

Abstract
There is widespread interest in circulating tumor cells (CTCs) in blood. Direct detection of CTCs (often < 1/ml) is complicated by a number of factors, but the presence of ~103 to 104 copies of target RNA per CTC, coupled with simple enrichments, can greatly increase detection capability. Here we used resonance frequency shifts induced by mass-amplifying gold nanoparticles, to detect a hybridization sandwich bound to functionalized nanowires. We selected PCA3 RNA as a marker for prostate cancer, optimized antisense binding sites and defined conditions allowing single nucleotide mismatch discrimination, and used a hybrid resonator integration scheme which combines elements of top-down fabrication with strengths of bottom-up fabrication, with a view to enable multiplexed sensing. Bound mass calculated from frequency shifts matched mass estimated by counting gold nanoparticles. This represents the first demonstration of use of such nanoresonators, which show promise of both excellent specificity and quantitative sensitivity.
Keywords: CTCs, PCA3 (DD3), prostate cancer, Nanoresonators
Background
The prognostic and diagnostic utility of detection of circulating tumor cells (CTCs) in cancer patients is being actively investigated 1-4, with potentially important significance for cancer treatment. A number of reports have clearly established a relationship between the number of circulating tumor cells and patient outcome for various cancers, particularly breast 5,6 and prostate 7-9 as well as other cancers . With respect to early markers for prostate cancer (PCa), standard screenings for potential biomarkers such as prostate-specific antigen are generally not effective and other molecular biomarkers are needed 10. However, a number of studies have established the utility of CTC levels as a prognostic indicator and/or a predictor of response to therapy in PCa 4,7-9,11,12.
CTCs have been detected using antibody-based methods targeting a global epithelial marker such as epCAM 4,13. Such “positive selections” are complicated by the fact that certain common types of cancers (e.g., normal-like breast cancers) do not express epCAM 14, and there is growing evidence that many CTCs have undergone the epithelial-to-mesenchymal transition, thereby losing expression of epCAM 15,16. More commonly, CTCs for a given cancer type have been identified using RT/PCR for pre-selected marker RNAs. In general, 2 markers appear to be superior to one for detection of CTCs by increasing sensitivity 17,18.
Drawbacks to the approach of identifying CTCs based on pre-chosen RNA markers are that the CTCs are being identified in patients only after they have been diagnosed with particular cancers, and that a relatively limited number of RNAs can be interrogated. In contrast, our approach is to develop a detection platform that would be useful for screening purposes, for early detection of multiple types of cancers, as well as for monitoring therapy. This approach is based on the original premise that metastasis is a relatively inefficient process 19-21, and there is anecdotal but growing support for the usefulness of CTC detection in diagnosis of early-stage disease. A general relationship between CTC levels and stage appears to exist in many different cancers 22 but significant CTC levels are generally found even in early-stage cancers. For example, in one early study of breast cancer patients, 68% of patients with Stage N1 disease had detectable CTCs, but 46% of patients with Stage N0 disease also had detectable CTCs17. Others have found similar CTC levels in localized vs. advanced PCa patients 23. Schmidt and co-workers 24 showed that even very small foci of PCa (0.2 cm3) give rise to CTCs, and recent work has also identified CTCs in PCa patients with “low-volume” tumors 25.
We intend to enrich CTCs from whole blood samples based on their decreased density using a simple porous membrane centrifugation device 11, and RNA is then purified from the enriched CTC-containing fraction. This provides CTC enrichment of ~400:1 with respect to white blood cells. The platform we are developing consists of a chip-based device, which utilizes antisense oligonucleotides (ASOs) covalently attached to metallic or silica-coated nanowires (NWs) to detect marker RNAs for various cancer types. NWs are first prepared off-chip and then assembled in a bottom-up manner from suspension and integrated to fabricate an array of NW-resonator (NR) devices. A hybridization “sandwich” is used, wherein target RNAs are bound to the ASO-derivatized NWs 26. A 2nd ASO, attached to a single 50 nm Au-nanoparticle (ASO:AuNP), is then hybridized to a different site on the bound target RNA; this requires a second stringent hybridization, which increases specificity, and also provides a very substantial increase in mass which is important for NR sensitivity. Detection of the hybridization sandwich attached to the NW is accomplished by optically measuring the shift in resonance frequency of the NRs. Related recent work towards NR biosensor design describes the evaluation of resonator clamp quality 27, elastic and dissipative properties of silicon 27, gold and rhodium 28, NW resonance in air 27 and programmed assembly of NWs 29.
We chose PCA3 (Gene ID#50652; also known as DD3) as a marker for PCa. PCA3 is an abundant, non-coding RNA; exon 4 is prostate–specific, and PCA3 is significantly up-regulated in PCa 30,31. CTCs in PCa serve as a predictive biomarker in patients with hormone-sensitive PCa 32 as well as a surrogate marker for outcome 33 and clinical management 12, and PCA3 is a prostate-specific marker for CTCs in peripheral blood 13, and is of interest as a PCa-specific marker in urine 34.
NRs are attractive for biosensing applications because they offer high mass sensitivity 35,36 and can be incorporated onto integrated circuit chips for electrical transduction and/or actuation 37. A recent critical review compares several nanobiosensor platforms 38. NRs, with their small mass and high resonance frequency can enable sub-attogram (10-20 g) limits of mass detection. Our efforts in this area have thus far focused on assembly and integration of NWs to predetermined locations on the chip, followed by integration to produce high density arrays of NR devices 27,29. This hybrid top-down/bottom-up fabrication method overcomes materials and chemical functionalization limitations imposed by traditional top-down fabrication while providing excellent NW positioning for high-yield integration with existing features on the chip 27,29.
Here we present data on the basic parameters of the PCA3 sandwich hybridizations using microarrays and functionalized NWs, as well as the resonant response of NRs prepared by our hybrid top-down/bottom-up fabrication method to the PCA3 complexes. The induced resonance frequency shift is about 1 kHz per hybridization complex suggesting that single molecule/particle detection sensitivity should be achievable. This represents the first demonstration of use of NRs for detection of a marker RNA sequence for PCa, as well as proof of the selectivity of the biorecognition that enables it. Based on RNA measurements and volumes involved, we estimate facile detection of ~ 1 CTC/10 ml blood.
Methods
Detailed Experimental Methods are presented in Supplementary Material.
Blood and tissue samples were obtained under IRB-approved protocols, with informed consent and following institutional guidelines.
DNA oligonucleotides used in this work (catalogued in Table 1 and Supplementary Material, Table 1) were purchased from Integrated DNA Technologies. Sequences used as ASOs were synthesized with either a primary amine group followed by a C6 or C12 carbon spacer at the 5′ end preceding the specific PCA3 ASO sequence, or with a 5'-thiol modification, followed by a tract of T's (T6-10) between the thiol group and the PCA3 ASO sequence.
Table 1.
ASOs used for Detection of PCA3 RNA transcripts.
| Name | Sequence (5’ to 3’) | Description |
|---|---|---|
| ASO468 | TGACCCAAGATGGCGGCCGG | ASO targeting PCA3 at the region immediately 3’ to nt 468 |
| ASO683 | TGCCATCAAGATTTTCTCGTC | ASO targeting PCA3 at the region immediately 3’ to nt 683 |
| ASO735 | GCT GCC TCA TGT CAT CAC AG | ASO targeting PCA3 at the region immediately 3’ to nt 735 |
| ASO735 MM1 | GCT GCC TCA TCT CAT CAC AG | Single-nt mismatch of ASO735 |
| ASO735 MM2A | GCT GCC TCA TCT CAT CAC GG | Two-nt mismatch of ASO735 |
| ASO735 MM2B | GCT GCC TCA TCG CAT CAC AG | Two-nt mismatch of ASO735 |
| ASO735 MM4 | GCT GCC TCA TCG AGT CAC AG | Four-nt mismatch of ASO735 |
| DNA mimic of PCA3 | CTG TGA TGA CAT GAG GCA GCT TTT TGA CGA GAA AAT CTT GAT GGC | 45-nt sequence containing the two portions of PCA3 that the ASOs were complementary to, separated by 5 T's |
| ASOT683 | (TTT) TTT TTT TTG CCA TCA AGA TTT TCT CGTC | ASO683 with 7 or 10 Ts added at the 5’ end as noted |
| ASOT735 | (TTT) TTT TTT TGC TGC CTC ATG TCA TCA CAG | ASO735 with 7 or 10 Ts added at the 5’ end as noted |
| ASOnon | CTC GTA TCT CAA CTC GTA TTT TTT TTT TTT | Non-complementary probe sequence |
Library Selection of Accessible sites in PCA3 for targeting ASOs
Total RNA was isolated from LNCaP cells (a human prostate cancer cell line), and a portion of the PCA3 transcript (nt 286-1005, which contains the prostate-specific exon 4) was cloned and used for in vitro transcription and purification of PCA3 RNA. We then performed a SELEX-based library selection protocol for identification of optimal binding sites, using a random library of N13 oligonucleotides and an RNase H-based protocol, and cut sites were identified by comparison with the hydrolysis ladders (Supplementary Material, Figure 1). ASOs targeted to the 3 best sites identified (designated 486, 683, and 735) were subsequently synthesized reverse complementary to the regions immediately 3’ to the RNase H cleavage sites (Table 1). These were tested for binding to PCA3 RNA is pairwise fashion to optimize detection of PCA3 RNA; the optimal pair was ASO683 and ASO735.
Preparation and Hybridization of MicroArrays with Fluorescent Detection
Identified 20-mer ASOs were synthesized with a primary amine group followed by a C6 or C12 carbon spacer at the 5’-end, preceding the specific antisense sequence. They were then coupled to Codelink slides to create microarrays for initial characterization of hybridization reactions.
PCA3 RNA was labeled with Alexafluor 546 dye (A546, or in some experiments A647) using standard methods (dye is generally incorporated every ~20 nt. Secondary ASO were labeled with AlexaFluor labels at the time of their manufacture, in such a way that 2 colors could be monitored.
PCA3 RNA (and total RNA where appropriate) was heated to 50°C and then cooled on ice prior to hybridization. The Alexafluor-labeled 2° ASOs were added at a molar ratio of ~40:1 PCA3 RNA), and prehybridized (1 hour at 37°C, in the dark) prior to hybridization to the array. In experiments that included total RNA extracted from 293T cells (which do not express PCA3 RNA), total RNA was also added to this prehybridization mixture at designated concentrations (at up to 1000X excess). Hybridizations were conducted with ArrayIt 1 × 16 Hybridization cassettes. Following hybridization under stringent conditions for 1 h at 47 C, arrays were rinsed (twice each for 30 sec at room temperature) in 2X SSC+ 0.2% SDS, 2X SSC, and 0.2X SSC (20X SSC = 3M NaCl, 0.3 M sodium citrate, pH 7.0). In some cases, arrays were finally rinsed in isopropanol (final rinse for NWs was in water). Slides were scanned using a Microarray scanner and software, and images were analyzed using the GeneSpring 7 software program.
Synthesis of ASO-Functionalized NWs and ASO:AuNPs for Hybridization Experiments
Rhodium and striped (bar-coded) Au/Ag NWs (ca. 320 nm in cross-sectional diameter and 4-6 microns in length, depending on the experiment) were synthesized by templated electrodeposition in commercial alumina membranes. These were then coated with a silica coating. For transmission electron microscopic (TEM) imaging, the Ag segments of the coated NWs were removed by etching and then thoroughly rinsed prior to attachment.
For ASO attachment to rhodium NWs, thiolated ASOs were cleaved with DTT and allowed to attach to the NWs via thiol self-assembly. For ASO attachment to the silica surface of the Au/Ag NWs, thiolated ASOs were covalently attached via a bifunctional coupling reagent after removal of the silica from the Ag segments.
For attachment of 2nd ASOs to 50 nm Au nanoparticles (AuNPs), DNA-derivatized AuNPs were prepared by adding ASOT735 (ASO735 containing T6 spacer) to 50 nm AuNPs and heating at 37°C overnight. After ~16 hours, sodium phosphate buffer was added, and samples were rinsed thoroughly.
Hybridization of ASO-Functionalized NWs and Fluorescently-labeled ASOs
The DNA-derivatized striped NWs were mixed together (10 μL of each type, with different patterns of Ag and Au which allow them to be distinguished). PCA3 target RNA was added, and parallel incubations were also conducted with a non-complementary RNA as a negative control. Samples were hybridized at 47°C for one hour in hybridization buffer, vortexed, and then rinsed as described for microarrays. Fluorescently-labeled 2nd ASOs were added, and samples were again hybridized, vortexed, and rinsed as in the previous step. Finally, the samples were resuspended in 2X SSC for imaging. Fluorescence imaging on NWs was performed with an inverted optical microscope using a plan fluor lens and Image Pro Plus software for imaging.
Hybridization of ASO-Functionalized NWs and ASO:AuNPs using TEM/FE-SEM
For hybridization experiments with 50 nm ASO:AuNP, samples were resuspended (separately) in hybridization buffer and mixed with either target PCA3 RNA or with a DNA “mimic” (Table 1). The DNA mimic was used instead of PCA3 RNA in initial experiments for ease of handling; this consisted on a 45mer, which contained the sequences to which the primary and secondary ASOs were targeted, separated by 5 Ts. Later experiments used in vitro transcribed PCA3 RNA (at 20X lower molar concentation). Samples were hybridized as described above, and ASO:AuNPs (~4.5 × 1010 particles) derivatized with the 2nd ASO were added to each sample, hybridized, and again rinsed as described. The hybridized conjugates were dried onto coated TEM grids, and a JEOL TEM or a Leo field-emission scanning electron microscope (FE-SEM) was used for imaging.
Resonance Measurements of NWs
The resonance peak frequency shift of NRs upon binding of the RNA target + ASO:AuNPs was measured using an optical scheme. A laser beam is focused at the NR tip, with light beams being reflected from the moving NR as well as the chip floor beneath. As the NR vibrates with respect to the chip, these reflected beams generate an optical interference signal tracking the frequency and amplitude of the NR. The optical interference signal is converted by a photodetector to an electrical signal. This signal is sent to a spectrum analyzer, which displays the resonance curve and allows evaluation of the peak frequency. The peak frequency decreases when the added mass of the target RNA + ASO:AuNPs are bound, and the frequency shift is measured. The measurements were conducted using a custom-designed setup (see Supplementary Material, Figure 5). The ability to resolve small shifts in the high resonance frequency (f) for the small mass (M) associated with the NRs makes it possible to achieve a sensing mass resolution (ΔM=2 M Δf/f ) down to subattogram (ag=10-18 g) levels at a modest vacuum (~0.1 Torr).
Results
We began by analyzing prostate tissue specimens, using conventional QPCR. We found that PCA3 RNA was present at approximately 6000 copies/cell in PCa specimens, which is ~100X higher than in noncancerous prostatic epithelium (see Supplementary Materials). Thus, the PCA3 marker provides us with an inherent amplification (equivalent to ~ 13 rounds of PCR) of a marker which is greatly increased in PCa epithelium. We also determined PCA3 transcript levels in 3 PCa cell lines. LNCaP cells expressed PCA3 transcripts at ~ 1300 copies/cell; 22RV1 cells had barely detectable levels, and PC3 cells did not express PCA3 transcripts.
We then obtained peripheral blood samples from 8 healthy volunteers, and fractionated them using the OncoQuick devices. QPCR (see Supplementary Materials for details) for PCA3 in the RNA from the “enriched” fraction showed that these control samples did not contain any significant levels of PCA3 transcripts (a value of 16 ± 24 copies/ml was observed, which did not differ significantly from 0). We then spiked in known numbers (from 4 to 500 cells/10 ml) of LNCap cells into blood, and then processed the blood using the OncoQuick system. We obtained essentially 100% recovery of the LNCaP cells, based on QPCR for cytokeratin markers 8 and 18 (see Supplementary Material, Figure 6; similar results were observed with 22RV1 PCa cells). We also spiked 1000 copies of PCA3 transcripts into the RNA isolated from the enriched fractions from normal volunteers; we obtained QPCR values which were equivalent to QPCR amplifications performed in the absence of control RNA, showing that RNA from controls (without PCA3) did not interfere with detection (see also below).
Selection of Optimal Antisense Detection Sites in PCA3 RNA
RNA adopts complex secondary and tertiary structures in solution 39, which presents a challenge for design of ASO probes to detect RNA. We identified ASO-accessible regions in the native folded RNA structure. This approach avoids potential difficulties in maintaining a fully denatured state and/or the extra step of fragmentation. PCA3 RNA was produced by in vitro transcription, and was subjected to a library selection protocol to identify optimal sites for binding of ASOs (Supplementary Material, Figure 1). Three strong sites of RNase H hydrolysis were identified, designated as 487, 684, and 736; ASO735 and ASO683 were selected for use after pair-wise testing of binding using the microarray format described below (Table 1).
Hybridization Studies using Selected ASO735 and ASO683
For microarray hybridization studies, ASO683 was synthesized with a NH2-C12 spacer at the 5’-end, and spotted/coupled to the Codelink glass slides using an array spotter and standard amine coupling chemistry. In vitro transcribed PCA3 RNA (500 ng, either unlabeled or labeled with an Alexafluor dye) was then prehybridized with Alexafluor-labeled ASO735 (at a ratio of 1:40) for 1 h at room temperature in the dark. This mixture was then hybridized to the spotted arrays for 1 h at 47° C, with stringent rinsing as described. Hybridizations also included reactions with spotted ASOs containing nucleotide mismatches (Table 1).
No hybridization was observed when the spotted primary ASO (specifically ASO735) was the same as the 2nd ASO735 used for detection (Figure 1A), and no binding of labeled PCA3 RNA was observed with even single-nucleotide mismatches in the spotted ASO683 sequence (Figure 1A, left panel). Similarly, single nucleotide mismatches in the spotted ASO683 sequence completely eliminated any sandwich hybridization signal from Alexafluor-labeled 2nd ASO (Figure 1A, right panel). Thus, the formation of the hybridization sandwich requires perfect matches between PCA3 RNA and both ASOs.
Figure 1. Sandwich Hybridization Specificity for PCA3 and Effects of Competing Cytoplasmic RNA.
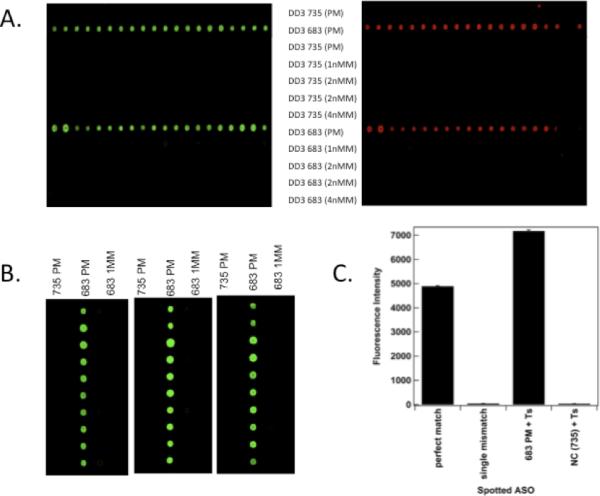
Panel A. Hybridization specificity for PCA3 RNA. ASOs were spotted onto Codelink slides as indicated (see Supplementary Material). Alexafluor546 (A546)-labeled PCA3 RNA (green fluorescence) was then pre-hybridized with A647-labeled ASO735 (red fluorescence) for 1 h at 47 C, and the mixture was then hybridized with the Arrays and rinsed. Panels show arrays containing rows of 10 identical spots of ASOs. The ASOs which were spotted are indicated in the center. PM indicates a perfect match, 1nMM a single nucleotide mismatch, 2 nMM two mismatches, and 4 nMM 4 mismatches (see Table 1). Left Section shows detection of A546-labeled PCA3 RNA, whereas the right section shows detection of the 2nd A647-labeled ASO735. The row containing spotted ASO683 shows strong hybridization, whereas there is no hybridization observed with the ASO683 containing even a single mismatch. No hybridization is observed with ASO735, since the 2nd ASO is directed against the same site as the spotted ASO.
Panel B. Effects of Competing Cytoplasmic RNA on PCA3 Hybridization. 100 ng of A546-labeled PCA3 RNA was hybridized with ASOT735, and then hybridized to spotted microarrays as before. Each section shows 10 duplicate spots in each of 3 columns, which differ by the sequence of ASO spotted onto the array, as indicated at the top (PM indicates perfect match, MM indicates 1 mismatched nucleotide). Sections from left to right had increasing amounts of competing 293T total cytoplasmic RNA added (0, 10, and 100 μg, respectively). Analogous results were also observed with A647-labeled 2nd ASO.
Panel C. Quantitative Measurement of Fluorescent Signal from Sandwich Hybridization Format. Hybridizations were performed as in Panel B (without competing cytoplasmic RNA), with comparison of ASO735 with and without the T-spacer tract as indicated.
Hybridization signal was not diminished by 1000X excess of competing (non-complementary) cytoplasmic RNA (Figure 1B). Quantitative evaluation of fluorescent signal showed that addition of a short stretch of T's as a spacer (designated ASOT735 and ASOT683; see Table I and Supplementary Material) increased signal by ~40%, and that background non-specific binding was < 0.5% (Figure 1C). We also assessed the effects of controlled fragmentation of RNA, with the idea that any residual secondary RNA structure would be reduced by the fragmentation. We did not observe any increase in hybridization following controlled fragmentation (Supplementary Material, Figure 2). These results indicate that short spacers increase accessibility, and that residual 2nd structure or excess non-complementary RNA do not diminish detection.
We then tested these hybridization parameters on ASO-derivatized NWs. We initially tested fluorescently-labeled reagents with functionalized NWs. ASOT683 was covalently coupled to amine-functionalized, silica-coated NWs via a 5′-thiol group. We used three different “barcoded” NWs 40 with distinct patterns of Au and Ag metal along their lengths (patterns 0011, 1001, and 0110, where 0 denotes an Au segment and 1 an Ag segment). The patterns can be visualized by illumination with 430 nm light, under which Ag appears more reflective than Au. Each batch of NWs was functionalized with a different ASO: ASOT683 on 0011-patterned wires; ASOT683MM, which has a single mismatch, on 1001-patterned wires; and a non-complementary ASOnon probe on 0110-patterned wires. The three NW populations were then mixed and incubated first with the target PCA3 RNA, rinsed, and then with fluorescently-labeled ASOT735, and reflectance and fluorescence optical microscope images were obtained after the hybridizations. All of the NW patterns were visible in the reflectance image. The corresponding fluorescence image showed bright fluorescence emission from the 0011 pattern wires. There was little or no fluorescence associated with the 1001 and 0110 wires (Supplementary Material, Figure 3), which were functionalized with single-mismatch-containing and non-complementary ASOs, respectively. These results demonstrate high selectivity for hybridizations on NWs.
Next, ASOT735 was covalently coupled to 50 nm AuNPs (ASOT735:AuNPs). ASO:AuNPs were used to increase the mass added to NRs after the hybridization reactions (a 50 nm AuNP has a mass ~3000 times greater than a PCA3 RNA molecule + ASOs). This increase in mass resulted in significant shifts of the resonance frequency of the suspended NRs, resulting in increased resolution of the frequency peaks, and significantly extending the lower limits of detection.
In addition to using the AuNPs to amplify the resonance frequency shift signal, it was also possible to visualize binding events directly via TEM and FE-SEM. In initial experiments, a 45mer DNA “mimic” for PCA3 was hybridized to the NWs, followed by hybridization with 50 nm ASOT735:AuNPs. TEM results (Figure 2) showed high specificity with the sandwich hybridization assay using AuNPs derivatized with the complementary ASOT735, with very weak or absent hybridization for NWs derivatized with non-complementary or mismatched ASOs. These results again indicate high specificity of the sandwich hybridization system.
Figure 2. TEM and FE-SEM images for Hybridization of PCA3 sequence to Functionalized NWs.

Panel A. Hybridizations using PCA3 mimic. NWs were functionalized with ASOT735, and AuNPs were functionalized with ASOT683. Top sections show TEM and FE-SEM images obtained using 45 nt DNA mimic of PCA3, and bottom sections show TEM and FE-SEM images obtained using a non-complementary control DNA. Left sections represent TEM images and right sections show FE-SEM images. Insets in the TEM images are 2X magnifications to show more detail on the SiO2 coating and attached AuNPs. Scale bars represent 500 nm.
Panel B. Hybridizations using authentic PCA3 RNA transcripts. Top sections show results with SiO2 coated NWs functionalized with complementary ASOT735, whereas bottom sections show results using NWs functionalized with a mismatched ASO. As in Panel A, left sections show TEM images, and right sections show FE-SEM images. RNA concentration was ~ 20X lower than that of the DNA mimic. Scale bars represent 500 nm.
AuNP surface coverages. as estimated from the FE-SEM images, were (1.4 ± 0.4) × 1010/cm2 (7100 nm2/particle) for the complementary target and (3 ± 2) × 107/cm2 (3.3 × 106 nm2/particle) for the noncomplementary control. Pre-hybridizing the target DNA to the ASO-coated AuNPs gave similar particle coverages compared with initial PCA3 hybridization to the NWs followed by hybridization to ASO:AuNP.
In similar experiments where the PCA3 mimic DNA sequence was replaced with 20 nM in vitro transcribed PCA3 RNA, we also observed excellent selectivity for PCA3 RNA over the noncomplementary control RNA (Figure 2B), although under these conditions lower surface densities of AuNPs were observed. Additionally, the wire-to-wire variability in AuNP coverage was higher for the experiments with authentic RNA samples (on the same order as the coverage; Figure 2B). The lower coverage for the PCA3 RNA as compared with the DNA mimic sequence could result from a number of factors. For example, multivalency is known to play a crucial role in the binding thermodynamics of DNA:AuNP to surfaces 41,42 , and reduced multivalency due to the larger size of the RNA molecules may be responsible for the lower coverage observed for PCA3 RNA (either for individual RNAs or the number of molecules involved in binding of each 50-nm AuNP). For CTC detection, multiple attachments per particle seem unlikely. Decreased coverage could also simply reflect RNA degradation, in which case RNAse inhibitors and stringent precautions will rectify the loss.
Optical Measurement of NW Resonance Frequency Shift
Biosensor transduction, involving the correlation between measured resonant frequency shift and ASO:AuNP target mass, was tested using Rh NRs. With the complementary PCA3-ASO:AuNPs in sandwich hybridization assays, we observed substantial binding of ASO:AuNPs (Figure 3 shows results for a representative Rh NR with significant numbers of bound particles and a measured frequency shift ~200 kHz lower). In comparison, following the same procedure with the non-complementary ASO:AuNPs, we observed a negligible frequency shift and no bound particles visible by SEM (Figure 4), indicating the high specificity of target capture by the ASO probes (background resonance frequency shifts were on the order of 1% of that induced by a single AuNP, but it was in the opposite (increased) direction, which presumably reflects slight alterations induced by the hybridization process itself and not non-specific binding).
Figure 3. Resonance frequency shift following sandwich hybridization assay with the complementary probe-PCA3 target.
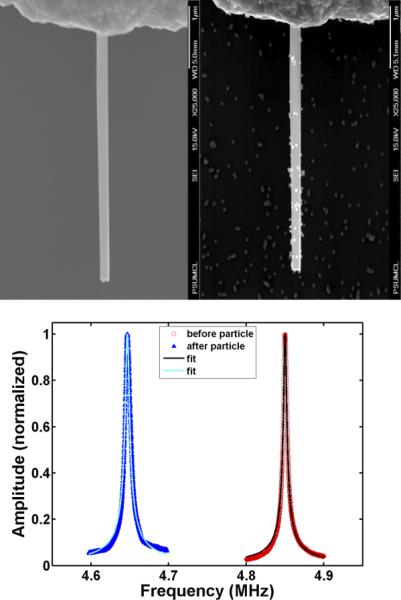
PCA3 target was used in a sandwich hybridization assay with the complementary ASO combination. Upper Left panel shows a scanning electron micrograph of a single clean RhNW before hybridization. Upper Right panel shows the same RhNW after the sandwich hybridization assay, with a substantial number of 50 nm AuNPs bound. Bottom panel shows a significant leftward shift (~200 kHz lower frequency) of the resonance peak upon particle binding.
Figure 4. Resonance frequency shift following sandwich hybridization assay with a non-complementary probe-PCA3 target.
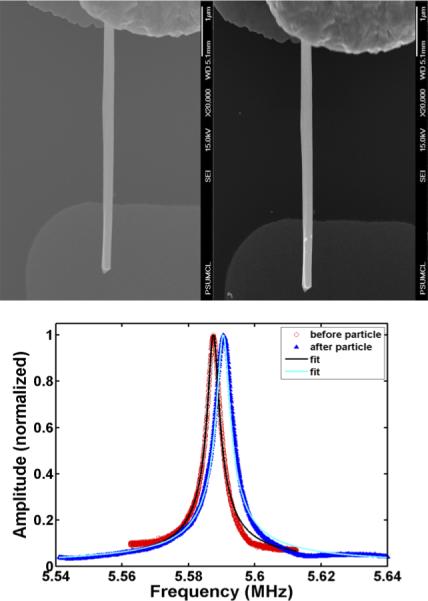
As a control, a non-complementary probe-target combination was used in the sandwich hybridization assay. Upper Left Panel shows a clean RhNW before hybridization. Upper Right Panel shows the same RhNW after hybridization with a non-complementary target. No AuNP binding was observed. We observed a negligible frequency shift (Bottom panel, requiring a different scale from Figure 3, to be visible), that represents <1% of the shift induced by a single AuNP, and this shift was an increase in resonance frequency, which presumably represents minor effects produced by the hybridization protocol.
To estimate bound mass, we used an SEM image count of the number of AuNPs x2, assuming an equal number of particles on the upper and lower halves of the NRs. For a lightly loaded, single-degree-of-freedom, linear harmonic resonator, the fractional mass change (added mass/NR mass) is twice the fractional change in frequency (frequency shift/NR resonance frequency). The best fit straight line through the origin was empirically found to have slope 2.0 (Figure 5), establishing that this simple resonator model performs mass detection very well (at least up to 15% mass loading, the maximum we have examined, which is far above any likely concentrations of PCA3 or other CTC marker RNAs to be tested).
Figure 5. Fractional increase in mass due to bound AuNPs.
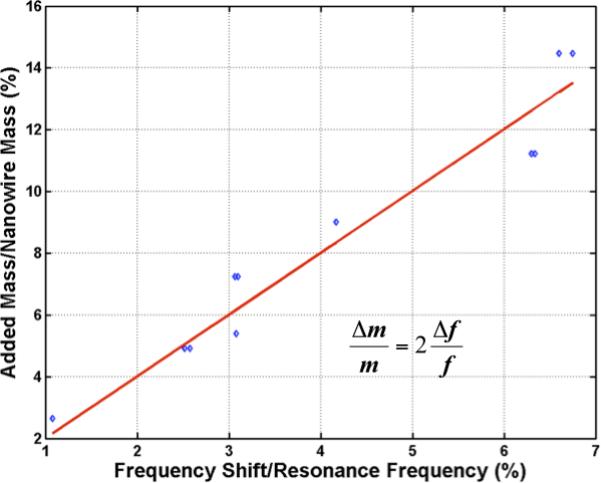
Following a sandwich hybridization, the number of AuNPs bound to NRs was counted by FE-SEM, and their mass is shown plotted against measured fractional change (left-shift) in frequency, for several RhNWs subjected to the sandwich hybridization assay with complementary probe-PCA3 target RNA combination. Data points and the best fit line through the origin show a slope of 2, in agreement with the expectation for small mass loading of a linear, single-degree-of-freedom resonator.
Discussion
Here we have determined optimal sites for ASO binding to PCA3, a PCa marker RNA, in a “sandwich” hybridization assay. Conditions were developed which showed essentially no non-specific binding, and target RNA binding was not adversely affected by addition of non-complementary competing cytoplasmic RNA (at 1000X excess).
Binding of ASO:AuNPs to PCA3 target molecules (either RNA or a “mimic” DNA) on silica-coated NWs retained the high specificity observed with spotted microarrays. These ASO:AuNP tags amplify the mass of the nucleic acids in the sandwich assay by a factor of ~3000, greatly amplifying resonance frequency shifts, which facilitates detection of very low levels of target RNA molecules. Specificity is excellent, since the sandwich hybridization format requires 2 high-specificity binding events. We found that binding of ASO:AuNPs to derivatized NRs induces a resonance frequency shift which is quantitatively proportional to the number of ASO:AuNPs bound, with each bound ASO:AuNP inducing a shift of ~ 1 kHz, which is easily measureable with standard equipment.
There is a growing interest in nanowire-based sensors 41 because of the many advantages they offer, including high surface-to-volume ratios, the relatively high densities of functionalization moieties such as nucleic acids (although the important contribution of “nanostructure” also influences capture efficiency; see 42), and their facile integration into addressable arrays. A variety of materials are suitable for NW synthesis, with particular interest in use of silicon 43, and many applications use optical sensing strategies 43,44. Here we used optical detection of resonance frequency shifts, in a manner compatible with bottom-up assembly of arrays. Others have also employed silicon NRs (in a top-down assembly scheme which is also compatible with CMOS fabrication) in conjunction with piezoresistive detection with impressive detection efficiency 45. Here, ASOs were displayed on NWs and AuNPs for RNA detection; the AuNPs greatly amplify NR resonance frequency shifts, although AuNPs are also finding wide use in a variety of other sensing schemes 46,47. Use of aptamers for detection of proteins or other targets proceeds in analogous manner 44.
By utilizing a bottom-up approach, we intend to array many (potentially hundreds) of different functionalized NRs, each containing different ASO sequences directed to regions found within distinct marker RNAs for many different cancers. Given many recent studies showing that even early-stage lesions can give rise to significant numbers of CTCs, such a functionalized array should enable screening for early cancers in a prospective manner. This approach has many advantages, including high specificity, sensitivity, and lack of need for amplification steps. It provides a viable alternative for other approaches, which have generally relied on positive selection using epCAM. For example, Chung et al. described a sensitive electrical biosensor which involved magnetic concentration, immunochemistry, and impedance spectroscopy for detection 48, and Stott et al. described a microvortexing chip also utilizing epCAM capture of CTCs 49. Other approaches, such as a “negative selection” protocol which depletes normal cells to achieve major CTC enrichment 50, although inherently more involved, are also of potential interest and compatible with our platform approach.
Supplementary Material
Acknowledgements
We would like to thank the DNA Microarray Core Facility at University Park for custom synthesis of microarrays, and the Genome Sciences Facility at Hershey.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The Authors declare no competing financial interests.
The Authors declare that they have no Conflicts of Interest.
REFERENCES
- 1.Gerges N, Rak J, Jabado N. New technologies for the detection of circulating tumour cells. Br Med Bull. 2010;94:49–64. doi: 10.1093/bmb/ldq011. [DOI] [PubMed] [Google Scholar]
- 2.Dawood S, Broglio K, Valero V, Reuben J, Handy B, Islam R, et al. Circulating tumor cells in metastatic breast cancer: from prognostic stratification to modification of the staging system? Cancer. 2008;113:2422–2430. doi: 10.1002/cncr.23852. [DOI] [PubMed] [Google Scholar]
- 3.Dotan E, Cohen SJ, Lapaugh KR, Meropol NJ. Circulating tumor cells: evolving evidence and future challenges. Oncologist. 2009;14:1070–1082. doi: 10.1634/theoncologist.2009-0094. [DOI] [PubMed] [Google Scholar]
- 4.Miller MC, Doyle GV, Terstappen LW. Significance of Circulating Tumor Cells Detected by the CellSearch System in Patients with Metastatic Breast Colorectal and Prostate Cancer. J Oncol. 2010;2010:617421. doi: 10.1155/2010/617421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smerage JB, Hayes DF. The prognostic implications of circulating tumor cells in patients with breast cancer. Cancer Invest. 2008;26:109–114. doi: 10.1080/07357900701784204. [DOI] [PubMed] [Google Scholar]
- 6.Cristofanilli M, Budd G, Ellis M, Stopeck A, Matera J, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004;351:824–826. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 7.Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med. 2010;2:25ra23. doi: 10.1126/scitranslmed.3000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helo P, Cronin AM, Danila DC, Wenski S, Gonzalez-Espinoza R, Anand A, et al. Circulating prostate tumor cells detected by reverse transcription-PCR in men with localized or castration-refractory prostate cancer: concordance with CellSearch assay and association with bone metastases and with survival. Clin Chem. 2009;55:765–773. doi: 10.1373/clinchem.2008.117952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scher H, Jia X, de Bono JS, Fleisher M, Pienta KJ, Raghaven D, et al. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer; a reanalysis of IMMC38 trial data. Lancet Oncol. 2009;10:233–239. doi: 10.1016/S1470-2045(08)70340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright JL, Lange PH. Newer potential biomarkers in prostate cancer. Rev Urol. 2007;9:207–213. [PMC free article] [PubMed] [Google Scholar]
- 11.Mitas M, Chaudhary U, Marshall D, Gattoni-Celli S. Detection of circulating tumor cells in the peripheral blood of patients with androgen-independent, advanced or metastatic prostate cancer. Am J Hematol. 2008;83:87. doi: 10.1002/ajh.21045. [DOI] [PubMed] [Google Scholar]
- 12.Kruck S, Gakis G, Stenzl A. Circulating and disseminated tumor cells in the management of advanced prostate cancer. Adv Urol. 2012;2012:135281. doi: 10.1155/2012/135281. (Epub) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jost M, Day JR, Slaughter R, Koreckij TD, Gonzales D, Kinnunen M, et al. Molecular assays for the detection of prostate tumor derived nucleic acids in peripheral blood. Mol Cancer. 2010;9:174–179. doi: 10.1186/1476-4598-9-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sieuwerts AM, Kraan J, Bolt J, van der Spoel P, Elstrodt F, Schutte M, et al. Anti-epithelial cell adhesion molecule antibodies and the detection of circulating normal-like breast tumor cells. J Natl Cancer Inst. 2009;101:61–66. doi: 10.1093/jnci/djn419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raimondi C, Gianni W, Cortesi E, Gazzaniga P. Cancer stem cells and epithelial-mesencymal transition: revisiting minimal residual disease. Curr Cancer Drug Targets. 2010;10:496–508. doi: 10.2174/156800910791517154. [DOI] [PubMed] [Google Scholar]
- 16.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, et al. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabisiewicz A, Kulik J, Kober P, Brewczynska E, Pienkowski T, Siedlecki J. Detection of circulating breast cancer cells in peripheral blood by a two-marker reverse transcriptase-polymerase chain reaction assay. Acta Biochim. Polonica. 2004;51:747–755. [PubMed] [Google Scholar]
- 18.Mocellin S, Del Fiore P, Guarnieri L, Scalerta R, Foletto M, Chiarion V, et al. Molecular detection of circulating tumor cells is an independent prognostic factor in patients with high-risk cutaneous melanoma. Int. J. Cancer. 2004;111:741–745. doi: 10.1002/ijc.20347. [DOI] [PubMed] [Google Scholar]
- 19.Liotta L, Stetler-Stevenson W. Tumor invasion and metastasis: an imbalance of positive and negative regulation. Cancer Research. 1991;51:5054s–5059s. [PubMed] [Google Scholar]
- 20.Weiss L. Metastatic inefficiency. Adv. Cancer Res. 1990;54:159–211. doi: 10.1016/s0065-230x(08)60811-8. [DOI] [PubMed] [Google Scholar]
- 21.Fidler I. Metastasis: Quantitative analysis of distribution and fate of tumor emboli labeled with 125I-5-iodo-deoxyuridine. J. Natl. Cancer Insti. 1970;45:773–782. [PubMed] [Google Scholar]
- 22.Nakagawa T, Martinez SR, Goto Y, Koyanagi K, Kitago M, Shingai T, et al. Detection of circulating tumor cells in early-stage breast cancer metastasis to axillary lymph nodes. Clin Cancer Res. 2007;13:4105–4110. doi: 10.1158/1078-0432.CCR-07-0419. [DOI] [PubMed] [Google Scholar]
- 23.Fizazi K, Morat L, Chauveinc L, Prapotnich D, De Crevoisier R, Escudier B, et al. High detection rate of circulating tumor cells in blood of patients with prostate cancer using telomerase activity. Annals Oncology. 2007;18:518–521. doi: 10.1093/annonc/mdl419. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt H, DeAngelis G, Eltze E, Gockel I, Semionow A, Brandt B. Asynchronous growth of prostate cancer is reflected by circulating tumor cells delivered from distinct, even small foci, harboring loss of heterozygosity of the PTEN gene. Cancer Res. 2006;66:8959–8965. doi: 10.1158/0008-5472.CAN-06-1722. [DOI] [PubMed] [Google Scholar]
- 25.Ali A, Furusato B, Ts'o PO, Lum ZP, Elsamanoudi S, Mohamed A, et al. Assessment of circulating tumor cells (CTCs) in prostate cancer patients with low-volume tumors. Pathol Int. 2010;60:667–672. doi: 10.1111/j.1440-1827.2010.02584.x. [DOI] [PubMed] [Google Scholar]
- 26.Su M, Li S, Dravid VP. Microcantilever resonance-based DNA detection with nanoparticle probes. Appl Phys Lett. 2003;82:3562–3564. [Google Scholar]
- 27.Li M, Bhiladvala RB, Morrow TJ, Sioss JA, Lew KK, Redwing JM, et al. Bottom-up assembly of large-area nanowire resonator arrays. Nature Nanotechnology. 2008;3:88–92. doi: 10.1038/nnano.2008.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li M, Mayer TS, Sioss JA, Keating CD, Bhiladvala RB. Template-grown metal nanowires as resonators: performance and characterization of dissipative and elastic properties. Nano Letters. 2007;7:3281–3284. doi: 10.1021/nl071416e. [DOI] [PubMed] [Google Scholar]
- 29.Morrow TJ, Li M, Kim JW, Mayer TS, Keating CD. Programmed assembly of DNA-coated nanowire arrays. Science. 2009;323:352. doi: 10.1126/science.1165921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klecka J, Holubec L, Pesta M, Topolcan O, Hora M, Eret V, et al. Differential display code 3 (DD3/PCA3) in prostate cancer diagnosis. Anticancer Res. 2010;30:665–670. [PubMed] [Google Scholar]
- 31.Floriano-Sanchez E, Cardenas-Rodiquez N, Castro-Marin M, Alvarez-Grave P, Lara-Padilla E. DD3(PCA3) gene expression in cancer and prostatic hyperplasia. Clin Invest Med. 2009;32:E258. [PubMed] [Google Scholar]
- 32.Goodman OBJ, Symanowski JT, Loudyi A, Fink LM, Ward DC, Vogelzang NJ. Circulating tumor cells as a predictive biomarker in patients with hormone-sensitive prostate cancer. Clin Genitourin Cancer. 2011;9:31–8. doi: 10.1016/j.clgc.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Doyen J, Alix-Panabieres C, Hofman P, Parks SK, Chamorey E, Naman H, et al. Circulating tumor cells in prostate cancer: A potential surrogate marker of survival. Crit Rev Oncol Hematol. 2011 doi: 10.1016/j.critrevonc.2011.05.004. Epub Jun 14 (PMID21680196) [DOI] [PubMed] [Google Scholar]
- 34.Durand X, Moutereau S, Xylinas E, de la Taille A. ProgensaTM PCA3 test for prostate cancer. Expert Rev Mol Diagn. 2011;11:137–144. doi: 10.1586/erm.10.122. [DOI] [PubMed] [Google Scholar]
- 35.Arlett JL, Myers EB, Roukes ML. Comparative advantages of mechanical biosensors. Nature Nanotech. 2011;6:203–215. doi: 10.1038/nnano.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waggoner PS, Tan CP, Craighead HG. Mirofluidic integration of nanomechanical resonators for protein analysis in serum. Sensors and Actuators B. 2010;150:550–555. [Google Scholar]
- 37.Ekinci KL. Electromechanical transducers at the nanoscale: actuation and sensing of motion in nanoelectromechanical systems (NEMS). Small. 2005;1:786–797. doi: 10.1002/smll.200500077. [DOI] [PubMed] [Google Scholar]
- 38.Erickson D, Mandal S, Yang AH, Cordovez B. Nanobiosensors: optofluidic, electrical and mechanical approaches to biomolecular detection at the nanoscale. Microfluidics Nanofluidics. 2008;4:33–52. doi: 10.1007/s10404-007-0198-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chai D. RNA structure and modeling: progress and techniques. Prog Nucleic Acid Res Mol Biol. 2008;82:71–100. doi: 10.1016/S0079-6603(08)00003-2. [DOI] [PubMed] [Google Scholar]
- 40.Sioss JA, Stoemer RL, Sha MY, Keating CD. Silica coated, Au/Ag striped nanowires for bioanalysis. Langmuir. 2007;23:11334–11341. doi: 10.1021/la7019846. [DOI] [PubMed] [Google Scholar]
- 41.Ramgir NS, Yang YL, Zacharias M. Nanowire-based sensors. Small. 2010;6:1705–1722. doi: 10.1002/smll.201000972. [DOI] [PubMed] [Google Scholar]
- 42.Bin X, Sargent EH, Kelley SO. Nanostructuring of sensors determines the efficiency of biomolecular capture. Anal Chem. 2010;82:5928–5931. doi: 10.1021/ac101164n. [DOI] [PubMed] [Google Scholar]
- 43.Wan Y, Sha J, Chen B, Fang Y, Wang Z, Wang Y. Nanodevices based on silicon nanowires. Recent Pat Nanotechnol. 2009;31:1–9. doi: 10.2174/187221009787003348. [DOI] [PubMed] [Google Scholar]
- 44.Wang G, Wang Y, Chen L, Choo J. Nanomaterial-assisted aptamers for optical sensing. Biosens Bioelectron. 2010;25:1859–1868. doi: 10.1016/j.bios.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 45.Mile E, Jourdan G, Bargatin I, Labarthe S, Marcoux C, NAndreucci P, et al. In-plane nanoelectromechanical resonators based on silicone nanowire piezoresistive detection. Nanotechnology. 2010;21:165504. doi: 10.1088/0957-4484/21/16/165504. [DOI] [PubMed] [Google Scholar]
- 46.Merkoci A. Nanoparticle-based strategies for DNA, protein and cell sensors. Biosens Bioelectron. 2010;26:1164–1177. doi: 10.1016/j.bios.2010.07.028. [DOI] [PubMed] [Google Scholar]
- 47.Jena BK, Ghosh S, Bera R, Dey DS, Das AK, Raj CR. Bioanalytical applications of au nanoparticles. Recent Pat Nanotechnol. 2010;4:41–52. doi: 10.2174/187221010790712075. [DOI] [PubMed] [Google Scholar]
- 48.Chung YK, Reboud J, Lee KC, Lim HM, Lim PY, Wang KY, et al. An electrical biosensor for the detection of circulating tumor cells. Biosens Bioelectron. 2010;26:2520–2526. doi: 10.1016/j.bios.2010.10.048. [DOI] [PubMed] [Google Scholar]
- 49.Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc Natl Acad Sci. 2010;107:18392–18397. doi: 10.1073/pnas.1012539107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang L, Lang JC, Balasubramanian P, Jatana KR, Schuller D, Agrawal A, et al. Optimization of an enrichment process for circulating tumor cells from the blood of head and neck cancer patients through depletion of normal cells. Biotechnol. Bioeng. 2008;102:521–534. doi: 10.1002/bit.22066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.