002;13:1238–1251. doi: 10.1091/mbc.01-10-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary JD, Wurmser AE, Bonangelino CJ, Weisman LS, Emr SD. Fab1p is essential for PtdIns(3)P 5-kinase activity and the maintenance of vacuolar size and membrane homeostasis. J Cell Biol. 1998;143:65–79. doi: 10.1083/jcb.143.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorner W, Schuller C, Ruis H. Being at the right place at the right time: the role of nuclear transport in dynamic transcriptional regulation in yeast. Biol Chem. 1999;380:147–150. doi: 10.1515/BC.1999.022. [DOI] [PubMed] [Google Scholar]
- Haas A, Conradt B, Wickner W. G-protein ligands inhibit in vitro reactions of vacuole inheritance. J Cell Biol. 1994;126:87–97. doi: 10.1083/jcb.126.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas A, Scheglmann D, Lazar T, Gallwitz D, Wickner W. The GTPase Ypt7p of Saccharomyces cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. EMBO J. 1995;14:5258–5270. doi: 10.1002/j.1460-2075.1995.tb00210.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Higa MM, Ullman KS, Prunuske AJ. Studying nuclear disassembly in vitro using Xenopus egg extract. Methods. 2006;39:284–290. doi: 10.1016/j.ymeth.2006.06.004. [DOI] [PubMed] [Google Scholar]
- Hoepfner D, van den Berg M, Philippsen P, Tabak HF, Hettema EH. A role for Vps1p, actin, and the Myo2p motor in peroxisome abundance and inheritance in Saccharomyces cerevisiae. J Cell Biol. 2001;155:979–990. doi: 10.1083/jcb.200107028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Jablonowski D, Taubert JE, Bar C, Stark MJ, Schaffrath R. Distinct subsets of Sit4 holophosphatases are required for inhibition of Saccharomyces cerevisiae growth by rapamycin and zymocin. Eukaryot Cell. 2009;8:1637–1647. doi: 10.1128/EC.00205-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Guo B, Arndt KT, Schmelzle T, Hall MN. TIP41 interacts with TAP42 and negatively regulates the TOR signaling pathway. Mol Cell. 2001;8:1017–1026. doi: 10.1016/s1097-2765(01)00386-0. [DOI] [PubMed] [Google Scholar]
- Jin N, et al. VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. EMBO J. 2008;27:3221–3234. doi: 10.1038/emboj.2008.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Fujioka Y, Suzuki NN, Inagaki F, Wullschleger S, Loewith R, Hall MN, Ohsumi Y. Tor2 directly phosphorylates the AGC kinase Ypk2 to regulate actin polarization. Mol Cell Biol. 2005;25:7239–7248. doi: 10.1128/MCB.25.16.7239-7248.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano F, Takenaka K, Yamamoto A, Nagayama K, Nishida E, Murata M. MEK and Cdc2 kinase are sequentially required for Golgi disassembly in MDCK cells by the mitotic Xenopus extracts. J Cell Biol. 2000;149:357–368. doi: 10.1083/jcb.149.2.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Herman PK, Emr SD. The fungal vacuole: composition, function, and biogenesis. Microbiol Rev. 1990;54:266–292. doi: 10.1128/mr.54.3.266-292.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop M, Schiffer HH, Rupp S, Wolf DH. Vacuolar/lysosomal proteolysis: proteases, substrates, mechanisms. Curr Opin Cell Biol. 1993;5:990–996. doi: 10.1016/0955-0674(93)90082-2. [DOI] [PubMed] [Google Scholar]
- Kuravi K, Nagotu S, Krikken AM, Sjollema K, Deckers M, Erdmann R, Veenhuis M, van der Klei IJ. Dynamin-related proteins Vps1p and Dnm1p control peroxisome abundance in Saccharomyces cerevisiae. J Cell Sci. 2006;119:3994–4001. doi: 10.1242/jcs.03166. [DOI] [PubMed] [Google Scholar]
- LaGrassa TJ, Ungermann C. The vacuolar kinase Yck3 maintains organelle fragmentation by regulating the HOPS tethering complex. J Cell Biol. 2005;168:401–414. doi: 10.1083/jcb.200407141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lempiainen H, Uotila A, Urban J, Dohnal I, Ammerer G, Loewith R, Shore D. Sfp1 interaction with TORC1 and Mrs6 reveals feedback regulation on TOR signaling. Mol Cell. 2009;33:704–716. doi: 10.1016/j.molcel.2009.01.034. [DOI] [PubMed] [Google Scholar]
- Li X, Gould SJ. The dynamin-like GTPase DLP1 is essential for peroxisome division and is recruited to peroxisomes in part by PEX11. J Biol Chem. 2003;278:17012–17020. doi: 10.1074/jbc.M212031200. [DOI] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Luzio JP, Poupon V, Lindsay MR, Mullock BM, Piper RC, Pryor PR. Membrane dynamics and the biogenesis of lysosomes. Mol Membr Biol. 2003;20:141–154. doi: 10.1080/0968768031000089546. [DOI] [PubMed] [Google Scholar]
- MacKintosh C, Beattie KA, Klumpp S, Cohen P, Codd GA. Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatases 1 and 2A from both mammals and higher plants. FEBS Lett. 1990;264:187–192. doi: 10.1016/0014-5793(90)80245-e. [DOI] [PubMed] [Google Scholar]
- Mancias JD, Goldberg J. Exiting the endoplasmic reticulum. Traffic. 2005;6:278–285. doi: 10.1111/j.1600-0854.2005.00279.x. [DOI] [PubMed] [Google Scholar]
- Mayer A, Scheglmann D, Dove S, Glatz A, Wickner W, Haas A. Phosphatidylinositol 4,5-bisphosphate regulates two steps of homotypic vacuole fusion. Mol Biol Cell. 2000;11:807–817. doi: 10.1091/mbc.11.3.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- Misteli T, Warren G. COP-coated vesicles are involved in the mitotic fragmentation of Golgi stacks in a cell-free system. J Cell Biol. 1994;125:269–282. doi: 10.1083/jcb.125.2.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlberg AB, Warnock DE, Schmid SL. Domain structure and intramolecular regulation of dynamin GTPase. EMBO J. 1997;16:6676–6683. doi: 10.1093/emboj/16.22.6676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller O, Neumann H, Bayer MJ, Mayer A. Role of the Vtc proteins in V-ATPase stability and membrane trafficking. J Cell Sci. 2003;116:1107–1115. doi: 10.1242/jcs.00328. [DOI] [PubMed] [Google Scholar]
- Nagotu S, Veenhuis M, van der Klei IJ. Divide et impera: the dictum of peroxisomes. Traffic. 2010;11:175–184. doi: 10.1111/j.1600-0854.2009.01019.x. [DOI] [PubMed] [Google Scholar]
- Newport JW, Forbes DJ. The nucleus: structure, function, and dynamics. Annu Rev Biochem. 1987;56:535–565. doi: 10.1146/annurev.bi.56.070187.002535. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Shaw JM. Mitochondrial morphology and dynamics in yeast and multicellular eukaryotes. Annu Rev Genet. 2005;39:503–536. doi: 10.1146/annurev.genet.38.072902.093019. [DOI] [PubMed] [Google Scholar]
- Osteryoung KW, McAndrew RS. The plastid division machine. Annu Rev Plant Physiol Plant Mol Biol. 2001;52:315–333. doi: 10.1146/annurev.arplant.52.1.315. [DOI] [PubMed] [Google Scholar]
- Ostrowicz CW, Meiringer CT, Ungermann C. Yeast vacuole fusion: a model system for eukaryotic endomembrane dynamics. Autophagy. 2008;4:5–19. doi: 10.4161/auto.5054. [DOI] [PubMed] [Google Scholar]
- Peplowska K, Markgraf DF, Ostrowicz CW, Bange G, Ungermann C. The CORVET tethering complex interacts with the yeast Rab5 homolog Vps21 and is involved in endo-lysosomal biogenesis. Dev Cell. 2007;12:739–750. doi: 10.1016/j.devcel.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Peters C, Baars TL, Buhler S, Mayer A. Mutual control of membrane fission and fusion proteins. Cell. 2004;119:667–678. doi: 10.1016/j.cell.2004.11.023. [DOI] [PubMed] [Google Scholar]
- Peters C, Bayer MJ, Buhler S, Andersen JS, Mann M, Mayer A. Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature. 2001;409:581–588. doi: 10.1038/35054500. [DOI] [PubMed] [Google Scholar]
- Pfaller R, Newport JW. Assembly/disassembly of the nuclear envelope membraneCharacterization of the membrane-chromatin interaction using partially purified regulatory enzymes. J Biol Chem. 1995;270:19066–19072. doi: 10.1074/jbc.270.32.19066. [DOI] [PubMed] [Google Scholar]
- Pieren M, Schmidt A, Mayer A. The SM protein Vps33 and the t-SNARE H(abc) domain promote fusion pore opening. Nat Struct Mol Biol. 2010;17:710–717. doi: 10.1038/nsmb.1809. [DOI] [PubMed] [Google Scholar]
- Prunuske AJ, Ullman KS. The nuclear envelope: form and reformation. Curr Opin Cell Biol. 2006;18:108–116. doi: 10.1016/j.ceb.2005.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor PR, Mullock BM, Bright NA, Gray SR, Luzio JP. The role of intraorganellar Ca(2+) in late endosome-lysosome heterotypic fusion and in the reformation of lysosomes from hybrid organelles. J Cell Biol. 2000;149:1053–1062. doi: 10.1083/jcb.149.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabouille C, Jokitalo E. Golgi apparatus partitioning during cell division. Mol Membr Biol. 2003;20:117–127. doi: 10.1080/0968768031000084163. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Misteli T, Watson R, Warren G. Reassembly of Golgi stacks from mitotic Golgi fragments in a cell-free system. J Cell Biol. 1995;129:605–618. doi: 10.1083/jcb.129.3.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese C, Heise F, Mayer A. Trans-SNARE pairing can precede a hemifusion intermediate in intracellular membrane fusion. Nature. 2005;436:410–414. doi: 10.1038/nature03722. [DOI] [PubMed] [Google Scholar]
- Reese C, Mayer A. Transition from hemifusion to pore opening is rate limiting for vacuole membrane fusion. J Cell Biol. 2005;171:981–990. doi: 10.1083/jcb.200510018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke A, Anderson S, McCaffery JM, Yates J, 3rd, Aronova S, Chu S, Fairclough S, Iverson C, Wedaman KP, Powers T. TOR complex 1 includes a novel component, Tco89p (YPL180w), and cooperates with Ssd1p to maintain cellular integrity in Saccharomyces cerevisiae. J Biol Chem. 2004;279:14752–14762. doi: 10.1074/jbc.M313062200. [DOI] [PubMed] [Google Scholar]
- Rong Y, et al. Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci USA. 2011;108:7826–7831. doi: 10.1073/pnas.1013800108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader M, Fahimi HD. Growth and division of peroxisomes. Int Rev Cytol. 2006;255:237–290. doi: 10.1016/S0074-7696(06)55005-3. [DOI] [PubMed] [Google Scholar]
- Shaw JM, Nunnari J. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 2002;12:178–184. doi: 10.1016/s0962-8924(01)02246-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Warren G. A role for the vesicle tethering protein, p115, in the post-mitotic stacking of reassembling Golgi cisternae in a cell-free system. J Cell Biol. 1999;146:57–70. doi: 10.1083/jcb.146.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J, Warren G. Golgi architecture and inheritance. Annu Rev Cell Dev Biol. 2002;18:379–420. doi: 10.1146/annurev.cellbio.18.030602.133733. [DOI] [PubMed] [Google Scholar]
- Storrie B, Desjardins M. The biogenesis of lysosomes: is it a kiss and run, continuous fusion and fission process? Bioessays. 1996;18:895–903. doi: 10.1002/bies.950181108. [DOI] [PubMed] [Google Scholar]
- Sturgill TW, Cohen A, Diefenbacher M, Trautwein M, Martin DE, Hall MN. TOR1 and TOR2 have distinct locations in live cells. Eukaryot Cell. 2008;7:1819–1830. doi: 10.1128/EC.00088-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweitzer SM, Hinshaw JE. Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell. 1998;93:1021–1029. doi: 10.1016/s0092-8674(00)81207-6. [DOI] [PubMed] [Google Scholar]
- Urban J, et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007;26:663–674. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- van der Goot FG, Gruenberg J. Intra-endosomal membrane traffic. Trends Cell Biol. 2006;16:514–521. doi: 10.1016/j.tcb.2006.08.003. [DOI] [PubMed] [Google Scholar]
- Vida TA, Emr SD. A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol. 1995;128:779–792. doi: 10.1083/jcb.128.5.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman LS. Yeast vacuole inheritance and dynamics. Annu Rev Genet. 2003;37:435–460. doi: 10.1146/annurev.genet.37.050203.103207. [DOI] [PubMed] [Google Scholar]
- Weisman LS, Bacallao R, Wickner W. Multiple methods of visualizing the yeast vacuole permit evaluation of its morphology and inheritance during the cell cycle. J Cell Biol. 1987;105:1539–1547. doi: 10.1083/jcb.105.4.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner W. Membrane fusion: five lipids, four SNAREs, three chaperones, two nucleotides, and a Rab, all dancing in a ring on yeast vacuoles. Annu Rev Cell Dev Biol. 2010;26:115–136. doi: 10.1146/annurev-cellbio-100109-104131. [DOI] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Yan M, Rayapuram N, Subramani S. The control of peroxisome number and size during division and proliferation. Curr Opin Cell Biol. 2005;17:376–383. doi: 10.1016/j.ceb.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Yoon Y, McNiven MA. Mitochondrial division: new partners in membrane pinching. Curr Biol. 2001;11:R67–R70. doi: 10.1016/s0960-9822(01)00011-2. [DOI] [PubMed] [Google Scholar]
- Yu L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]