

The endoplasmic reticulum (ER) unfolded protein response (UPR) is correlated with changes in unfolded secretory levels. A novel fluorescence biosensor now reports changes in the unfolded protein burden. This reporter reveals a form of ER stress—inositol withdrawal—that stimulates the UPR without changes in unfolded protein levels.
Abstract
Accumulation of misfolded secretory proteins in the endoplasmic reticulum (ER) activates the unfolded protein response (UPR) stress pathway. To enhance secretory protein folding and promote adaptation to stress, the UPR upregulates ER chaperone levels, including BiP. Here we describe chromosomal tagging of KAR2, the yeast homologue of BiP, with superfolder green fluorescent protein (sfGFP) to create a multifunctional endogenous reporter of the ER folding environment. Changes in Kar2p-sfGFP fluorescence levels directly correlate with UPR activity and represent a robust reporter for high-throughput analysis. A novel second feature of this reporter is that photobleaching microscopy (fluorescence recovery after photobleaching) of Kar2p-sfGFP mobility reports on the levels of unfolded secretory proteins in individual cells, independent of UPR status. Kar2p-sfGFP mobility decreases upon treatment with tunicamycin or dithiothreitol, consistent with increased levels of unfolded proteins and the incorporation of Kar2p-sfGFP into slower-diffusing complexes. During adaptation, we observe a significant lag between down-regulation of the UPR and resolution of the unfolded protein burden. Finally, we find that Kar2p-sfGFP mobility significantly increases upon inositol withdrawal, which also activates the UPR, apparently independent of unfolded protein levels. Thus Kar2p mobility represents a powerful new tool capable of distinguishing between the different mechanisms leading to UPR activation in living cells.
INTRODUCTION
Proper secretory protein folding in the endoplasmic reticulum (ER) is essential for the maintenance of homeostasis and cell viability. The eukaryotic cell has evolved machinery and signaling pathways to enhance secretory protein folding and stability (Brodsky and Skach, 2011). In the ER, various chaperones directly assist folding of nascent peptides as they enter the ER lumen (Ma and Hendershot, 2004). The quality control (QC) machinery remodels the newly synthesized protein via multiple posttranslational modifications, including formation of disulfide bonds (Feige and Hendershot, 2011) and N-linked glycosylation (Hulsmeier et al., 2011). Failure to correctly complete these tasks can result in a misfolded protein. If accumulation of misfolded protein exceeds the capacity of the ER QC machinery, the cell enters a state of ER stress. To cope with accumulation of misfolded proteins, the cell can activate an adaptive program termed the unfolded protein response (UPR; Ron and Walter, 2007). UPR sensors monitor the ER lumen for increased levels of misfolded proteins and then initiate the UPR (Kimata and Kohno, 2011). In yeast, the sole UPR sensor is the endoribonuclease Ire1p (Mori, 2009). During UPR activation, Kar2p releases from the luminal domain of Ire1p, enabling Ire1p dimerization (Oikawa et al., 2005). The Ire1p dimers undergo transautophosphorylation, assemble into clusters (Kimata et al., 2007; Aragon et al., 2009), and activate the site-specific RNase domain of the protein (Sidrauski and Walter, 1997). Ire1p cleaves HAC1 mRNA to remove a 252-nucleotide intron to generate the spliced functional form of the mRNA (Cox and Walter, 1996; Ruegsegger et al., 2001). The resulting translated Hac1p transcription factor up-regulates numerous cell functions, among them the transcription of genes encoding ER QC machinery, including chaperones such as Kar2p (Harding et al., 1999) and ER-associated protein degradation (ERAD) components (Yoshida et al., 2003). Hac1p also stimulates ER membrane expansion (Bernales et al., 2006; Schuck et al., 2009). These changes increase the folding capacity of the ER and can enable a return to homeostasis. Therefore UPR signaling is critical during ER stress (Chawla et al., 2011; Rubio et al., 2011), and failure to generate an adaptive UPR can result in cell death (Hetz et al., 2006; Tabas and Ron, 2011).
Cells are described as experiencing ER stress if they exhibit activation of the UPR components. Binding of accumulated misfolded proteins by Ire1p has been shown to directly activate the UPR in yeast (Gardner and Walter, 2011; Promlek et al., 2011). The classic markers of ER stress include Ire1p activation, HAC1 splicing, and increased Kar2p levels (Kimata and Kohno, 2011). In contrast to these readily measurable parameters, it has been proven to be difficult to quantitate the global levels of unfolded protein accumulating in the ER lumen during the UPR. Moreover, most reporters rely on aspects of UPR sensor function and therefore cannot measure ER stress in cells with compromised UPR signaling. Our lab recently developed a method to measure the unfolded protein burden in living cells that exploits the ability of the chaperone BiP (the mammalian Kar2p homologue) to recognize and directly bind unfolded proteins (Lai et al., 2010; Lajoie and Snapp, 2011). Using the technique of fluorescence recovery after photobleaching (FRAP), we demonstrated that green fluorescent protein (GFP)–tagged BiP diffusional mobility correlates with unfolded protein accumulation independent of the status of the UPR. In mammalian cells, this assay is complicated by the presence of endogenous BiP, which probably masks the full extent of BiP occupancy. Here we circumvented this limitation by chromosomal tagging of KAR2 in budding yeast with the superfolder variant of GFP (sfGFP; Janke et al., 2004; Pedelacq et al., 2006). This new endogenous reporter allowed us to 1) confirm the appropriate localization and function of Kar2p-sfGFP, 2) directly quantify unfolded protein accumulation in cells independent of UPR activation, and 3) investigate the effects of various forms of ER stress on the unfolded protein burden in living cells independent of and relative to UPR activation.
RESULTS
Generation of endogenous Kar2p-sfGFP
To generate an endogenous fluorescent Kar2p variant, we exploited the high efficiency of homologous recombination in yeast and the ability to perform chromosomal tagging of endogenous genes with fluorescent proteins (Huh et al., 2003). Two important modifications to previous efforts involving GFP tagging were incorporated. First, our lab recently established that the improved folding capacity of sfGFP prevents significant misfolding of GFP via inappropriate disulfide bonds and minimizes the formation of nonfluorescent species in oxidizing environments, including the ER (Aronson et al., 2011). Therefore we used sfGFP for our tagging efforts. Second, it was previously reported that yeast cells carrying Kar2p-GFP were viable, but the cellular localization of the fusion protein was ambiguous (Huh et al., 2003). This result could be partially explained by disruption of the ER retrieval motif, HDEL, on the resulting fusion protein (Munro and Pelham, 1987; Snapp, 2009). Therefore we added an HDEL motif to the C-terminus of the sfGFP to restore HDEL-mediated slowing of the loss of Kar2p from the ER (Figure 1A). The sfGFP-HDEL construct, along with a HIS3-selectable marker, was integrated at the KAR2 locus. Successful tagging of the gene and production of the Kar2p-sfGFP fusion protein was confirmed by immunoblot for both Kar2p and sfGFP (Figure 1C). Imaging of live cells expressing Kar2p-sfGFP revealed a typical pattern of peripheral and nuclear ER fluorescence expected for an ER-localized protein (Figure 1B).
FIGURE 1:

Expression of Kar2p-sfGFP in yeast. (A) Schematic of sfGFP-HDEL vector used to chromosomally tag Kar2p with sfGFP. Arrows indicate distinct transcription of fusion protein and selection marker. (B) Representative fluorescent image of wild-type yeast expressing Kar2p-sfGFP in a typical ER pattern. Bar, 10 μm. (C) Immunoblots of extracts from yeast cells expressing either the endogenous Kar2p or Kar2p-sfGFP. The bands below the Kar2p-sfGFP lane in the anti-Kar2p immunoblot are also evident in a long exposure of the anti-GFP blot, indicating that these are degradation products that still contain the fused sfGFP and are likely due to the high levels of proteases in yeast lysates (North and Beynon, 2001). (D) Wild-type yeast strains expressing either endogenous or Kar2p-sfGFP were diluted to OD600 nm = 0.5, serially diluted (10-fold), and spotted on plates containing 0 or 1.0 μg/ml Tm and grown at 30°C. Alternatively, untreated cells were grown at 37°C to assay temperature sensitivity of the various strains. (E) Yeast cells expressing either wild-type Kar2p or Kar2p-sfGFP and the UPR-mCherry reporter were treated with 5 mM or no DTT for 1 h and analyzed by flow cytometry. Median fluorescence intensity (MFI) for UPR-mCherry is shown for three biological replicates. (F) RNA from yeast cells expressing either wild-type Kar2p or Kar2p-sfGFP was isolated, and HAC1 splicing was analyzed by Northern blot. Unspliced (uHAC1) and spliced (sHAC1) products are indicated.
Next we established the functionality of the Kar2p-sfGFP protein. KAR2 is essential for cell viability (Normington et al., 1989). KAR2 conditional mutants are unable to rescue cells from low levels of ER stressors such as tunicamycin (Tm), an inhibitor of N-glycosylation (Rose et al., 1989; Kimata et al., 2003), or elevated temperature, that is, 37°C. The Kar2p-sfGFP strain grew well on synthetic media and grew only slightly less well on Tm-containing media relative to the wild-type strain (Figure 1D). For comparison, ire1Δ cells, which cannot induce the UPR, were unable to survive on Tm. Moreover, when cultured at 37°C, Kar2p-sfGFP cells grew robustly (Figure 1D). This contrasts with temperature-sensitive KAR2 mutants, which grow robustly at the permissive temperature of 22°C and grow poorly at higher temperatures (Polaina and Conde, 1982; Kimata et al., 2007).
To further characterize the Kar2p-sfGFP fusion protein, we investigated whether the sfGFP fusion on Kar2p affected UPR regulation. A plasmid containing the fluorescent UPR reporter UPR-mCherry (Merksamer et al., 2008) was introduced into wild-type and Kar2p-sfGFP yeast strains. The reporter mCherry expression is driven by a minimal CYC1 promoter and four tandem UPR elements and thus should be expressed only during ER stress. The fluorescence intensity of UPR-mCherry was recorded by flow cytometry (Figure 1E) in untreated and dithiothreitol (DTT)-treated (which prevents the formation of disulfide bonds in nascent secretory proteins) strains. In addition, the splicing of HAC1 mRNA in response to DTT-mediated UPR activation was assessed directly by Northern analysis (Figure 1F). Together these results established two important features of the chromosomally tagged Kar2p-sfGFP strain. First, the GFP fusion does not constitutively activate the UPR at steady state. Second, Kar2p-sfGFP cells can induce a robust UPR in response to unfolded protein stress. Note that efficient release of Kar2p from Ire1p is required for UPR activation and Kar2p binding, and release of substrates correlates with regulated UPR activation (Kimata et al., 2003). However, Ire1p mutants defective in Kar2p binding do not exhibit constitutive UPR, suggesting that Kar2p buffers against unfolded protein levels and modulates the stress response rather than directly regulates Ire1p activation (Kimata et al., 2004; Oikawa et al., 2009; Pincus et al., 2010).
Next we asked whether we could monitor Kar2p-sfGFP levels in living cells during ER stress as a proxy for UPR activation. KAR2 is a classic UPR target gene, and its expression increases following treatment with ER stressors, including DTT and Tm (Travers et al., 2000). We treated Kar2p-sfGFP cells with either ER stressor and quantified the fluorescence levels by microscopy and flow cytometry. The Kar2p-sfGFP signal increased nearly 10-fold after a 4 h treatment either Tm or DTT (Figure 2, A and B). The increase in Kar2p-sfGFP signal was dependent on stress activation of the UPR, since no change in Kar2p-sfGFP fluorescence intensity was observed in an ire1Δ strain (Figure 2C). Moreover, the change in Kar2p-sfGFP fluorescence signal was specific, since the fluorescence intensity of the inert reporter ER-sfGFP-HDEL did not change in response to DTT treatment (Figure 2D). Finally, the comparable accumulation of wild-type and Kar2p-sfGFP proteins in response to ER stress indicates that the addition of the sfGFP tag preserves the UPR regulation of target genes, including Kar2p (Figure 2E). These results demonstrate that the Kar2p-sfGFP fusion protein robustly reports on UPR activation by both standard microscopy and by high-throughput methods, including flow cytometry.
FIGURE 2:

Increased expression of Kar2p-sfGFP following ER stress. (A) Representative phase and fluorescence images of wild-type and ire1Δ yeast strains expressing Kar2p-sfGFP untreated or treated with 5.0 μg/ml Tm for 4 h. (B) Yeast cells expressing wild-type Kar2p or Kar2p-sfGFP were treated with either 0 or 5 mM DTT for 4 h and analyzed by flow cytometry. Increased GFP fluorescence was detected in stressed cells. (C) Wild-type and ire1Δ yeast strains expressing Kar2p-sfGFP were treated with either DTT (1 or 5 mM) or Tm (1 or 5 μg/ml) and MFI values were measured at different times by flow cytometry. A functional UPR was required for stress-induced increases in Kar2p-sfGFP levels. (D) Yeast strains expressing either Kar2p-sfGFP or ER-sfGFP-HDEL were treated with 0 or 5 mM DTT for 2 h and analyzed by flow cytometry. The sfGFP MFI values are plotted. The expression of ER-sfGFP-HDEL does not change upon DTT treatment. (E) Immunoblot for Kar2p shows increased levels of both endogenous and Kar2p-sfGFP following treatment with 5 μg/ml Tm for 0, 2, and 4 h. Rpl5p served as a loading control. Bar, 20 μm.
ER lumen unfolded protein burden and Kar2p availability
We previously used fluorescence microscopy techniques to establish in mammalian cells that the decrease of BiP-GFP mobility reflects binding of the chaperone to unfolded protein substrates (Lai et al., 2010; Lajoie and Snapp, 2011). We asked whether Kar2p-sfGFP would function comparably as a reporter of unfolded protein accumulation. First, the mobility of Kar2p-sfGFP was assessed using fluorescence loss in photobleaching (FLIP; Ellenberg et al., 1997). For this technique, a discrete region of interest within the cells is repeatedly photobleached while images are acquired. If the protein is mobile within a continuous compartment, the total fluorescence within this compartment will eventually be depleted by FLIP. In unstressed cells, Kar2p-sfGFP is mobile throughout the ER (Figure 3A, top), similar to our previous results with BiP-GFP in mammalian cells (Lai et al., 2010). Total cellular fluorescence was homogeneously depleted within a short time window (Figure 3A), indicating that Kar2p-sfGFP is not immobilized or enriched in ER subdomains. Treatment with ER stressors—Tm or DTT—significantly decreased the mobility of Kar2p-sfGFP as demonstrated by the longer time interval required to deplete 50% of the GFP fluorescence (control, 125 s; +Tm, 230 s; +DTT, 315 s; (Figure 3, A, bottom, and B). Of importance, the GFP fluorescence was ultimately homogeneously depleted in the Kar2p-sfGFP strain, excluding the possibility that Kar2p-sfGFP becomes trapped or incorporated into a chaperone matrix following acute ER stress (Pfeffer and Rothman, 1987).
FIGURE 3:

Kar2p is mobile under both homeostatic and stress states. FLIP series of cells repeatedly bleached in the region of interest (white box). (A) FLIP of Kar2p-sfGFP expressing yeast in early log phase untreated (top) or treated with either 5 mM DTT for 1 h or 1 μg/ml Tm for 2 h (bottom). Although Kar2p-sfGFP appears to be mobile throughout the ER, stressor-treated cells were depleted of fluorescence at much slower rates. (B) Plot of the mean Kar2p-sfGFP intensities during FLIP shows the faster depletion of fluorescence in untreated cells compared with DTT- and Tm-treated cells (C) FLIP of ER-sfGFP-HDEL–expressing yeast untreated or treated as in A. Depletion of cellular fluorescence is significantly faster than for Kar2p-sfGFP. (D) Plot of the mean ER-sfGFP-HDEL intensities during FLIP shows the faster depletion of fluorescence in untreated cells compared with DTT- and Tm-treated cells. Bar, 10 μm.
A potential caveat for interpreting these measurements is that soluble protein mobility is affected both by the size of the molecule and the viscosity of its environment (Einstein, 1905). ER luminal viscosity in living cells can be assessed with inert fluorescent protein probes (Snapp et al., 2006; Lai et al., 2010), such as ER-targeted sfGFP, which has no known interacting partners. In this case, treatment with Tm or DTT modestly reduces mobility of ER-sfGFP-HDEL (Figure 3, C and D). The time interval to deplete 50% of the GFP fluorescence was 20 s for the control and 30 s for both Tm- and DTT-treated cells. Thus treatment with ER stressors does not induce gross changes in the ER environment that disrupt ER continuity or immobilize pools of ER luminal proteins. Of importance, the FLIP loss of fluorescence of ER-sfGFP-HDEL in treated cells was significantly more rapid than for the much larger Kar2p-sfGFP. These data are consistent with our previous finding in mammalian cells, in which slower BiP-GFP mobility correlated with binding of the chaperone to substrate.
To further characterize Kar2p substrate binding in yeast, we quantified the mobility of Kar2p-sfGFP using FRAP. During FRAP experiments, changes in the mobility or molecular availability of the fluorescently tagged protein are reflected by the effective diffusion coefficient (D). Changes in D report on changes in the environment viscosity, size of the molecule, or its incorporation into or release from molecular complexes (Snapp et al., 2003). In mammalian cells, the mobility of BiP-GFP was shown to increase after substrate depletion by translational inhibition and with loss of function in the BiP-GFP substrate-binding domain (Lai et al., 2010). FRAP analysis of Kar2p mobility in yeast ER was similarly responsive to substrate depletion (Figure 4A). A significant increase in D was observed following treatment of yeast with the translational inhibitor cycloheximide (CHX) for 30 min (Figure 4B), consistent with depletion of substrate and increased Kar2p-sfGFP availability. We hypothesized that if Kar2p-sfGFP mobility reflects binding of Kar2p to misfolded proteins, then D should decrease as the unfolded protein burden increases. To test this hypothesis, cells were treated with Tm for 2 h and analyzed by FRAP. Indeed, there was a significant decrease in D following Tm treatment (Figure 4B). Of interest, when cells were treated with Tm for 90 min and CHX was added for the last 30 min of treatment, we observed a significant increase in D (Figure 4B). This result indicates that whereas Tm prevents glycosylation of nascent proteins, the inhibition of protein synthesis can decrease the Tm-induced misfolded protein burden and result in increased Kar2p-sfGFP mobility in stressed cells. These data support a role for Kar2p substrate levels as a major contributing factor in affecting Kar2p-sfGFP mobility.
FIGURE 4:

Kar2p-sfGFP availability quantitatively decreases during unfolded protein stress. D values of single cells analyzed by FRAP. (A) Representative FRAP series of cells expressing Kar2p-sfGFP. Bar, 10 μm. (B) D values of single Kar2p-sfGFP untreated cells or cells treated with 10 μg/ml CHX for 30 min (+CHX), 1 μg/ml Tm for 2h (+Tm), or 1 μg/ml Tm for 2 h including 10 μg/ml CHX for the last 30 min (+Tm +CHX). (C) Reversibility of stress induced decreased Kar2p-sfGFP mobility. D values of single Kar2p-sfGFP untreated cells or cells treated with 5 mM DTT for 30 min followed by 4 h washout. (D) Small but significant decrease in mobility of ER-sfGFP-HDEL was observed with stress, suggesting an altered ER viscosity. D values of single ER-sfGFP-HDEL–expressing cells untreated or treated with 1.0 μg/ml Tm for 2 h, 5 mM DTT for 30 min, or 10 μg/ml CHX for 30 min. (E) Kar2p-sfGFP mobility is lower in cells without a functional UPR. D values of single wild-type or ire1Δ Kar2p-sfGFP cells untreated or treated with either 0.025 or 1.0 μg/ml Tm for 2 h are plotted. (F) Wild-type and ire1Δ yeast strains expressing ER-sfGFP-HDEL were treated with 1.0 μg/ml Tm for 2 h and then analyzed by FRAP. No significant changes in D values were observed between untreated strains. Both strains exhibited similar significant decreases in D following Tm treatment. *p < 0.05, **p < 0.001
Next the reversibility of Kar2p-sfGFP substrate binding was assessed using DTT treatment, which induces reversible protein unfolding (Braakman et al., 1992; Lai et al., 2010). Washout of DTT permits unfolded secretory proteins to release from ER chaperones and refold (Simons et al., 1995) and is accompanied by a gradual loss of UPR signaling as reported by HAC1 mRNA splicing and Ire1p clustering (Pincus et al., 2010). The mobility of Kar2p-sfGFP decreased after DTT treatment to a comparable extent as detected with Tm treatment (Figure 4C). Consistent with the reversibility of the DTT-induced protein misfolding, Kar2p-sfGFP mobility was restored after washout of DTT, approaching the mobility level detected in untreated cells.
The increase in ER chaperone and other UPR targeted proteins in response to ER stress (e.g., the 10-fold increase in Kar2p protein levels; Figure 2, C and D) has the potential to alter ER lumen crowdedness and affect ER-sfGFP-HDEL mobility. Although the FLIP results (Figure 3, C and D) did not indicate a gross change in the mobility of the inert ER-sfGFP-HDEL reporter in response to ER stress, we quantified the effect of ER stress on ER environment viscosity using FRAP using the ER-sfGFP strain. Treatment with either DTT or Tm (Figure 4D) decreased ER-sfGFP-HDEL mobility by ∼25%. A comparable decrease was detected in ire1Δ cells (Supplemental Figure S3), indicating that the effect is independent of the canonical UPR. The small diameter of ER tubules is conserved from yeast to mammalian cells (40–70 nm; Voeltz et al., 2002) and may limit the diffusional mobility of luminal proteins once unfolded proteins accumulate in response to Tm or DTT treatment. Indeed, we observed no changes in ER-sfGFP-HDEL mobility in most mammalian cell lines during ER stressor treatment (Lai et al., 2010; Lajoie and Snapp, 2011). Nonetheless, the mobility decrease observed for Kar2p-sfGFP (>70%) is significantly greater than the decrease in D of the inert reporter (∼35%). Moreover, Kar2p mobility is sensitive to CHX (Figure 4B), whereas ER-sfGFP-HDEL mobility is not. Together these data argue that changes in Kar2p-sfGFP mobility predominantly reflect its binding to substrate.
One challenge to dissecting the various cellular events that affect UPR activation has been the ability to robustly quantify changes in unfolded protein accumulation in the ER lumen at any given time. Our studies in mammalian cells using the BiP-GFP mobility assay demonstrated that changes in unfolded protein burden can be detected independent of the UPR pathway (Lai et al., 2010). These analyses, however, were limited to cells with functional UPR machinery. The Kar2p-sfGFP mobility assay in Saccharomyces cerevisiae allowed us to assess changes in the ER unfolded protein burden in the absence of a functional UPR (ire1Δ cells). In unstressed ire1Δ cells, Kar2p-sfGFP mobility was significantly lower than that of wild-type cells (Figure 4E). There was no difference in ER-sfGFP-HDEL mobility (Figure 4F). The viability of ire1Δ cells and their lack of gross growth defects under nonstressful conditions (Figure 1D) suggest that the decrease in Kar2p-sfGFP mobility in the untreated ire1Δ cells reflects higher but nonlethal steady-state levels of unfolded protein. Treatment of ire1Δ cells with 0.025 μg/ml Tm further decreased Kar2p-sfGFP mobility (Figure 4E), consistent with their hypersensitivity to ER stressors (Figure 1D). Kar2p-sfGFP mobilities were indistinguishable between wild-type and mutant cells treated with a high dose of Tm (Figure 4E). Of importance, the Kar2p-sfGFP mobility assay enabled quantitation of the misfolded protein burden in the ER of cells with compromised UPR signaling. The only measure of ER stress independent of UPR signaling has been the quantitation of ER redox potential using a GFP reporter (Merksamer et al., 2008). The Kar2p-sfGFP mobility assay allows not only detection of unfolded protein levels under various conditions, but also reports on differences in unfolded protein levels in unstressed cells of various genetic backgrounds (Merksamer et al., 2008; Pincus et al., 2010).
Just as it is important for cells to cope with an unfolded secretory protein stress, the ability of cells to resolve the UPR, itself, is critical for survival. Yeast demonstrably overcome chemically mediated, unfolded secretory protein stresses and attenuate the UPR (Pincus et al., 2010; Chawla et al., 2011; Rubio et al., 2011). However, yeast expressing Ire1p mutants unable to attenuate the UPR are hypersensitive to tunicamycin and DTT stressors (Chawla et al., 2011; Rubio et al., 2011). Implicit to these problems is whether there is a correlation between resolution of the unfolded protein stress and attenuation of the UPR. That is, is the UPR attenuated at the time that the unfolded protein burden has been resolved? Therefore we investigated Kar2p availability before and after UPR attenuation.
To assess UPR status in live cells, we used a fluorescent reporter (splicing reporter [SR]-GFP) that directly reports on the endonuclease activity of Ire1p (Pincus et al., 2010). The increasing fluorescence signal in stressed cells, expressing the reporter, was measured by flow cytometry (Figure 5A). Cells were treated with 1 μg/ml Tm, a dose sufficient to activate the UPR. Wild-type cells can attenuate the UPR from this Tm dose ∼4 h later and continue growing (Figure 1D), consistent with adaptation to this level of unfolded protein stress (Chawla et al., 2011). After 4 h, the median GFP fluorescence reached an asymptote (Figure 5A). The plateau in intensity indicates no additional stress has been detected and signaled (Pincus et al., 2010). The GFP signal persists for several hours as a consequence of the long half-life of GFP. Thus a plateau reports on attenuation of Ire1p endonuclease activity and restoration of the folding capacity of the ER. FRAP at 4 h of Tm treatment revealed significantly reduced Kar2p-sfGFP mobility. After this time, no significant increase in UPR reporter fluorescence was observed. In contrast, it was only after 16 h of Tm treatment that Kar2p-sfGFP mobility returned to D values comparable with those of untreated cells (Figure 5B). The restored mobility is unlikely to reflect the loss of Tm activity, since the media of Tm-treated cells can be used to induce UPR in naive cells (Chawla et al., 2011). Therefore we conclude that the ER folding capacity was restored. UPR-mediated increases in levels of chaperones, ERAD and secretion components, and the ALG7 gene product (the target of Tm) and up-regulation of proteasomal activity probably all account for the decrease in ER misfolded protein in adapted cells.
FIGURE 5:
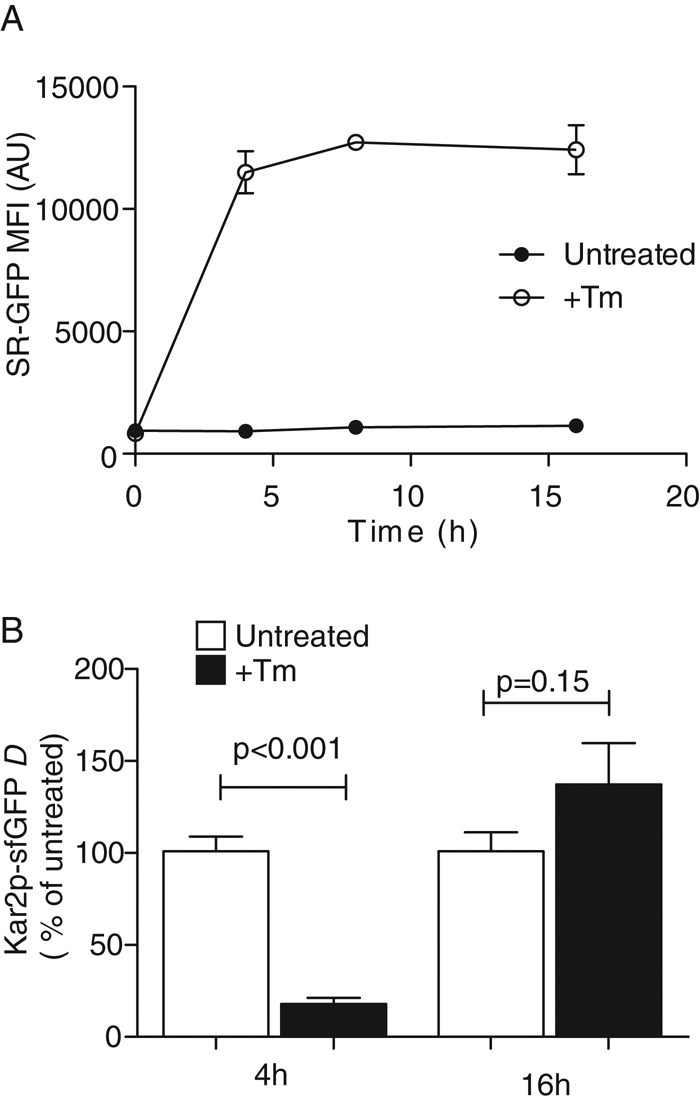
Kar2p availability reveals changes in the ER misfolded protein during adaptation. (A) Wild-type cells expressing the fluorescent splicing reporter (SR) consisting of GFP replacing the HAC1 open reading frame produce a fluorescence signal only when spliced by ire1. SR-GFP–expressing cells were treated with 1 μg/ml Tm, and GFP signal was measured over time using flow cytometry. (B) Mobility of Kar2p-sfGFP untreated cells or cells treated with 1 μg/ml Tm for 4 or 16 h was measured by FRAP. Data were normalized to the mean D values of untreated cells for both time points. At 4 h, Tm induces significant decrease in Kar2p-sfGFP mobility. Once stress is resolved and folding capacity of the ER is restored, the Kar2p-sfGFP mobility returned to the unstressed D values.
These results lead to the somewhat surprising conclusion that UPR inactivation occurs even while a substantial unfolded protein burden remains. We hypothesize that UPR attenuation likely occurs when the stressed ER achieves a small (below our limit of detection) increase in the available pool of Kar2p and potentially other quality control machinery components. Simultaneously or in parallel, Ire1p regulatory attenuators, such as the phosphatase Ptc2p (Welihinda et al., 1998), may achieve a sufficient level of activity to turn off the UPR. Although these matters remain speculative, the results suggest that tools capable of measuring the instantaneous status of UPR activity will be needed to understand the temporal and mechanistic aspects of UPR attenuation.
Kar2p mobility distinguishes various forms of UPR
Finally, we investigated the ability of our FRAP assay to distinguish between different types of UPR activation. The UPR is generally considered to depend on increased levels of unfolded proteins (Credle et al., 2005; Kimata et al., 2007; Kimata and Kohno, 2011; Gardner and Walter, 2011). Recently, however, it was shown that UPR activation by either inositol depletion or deletion of lipid biosynthetic genes does not require the luminal unfolded peptide-binding portion of Ire1p (Promlek et al., 2011). Thus lipid imbalance can lead to UPR activation independent of the accumulation of unfolded proteins, distinct from the response to the classic ER stressors Tm and DTT. We asked how the Kar2p-sfGFP and UPR-mCherry reporters respond to perturbation of lipid metabolism. In the absence of inositol, the phosphatidic acid levels on the ER are high and genes involved in inositol biosynthesis and phospholipid metabolism are actively transcribed (Carman and Henry, 2007). Inositol depletion increased expression of both Kar2p-sfGFP and UPR-mCherry reporters, indicating UPR activation (Figure 6, A and B). Next the effect of inositol depletion on Kar2p-sfGFP mobility was examined by FRAP. Despite strong evidence of UPR activation, inositol depletion did not decrease Kar2p-sfGFP mobility, unlike the effect of Tm treatment (Figure 6B). On the contrary, the Kar2p-sfGFP mobility increased, paralleling the increase in Kar2p-sfGFP intensity (Figure 6B). The increased mobility may reflect a higher Kar2p/substrate ratio that leads to an increase in unbound Kar2p. Inositol depletion did not significantly change the mobility of the inert ER-sfGFP-HDEL, indicating that the increase in Kar2p mobility was independent of ER viscosity (Figure 6C). Therefore various stresses appear to activate Ire1p and elicit the UPR by distinct mechanisms. Kar2p-sfGFP mobility represents a novel noninvasive assay to distinguish between these different forms of UPR in intact cells.
FIGURE 6:

Kar2p availability distinguishes different forms of ER stresses. (A) Wild-type yeast expressing Kar2p-sfGFP and UPR-mCherry were incubated in inositol-minus media for indicated times and analyzed by flow cytometry. MFI for the GFP (left) and mCherry (right) channels is plotted. Data are expressed as percentage of maximum MFI of cells treated with a robust ER stress, 1 μg/ml Tm for 4 h. (B) Time course of D values obtained by FRAP of single Kar2p-sfGFP cells after transfer to inositol-minus media. The reference for ER stress is 2 h treatment with 1.0 μg/ml Tm. (C) D values obtained by FRAP of single ER-sfGFP-HDEL–expressing cells 6 h after transfer to inositol-minus media.
DISCUSSION
The ability to evaluate unfolded protein accumulation in the ER, independent of the UPR, should now enable novel analyses of how cells sense and cope with different environmental stresses. Our Kar2p-sfGFP reporter has the advantage of reporting simultaneously on both the activation of UPR and changes in the unfolded protein burden. In this study, Kar2p-sfGFP allowed us to distinguish between two distinct forms of stress capable of activating the UPR (Figure 7). This study and the work of other groups suggest that lipid imbalance can lead to activation of the yeast UPR (Pineau et al., 2009; Deguil et al., 2011; Promlek et al., 2011). However, the mechanisms by which changes in ER membrane lipid composition lead to Ire1p activation remain unclear. A recent study described a mutant Ire1p (ΔIII mutant) that was unable to bind misfolded peptides and was therefore insensitive to DTT or tunicamycin treatment but could still be activated upon inositol depletion (Promlek et al., 2011). Our study and that of Promlek et al. (2011) appear to conflict with another study reporting that activation of the UPR by accumulation of fatty acids occurs via accumulation of misfolded protein. The argument was supported by the ability to block fatty acid–induced UPR with the chemical chaperone 4-phenyl butyrate (Pineau et al., 2009). We argue that our direct measure of the unfolded protein burden combined with the data from the Promlek et al. (2011) study strongly support the existence of a novel activation mechanism for Ire1p, at least for some stresses. Thus inositol depletion–stimulated Ire1p activation represents at least one alternative pathway that differs from the classic Ire1p activation pathway involving direct binding of the stress sensor to misfolded peptides (Kimata et al., 2007; Gardner and Walter, 2011). An important implication of this study is that either Ire1p or a hypothetical binding partner contains at least one additional regulatory site for activation.
FIGURE 7:

Conceptual model of Kar2p availability during different ER stress conditions. Decreased Kar2p availability is induced by accumulation of misfolded proteins in the ER lumen. This measurement is independent of UPR activation status and can be performed in cells lacking a functional UPR (i.e., ire1Δ cells). Kar2p-sfGFP occupancy defines different forms of UPR based on the ER unfolded secretory protein burden. The classic ER stressors Tm and DTT cause massive accumulation of unfolded protein, increase Kar2p occupancy, and lower Kar2p mobility. In contrast, depletion of inositol induces a UPR without increasing the unfolded protein burden. Kar2p mobility actually increases, likely due to increased Kar2p levels, without an increase in substrate levels.
Understanding how changes in inositol and other lipids regulate Ire1p will be important for understanding the fundamental biology of the UPR and may have implications for drug development. Therapeutic targets for positive and negative regulation of UPR sensors could be present at this regulatory site. Up-regulation of Kar2p-sfGFP fluorescence provides a noninvasive method to monitor UPR activation in living cells and is suitable for high-throughput screens. Our Kar2p-sfGFP reporter should enable novel interrogation of cells to characterize modulators of the ER folding capacity relative to UPR activation under homeostatic, stressed, and adapted states in various genetic backgrounds.
MATERIALS AND METHODS
Drugs
Stock solutions of DTT (1 M in water; Fisher Scientific, Pittsburgh, PA), Tm (5 mg/ml in DMSO; Calbiochem, La Jolla, CA), and CHX (5 mg/ml in water; Sigma-Aldrich, St. Louis, MO) were prepared and used at the concentrations and times indicated.
Strains and cell growth
See Table 1 for all yeast strains used in this study. SfGFP was chromosomally integrated into KAR2 in wild-type and mutant strains using standard PCR methodology (Janke et al., 2004). All yeast strains were grown in synthetic complete media supplemented with 400 μM inositol and appropriate amino acids at 30°C. Yeast strains were grown overnight to early log phase (OD600 nm ≈ 0.5) for analysis. For DTT-washout experiments, cells were incubated with 5 mM DTT for 30 min, then pelleted and resuspended in fresh media and incubated for 4 h before processing for FRAP. For inositol depletion experiments, cells were pelleted, washed twice with inositol-free media, and incubated for various amounts of time in absence of inositol.
TABLE 1:
Yeast strains used in this study.
| Yeast strain | Description | Strain name |
|---|---|---|
| BY4741 Mata ura3Δ0 leu2Δ0 his3Δ1 met15Δ0 | ||
| Deletion array | BY4741 ire1Δ::KanMX4 | |
| This work | BY4741 Kar2p-sfGFP::HIS | YPL001 |
| This work | BY4741 ire1Δ::KanMX4 Kar2p-sfGFP::HIS | YPL002 |
| This work | BY4741 UPR-mCherry::URA | YPL003 |
| This work | BY4741 Kar2-sfGFP::HIS UPR-mCherry::URA | YPL004 |
| This work | BY4741 ER-sfGFP-HDEL::LEU | YPL005 |
| This work | BY4741 ire1Δ::KanMX4 ER-sfGFP-HDEL::LEU | YPL006 |
| This work | BY4741 SR-GFP::LEU | YPL007 |
Plasmid construction
UPR-mCherry (Merksamer et al., 2008) was obtained from AddGene (Cambridge, MA). SR-GFP was obtained from Peter Walter (University of California, San Francisco). The KDEL retrieval motif inserted at the C-terminus of the sfGFP-N1 plasmid (Pedelacq et al., 2006; Lai et al., 2010) was modified to contain the yeast HDEL motif using the following primers:
Forward, GGACGAGCTGTACAAGGATGAATTGTAAGCG
Reverse, CGCTTACAATTCATCGTGGTACAGCTCGTCC
An amplifiable cassette of sfGFP-HDEL was generated in the yeast PCR plasmid pYM28 (Janke et al., 2004) by substituting sfGFP-HDEL for EGFP at SalI/BamHI sites. The following primers were used to amplify the sfGFP-HDEL cassette from sfGFP–N1:
Forward, GATCGTCGACATGGTGAGCAAGGGC
Reverse, GATCGGATCCTTACAATTCATCGTG
The sfGFP-HDEL module can be amplified by using the original S2 and S3 primers and the original HIS3MX6 selection marker.
ER-sfGFP-HDEL was made by inserting the first 135 base pairs of the Kar2p coding sequence (Rossanese et al., 2001) fused into the BglII/AgeI sites of sfGFP-HDEL using the following primers:
Forward, GATCAGATCTCTAAAAATGTTTTTCAACAGAC
Reverse, GATCACCGGTCCATCATCGGCACCTCTAAC
The ER-sfGFP-HDEL fragment was subsequently amplified using the following primers:
Forward, GCAAATGGGCGGTAGGCG
Reverse, GATCGGATCCTTACAATTCATCGTG
The resulting fragment was digested with BglII and BamHI and cloned into the BamHI site of pRS415-GPD (a generous gift from Elizabeth Miller, Columbia University, New York, NY).
Fluorescence microscopy, FRAP, and FLIP
After incubation with various stressors, log-phase cells were placed in eight-well Lab-Tek chambers (ThermoFisher Scientific, Waltham, MA) and allowed to settle for 5 min before imaging. Cells were imaged an Axiovert 200 wide-field fluorescence microscope (Carl Zeiss Microimaging, Jena, Germany) with a 63×, numerical aperture 1.4 oil objective, a 450- to 490-nm excitation/500- to 550-nm emission bandpass filter, and a Retiga 2000R camera. Image analysis was performed with ImageJ (National Institutes of Health, Bethesda, MD).
For FRAP and FLIP, live cells were imaged on a Duoscan confocal microscope system (Carl Zeiss Microimaging) with a 63×, numerical aperture 1.4 oil objective and a 489-nm, 100-mW diode laser with a 500- to 550-nm bandpass filter for GFP. FRAP and FLIP experiments were performed by photobleaching a region of interest at full laser power of the 489-nm line and monitoring fluorescence loss or recovery over time. No photobleaching of the adjacent cells during the processes was observed. D measurements were made using an inhomogeneous diffusion simulation, as described previously (Siggia et al., 2000; Snapp et al., 2003).
Immunoblots
Early-log-phase Kar2p and Kar2p-sfGFP strains, untreated or treated with Tm, were pelleted and total protein extracted by alkaline lysis (Kushnirov, 2000). Lysates were separated on 7.5% SDS–PAGE tricine gels, transferred to nitrocellulose membranes, and detected with anti-GFP (from Ramanujan Hegde, MRC Laboratory of Molecular Biology, Cambridge, United Kingdom), anti-Kar2p (from Peter Walter, University of California, San Francisco), anti-Rpl5p (from Jonathan Warner, Albert Einstein College of Medicine, New York, NY), or horseradish peroxidase–labeled anti-rabbit (Jackson ImmunoResearch Laboratories, West Grove, PA).
Northern blot
RNA extraction, Northern analysis, and quantitation were as previously described (Li et al., 2000). Early-log-phase Kar2p and Kar2p-sfGFP yeast strains, untreated or treated with DTT, were pelleted on ice to preserve RNA intermediates and quickly frozen. Total RNA was extracted by the glass-bead, hot-phenol method (O’ Connor and Peebles, 1991). RNA was analyzed on 5% agarose gels containing 6% formaldehyde. After transfer to Nytran Plus membranes, blots were probed with 32P-labeled oligonucleotides, washed, and imaged with a Storm 820 Phosphorimager (GE Healthcare, Pittsburgh, PA).
Flow cytometry
Early-log-phase cells expressing the various plasmids were treated with ER stressors as described. Cells were pelleted, resuspended in phosphate-buffered saline containing 2% glucose, and analyzed by flow cytometry using an LSR II flow cytometer (BD Biosciences, San Diego, CA) equipped with a 488-nm laser with a 525/50 bandpass filter for sfGFP and a 561-nm laser with a 610/20 bandpass filter for mCherry and FACSDIVA software to compile .fcs files. The files were analyzed using FloJo (Tree Star Ashland, OR). No gates were applied. Median fluorescence intensities were calculated for each channel and plotted in Prism, version 5.0c (GraphPad Software, La Jolla, CA). Errors bars represent the SD of the median of three biological replicates.
Acknowledgments
We thank Ramanujan Hegde for the anti-GFP antibody, Peter Walter for the anti-Kar2p antibody and the SR-GFP vector, Jonathan Warner for the anti-Rpl5p antibody, and Elizabeth Miller for the p415-GPD vector. This study was supported by National Institutes of Health, National Institute of General Medical Sciences, Grants RO1GM086530-01 to E.L.S. and RO1GM085177 to I.M.W. and National Cancer Institute Center Support Grant P30CA013330 to the Einstein Flow Cytometry Core Facility.
Abbreviations used:
- CHX
cycloheximide
- D
effective diffusion coefficient
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- ERAD
ER–associated degradation
- FLIP
fluorescence loss in photobleaching
- FRAP
fluorescence recovery after photobleaching
- GFP
green fluorescent protein
- MFI
median fluorescence intensity
- QC
quality control
- sfGFP
superfolder GFP
- Tm
tunicamycin
- UPR
unfolded protein response
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-12-0995) on January 4, 2012.
REFERENCES
- Aragon T, van Anken E, Pincus D, Serafimova IM, Korennykh AV, Rubio CA, Walter P. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronson DE, Costantini LM, Snapp EL. Superfolder GFP is fluorescent in oxidizing environments when targeted via the Sec translocon. Traffic. 2011;12:543–548. doi: 10.1111/j.1600-0854.2011.01168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, McDonald KL, Walter P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006;4:e423. doi: 10.1371/journal.pbio.0040423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I, Helenius J, Helenius A. Manipulating disulfide bond formation and protein folding in the endoplasmic reticulum. EMBO J. 1992;11:1717–1722. doi: 10.1002/j.1460-2075.1992.tb05223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky JL, Skach WR. Protein folding and quality control in the endoplasmic reticulum: recent lessons from yeast and mammalian cell systems. Curr Opin Cell Biol. 2011;23:464–475. doi: 10.1016/j.ceb.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman GM, Henry SA. Phosphatidic acid plays a central role in the transcriptional regulation of glycerophospholipid synthesis in Saccharomyces cerevisiae. J Biol Chem. 2007;282:37293–37297. doi: 10.1074/jbc.R700038200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Chakrabarti S, Ghosh G, Niwa M. Attenuation of yeast UPR is essential for survival and is mediated by IRE1 kinase. J Cell Biol. 2011;193:41–50. doi: 10.1083/jcb.201008071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87:391–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. On the mechanism of sensing unfolded protein in the endoplasmic reticulum. Proc Natl Acad Sci USA. 2005;102:18773–18784. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguil J, Pineau L, Rowland Snyder EC, Dupont S, Beney L, Gil A, Frapper G, Ferreira T. Modulation of lipid-induced ER stress by fatty acid shape. Traffic. 2011;12:349–362. doi: 10.1111/j.1600-0854.2010.01150.x. [DOI] [PubMed] [Google Scholar]
- Einstein A. Über die von der molekularkinetischen Theorie der Wärme geforderte Bewegung von in ruhenden Flüssigkeiten suspendierten Teilchen. Ann Phys. 1905;17:549–560. [Google Scholar]
- Ellenberg J, Siggia ED, Moreira JE, Smith CL, Presley JF, Worman HJ, Lippincott-Schwartz J. Nuclear membrane dynamics and reassembly in living cells: targeting of an inner nuclear membrane protein in interphase and mitosis. J Cell Biol. 1997;138:1193–1206. doi: 10.1083/jcb.138.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige MJ, Hendershot LM. Disulfide bonds in ER protein folding and homeostasis. Curr Opin Cell Biol. 2011;23:167–175. doi: 10.1016/j.ceb.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333:1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Hetz C, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Hulsmeier AJ, Welti M, Hennet T. Glycoprotein maturation and the UPR. Methods Enzymol. 2011;491:163–182. doi: 10.1016/B978-0-12-385928-0.00010-9. [DOI] [PubMed] [Google Scholar]
- Janke C, et al. A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Ishiwata-Kimata Y, Ito T, Hirata A, Suzuki T, Oikawa D, Takeuchi M, Kohno K. Two regulatory steps of ER-stress sensor Ire1 involving its cluster formation and interaction with unfolded proteins. J Cell Biol. 2007;179:75–86. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Kimata YI, Shimizu Y, Abe H, Farcasanu IC, Takeuchi M, Rose MD, Kohno K. Genetic evidence for a role of BiP/Kar2 that regulates Ire1 in response to accumulation of unfolded proteins. Mol Biol Cell. 2003;14:2559–2569. doi: 10.1091/mbc.E02-11-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimata Y, Kohno K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr Opin Cell Biol. 2011;23:135–142. doi: 10.1016/j.ceb.2010.10.008. [DOI] [PubMed] [Google Scholar]
- Kimata Y, Oikawa D, Shimizu Y, Ishiwata-Kimata Y, Kohno K. A role for BiP as an adjustor for the endoplasmic reticulum stress-sensing protein Ire1. J Cell Biol. 2004;167:445–456. doi: 10.1083/jcb.200405153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000;16:857–860. doi: 10.1002/1097-0061(20000630)16:9<857::AID-YEA561>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Lai CW, Aronson DE, Snapp EL. BiP availability distinguishes states of homeostasis and stress in the endoplasmic reticulum of living cells. Mol Biol Cell. 2010;21:1909–1921. doi: 10.1091/mbc.E09-12-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie P, Snapp E. Changes in BiP availability reveal hypersensitivity to acute ER stress in cells expressing mutant huntingtin. J Cell Sci. 2011;124:332–43. doi: 10.1242/jcs.087510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Moir RD, Sethy-Coraci IK, Warner JR, Willis IM. Repression of ribosome and tRNA synthesis in secretion-defective cells is signaled by a novel branch of the cell integrity pathway. Mol Cell Biol. 2000;20:3843–3851. doi: 10.1128/mcb.20.11.3843-3851.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. ER chaperone functions during normal and stress conditions. J Chem Neuroanat. 2004;28:51–65. doi: 10.1016/j.jchemneu.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Merksamer PI, Trusina A, Papa FR. Real-time redox measurements during endoplasmic reticulum stress reveal interlinked protein folding functions. Cell. 2008;135:933–947. doi: 10.1016/j.cell.2008.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146:743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- Munro S, Pelham HR. A C-terminal signal prevents secretion of luminal ER proteins. Cell. 1987;48:899–907. doi: 10.1016/0092-8674(87)90086-9. [DOI] [PubMed] [Google Scholar]
- Normington K, Kohno K, Kozutsumi Y, Gething MJ, Sambrook J. S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell. 1989;57:1223–1236. doi: 10.1016/0092-8674(89)90059-7. [DOI] [PubMed] [Google Scholar]
- North MJ, Beynon RJ. Prevention of unwanted proteolysis. In: Proteolytic Enzymes: A Practical Approach, eds. R. Beynon and J.S. Bond, Oxford, UK: IRL Press Oxford. 2001:211–233. [Google Scholar]
- O’Connor JP, Peebles CL. In vivo pre-tRNA processing in Saccharomyces cerevisiae. Mol Cell Biol. 1991;11:425–439. doi: 10.1128/mcb.11.1.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa D, Kimata Y, Kohno K, Iwawaki T. Activation of mammalian IRE1alpha upon ER stress depends on dissociation of BiP rather than on direct interaction with unfolded proteins. Exp Cell Res. 2009;315:2496–2504. doi: 10.1016/j.yexcr.2009.06.009. [DOI] [PubMed] [Google Scholar]
- Oikawa D, Kimata Y, Takeuchi M, Kohno K. An essential dimer-forming subregion of the endoplasmic reticulum stress sensor Ire1. Biochem J. 2005;391:135–142. doi: 10.1042/BJ20050640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- Pfeffer SR, Rothman JE. Biosynthetic protein transport and sorting by the endoplasmic reticulum and Golgi. Annu Rev Biochem. 1987;56:829–852. doi: 10.1146/annurev.bi.56.070187.004145. [DOI] [PubMed] [Google Scholar]
- Pincus D, Chevalier MW, Aragon T, van Anken E, Vidal SE, El-Samad H, Walter P. BiP binding to the ER-stress sensor Ire1 tunes the homeostatic behavior of the unfolded protein response. PLoS Biol. 2010;8:e1000415. doi: 10.1371/journal.pbio.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineau L, Colas J, Dupont S, Beney L, Fleurat-Lessard P, Berjeaud JM, Berges T, Ferreira T. Lipid-induced ER stress: synergistic effects of sterols and saturated fatty acids. Traffic. 2009;10:673–690. doi: 10.1111/j.1600-0854.2009.00903.x. [DOI] [PubMed] [Google Scholar]
- Polaina J, Conde J. Genes involved in the control of nuclear fusion during the sexual cycle of Saccharomyces cerevisiae. Mol Gen Genet. 1982;186:253–258. doi: 10.1007/BF00331858. [DOI] [PubMed] [Google Scholar]
- Promlek T, Ishiwata-Kimata Y, Shido M, Sakuramoto M, Kohno K, Kimata Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol Biol Cell. 2011;22:3520–3532. doi: 10.1091/mbc.E11-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Rose MD, Misra LM, Vogel JP. KAR2, a karyogamy gene, is the yeast homolog of the mammalian BiP/GRP78 gene. Cell. 1989;57:1211–1221. doi: 10.1016/0092-8674(89)90058-5. [DOI] [PubMed] [Google Scholar]
- Rossanese OW, Reinke CA, Bevis BJ, Hammond AT, Sears IB, O’Connor J, Glick BS. A role for actin, Cdc1p, and Myo2p in the inheritance of late Golgi elements in Saccharomyces cerevisiae. J Cell Biol. 2001;153:47–62. doi: 10.1083/jcb.153.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio C, Pincus D, Korennykh A, Schuck S, El-Samad H, Walter P. Homeostatic adaptation to endoplasmic reticulum stress depends on Ire1 kinase activity. J Cell Biol. 2011;193:171–184. doi: 10.1083/jcb.201007077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegsegger U, Leber JH, Walter P. Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell. 2001;107:103–114. doi: 10.1016/s0092-8674(01)00505-0. [DOI] [PubMed] [Google Scholar]
- Schuck S, Prinz WA, Thorn KS, Voss C, Walter P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J Cell Biol. 2009;187:525–536. doi: 10.1083/jcb.200907074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- Siggia ED, Lippincott-Schwartz J, Bekiranov S. Diffusion in inhomogeneous media: theory and simulations applied to whole cell photobleach recovery. Biophys J. 2000;79:1761–1770. doi: 10.1016/S0006-3495(00)76428-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons JF, Ferro-Novick S, Rose MD, Helenius A. BiP/Kar2p serves as a molecular chaperone during carboxypeptidase Y folding in yeast. J Cell Biol. 1995;130:41–49. doi: 10.1083/jcb.130.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp EL. Fluorescent proteins: a cell biologist's user guide. Trends Cell Biol. 2009;19:649–655. doi: 10.1016/j.tcb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp EL, Altan N, Lippincott-Schwartz J. Measuring protein mobility by photobleaching GFP chimeras in living cells. Curr Protoc Cell Biol. 2003 doi: 10.1002/0471143030.cb2101s19. Chapter 21 Unit 21.1. [DOI] [PubMed] [Google Scholar]
- Snapp EL, Sharma A, Lippincott-Schwartz J, Hegde RS. Monitoring chaperone engagement of substrates in the endoplasmic reticulum of live cells. Proc Natl Acad Sci USA. 2006;103:6536–6541. doi: 10.1073/pnas.0510657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Voeltz GK, Rolls MM, Rapoport TA. Structural organization of the endoplasmic reticulum. EMBO Rep. 2002;3:944–950. doi: 10.1093/embo-reports/kvf202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welihinda AA, Tirasophon W, Green SR, Kaufman RJ. Protein serine/threonine phosphatase Ptc2p negatively regulates the unfolded-protein response by dephosphorylating Ire1p kinase. Mol Cell Biol. 1998;18:1967–1977. doi: 10.1128/mcb.18.4.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell. 2003;4:265–271. doi: 10.1016/s1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]