

Abstract
Objective
Innate immune responses activate synoviocytes and recruit inflammatory cells into the rheumatoid joint. Type I interferons (IFN) play a role in autoimmunity and IFN gene transcription is activated by IFN-regulatory factors (IRF) in response to innate sensor recognition. This study examined the effect of genetic deficiency of IRF7 in a passive K/BxN serum transfer model of arthritis.
Methods
Passive transfer arthritis was induced in Irf7−/− mice and additional groups were treated with IFNβ or poly (I-C). Clinical arthritis scores, histology, microcomputed tomography, and synovial tissue Q-PCR were performed. Mouse serum was analyzed by ELISA.
Results
In the K/BxN passive model, arthritis severity was significantly increased in Irf7−/− mice compared with wild type (WT). In addition, synovial and serum IFNβ expression was decreased, potentially contributing to increased arthritis. Irf7−/− mice injected with replacement IFNβ had a decrease in arthritis. Poly (I-C) treatment diminished arthritis in Irf7−/− mice, restored synovial IFNβ gene expression, and increased serum IFNβ. In vitro studies demonstrated that poly (I-C) stimulation of Irf7−/− fibroblast-like synoviocytes (FLS) resulted in increased induction of pro-inflammatory gene expression compared with WT FLS; however, IFNβ expression was not significantly different. In contrast, Irf7−/− macrophages showed significantly less induction of IFNβ in response to poly (I-C) stimulation.
Conclusions
IRF7 deficiency exacerbated arthritis and replacement treatment with IFNβ or poly (I-C) decreased arthritis severity. Both macrophage and synoviocyte specific roles for IRF7 likely contribute to increased arthritis. IRF7 might play an anti-inflammatory role in passive transfer arthritis through regulation of macrophage IFNβ production.
Keywords: transcription factors, signal transduction, inflammation, rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA) is characterized by critical alterations in synovial inflammation, matrix degradation, and cellular proliferation. This destructive process is controlled by a number of intracellular signaling pathways that regulate gene expression in the rheumatoid synovial intimal lining (1, 2). Innate sensors induce secretion of type I interferons (IFNα and IFNβ) that serve as key sentinel cytokines and play a complex role in autoimmunity (3). Type I IFN can induce expression of hundreds of IFN-stimulated genes that can both promote immunity and other functions that suppress inflammation and immune responses. The IFN-regulatory factors (IRF) are involved in innate receptor signaling and are mainly responsible for induction of the type I IFN response downstream of pattern recognition receptors (4).
Type I IFN can promote immunity but can also suppress immunity and inflammation. IFNβ inhibits inflammatory arthritis in mouse models. Mice with collagen-induced arthritis injected with fibroblasts expressing IFNβ have decreased arthritis severity and bone and cartilage destruction (5). IFNβ also decreases matrix metalloproteinases (MMP), IL-1, and TNF production by fibroblast-like synoviocytes (FLS) (6). However, a clinical trial using IFNβ in patients with RA showed minimal efficacy despite decreased synovial IL-1, IL-6, and MMP1 (7). Similar to the IFN response, innate receptor activation can also have multiple, diverse effects depending on dose and administration of the ligand. For example, TLR activation has been shown to contribute to inflammation in autoimmune diseases; however, repeated exposure to a TLR agonist such as polyinosinic polycytidylic acid [poly (I-C)] induces a hypo-responsiveness to further TLR stimulation (8, 9). In addition, poly (I-C) has been previously studied in the K/BxN passive antibody-mediated arthritis model and resulted in a decrease in arthritis that was dependent on IFNα/β receptor signaling (9, 10). Of interest, IKKε, an IKK-related kinase that activates IRF3 and IRF7 in response to dsRNA innate receptor ligation, was studied in murine passive K/BxN transfer arthritis (10). Low dose IFNβ treatment enhanced the anti-inflammatory effect of IKKε deficiency possibly by increasing production of IL-1 receptor antagonist (IL-1RA).
IRF3 and IRF7 are key regulators of type I IFN gene expression (11). We previously demonstrated that IRF3 is expressed and activated in rheumatoid synovium, suggesting the potential importance of IRF3 in RA (12). In vitro siRNA knockdown indicated that IRF3, but not IRF7, played a key role in human FLS type I IFN responses as well as traditionally AP-1 regulated gene expression (13). In mouse embryonic fibroblasts and plasmacytoid dendritic cells, IRF7 is considered the master regulator of type I IFN immune responses (14). Constitutive and ubiquitous IRF3 mainly controls initial IFNβ production, whereas basal expression of IRF7 is low in most cell populations, but induced by type I IFN. Nuclear translocation of IRF7 can induce IFNβ gene expression and also numerous subtypes of IFNα. We hypothesized that IRF7 regulates synovial inflammation and contributes to innate immunity and the clinical course of arthritis, possibly by altering IFNβ and other IFN-regulated genes in a cell specific manner. Further understanding of this regulatory mechanism will have major implications for targeting the IRF transcription factor family to treat autoimmune and inflammatory diseases.
Materials and Methods
Mice
Irf7−/− transgenic mice were a kind gift from Dr. T. Taniguchi (University of Tokyo, Tokyo Japan) and were backcrossed 10 generations onto C57BL/6 background (14). KRN T cell receptor (TCR) transgenic mice were a gift from Drs. D. Mathis and C. Benoist (Harvard Medical School, Boston, MA) and Institut de Génétique et de Biologie Moléculaire et Cellulaire (Strasbourg, France) (15, 16). C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The mice were bred and maintained under standard conditions in the UCSD Animal Facility accredited by the American Association for Accreditation of Laboratory Animal Care. All animal experiments were carried out according to protocols approved by the Institutional Animal Care and Use Committee.
Passive K/BxN serum transfer arthritis
Arthritic adult K/BxN mice were bled and the sera were pooled. Groups of 8 to 10 recipient mice were injected with 75μL pooled sera intraperitoneally (IP) on day 0 and day 2. The standard 150μL amount was decreased 75μL to allow detection of the clinical effect. Some groups also received 1000 IU of intraperitoneal IFNβ (Chemicon, Temecula, CA) daily, 200μg of poly (I-C) (Sigma, St. Louis, MO) on day 0 and day 2, or PBS. Clinical arthritis scores were evaluated using a scale of 0–4 for each paw for a total of 16. Ankle thickness was measured with a caliper (Mitutoyo Corporation, Japan) in mm.
Microcomputed tomography (micro-CT) of the calcaneus
Ankle joints of arthritic WT and Irf7−/− mice were imaged on day 12 using the Skyscan 1076 microCT-40 system (Kontich, Belgium). Samples were scanned at 9μm voxel size, applying an electric potential of 50 kVp and current of 200 uA, using a 0.5mm aluminum filter. Skyscan software, Dataviewer, CTAn, CT Vol (Kontich, Belgium) was used for orientation and analysis to determine bone histomorphometric parameters. Cortical and trabecular bone analysis was performed on the calcaneus.
Gene Expression
The ankles of arthritic mice were snap frozen and pulverized. FLS and macrophage mRNA from non-arthritic mice was isolated with RNA STAT-60 (Tel Test Inc., Friendswood, TX). Total RNA isolation with RNA STAT and cDNA synthesis by RT-PCR were performed as previously described (17). Forward and reverse primers as well as fluorogenic TaqMan FAM/TAMRA-labeled hybridization probes were used for IFNβ, TGFβ, IL-1RA, IL-6, RANTES, MIP-1α, IFNγ-inducible protein 10 (IP-10), matrix metalloproteinase-3 (MMP-3), and vascular endothelial grow factor alpha (VEGFα) (Assay on Demand; Applied Biosystems, Foster City, CA) and normalized to hypoxanthine-guanine phosphoribosyltransferase 1 (HPRT). Data are expressed as the ratio between gene of interest CE and HPRT1 CE, yielding the relative gene expression (RE).
Cytokine Measurement
Serum from whole blood was obtained from wild type (WT) and Irf7−/− mice and assayed for IFNβ, IL-1RA, and IL-10 using a commercial capture ELISA (R&D Systems, Minneapolis, MN).
Murine synoviocytes and peritoneal macrophages
FLS and peritoneal macrophages were isolated from IRF7 deficient mice. We derived murine Irf7−/− FLS from these animals by dissecting synovium, mincing the tissue and enzymatically dispersing the cells with 1 mg/ml collagenase type VIII in serum-free RPMI 1640 for 1 hour at 37° C. FLS were used from passage 3 through 8 during which time they were a homogeneous population of cells (<1% CD11b positive, <1% phagocytic, and <1% FcR II and FcR III receptor positive) (12). In addition, we isolated peritoneal macrophages via intraperitoneal lavage with PBS. FLS and macrophages were stimulated with 10μg/mL poly (I-C) (Sigma, St. Louis, MO) for 18h. Cells were harvested with 1mL RNA STAT-60 (Tel Test Inc., Friendswood, Texas, USA) per sample.
Statistics
Data are expressed as a mean ± SEM. Comparisons of PCR gene expression data and ELISA cytokine measurements were performed by Student's t test. Areas under the curve were calculated from delta paw swelling. Two-way ANOVA was used for multiple comparisons. P values of < 0.05 were considered statistically significant.
Results
IRF7 deficiency increases the clinical severity of K/BxN serum transfer arthritis
The type I IFN response plays a critical role in inflammation and is regulated by several transcription factors as part of the IFN enhanceosome (18). To evaluate the in vivo contribution of IRF7 to synovial inflammation and arthritis, we used the passive transfer K/BxN arthritis model in Irf7−/− mice. Deficiency of IRF7 resulted in increased arthritis in the K/BxN antibody-mediated passive transfer model after day 8 (Figure 1A) shown as change in paw swelling in Irf7−/− and WT mice (average change in paw measurement in mm). On days 8 and 12, there was increased arthritis observed in the Irf7−/− mice suggesting a counter-regulatory or anti-inflammatory role for IRF7 in this model. Arthritis severity in Irf7−/− mice was significantly increased on day 8 (average change in paw measurement 5.5mm WT vs. 9.9mm Irf7−/−, p<0.02) and on day 12 (3.5mm WT vs. 8.6mm Irf7−/−, p<0.01) compared with WT (Figure 1A, left). Total area under the curve (AUC) was significantly increased in Irf7−/− mice compared with WT (Figure 1A, right, p<0.05). No significant difference in synovial histology was detected in the Irf7−/− ankle tissue compared with WT (data not shown). The lack of effect on synovial histology despite increased paw volume suggested that IRF7 might also play a role in vascular permeability and subsequent tissue edema contributing to paw measurement differences as noted in other arthritis models (19, 20). In addition, clinical score and paw measurements are more sensitive compared with histological score so it might be difficult to detect a significant difference with this degree of arthritis and joint inflammation.
Figure 1.
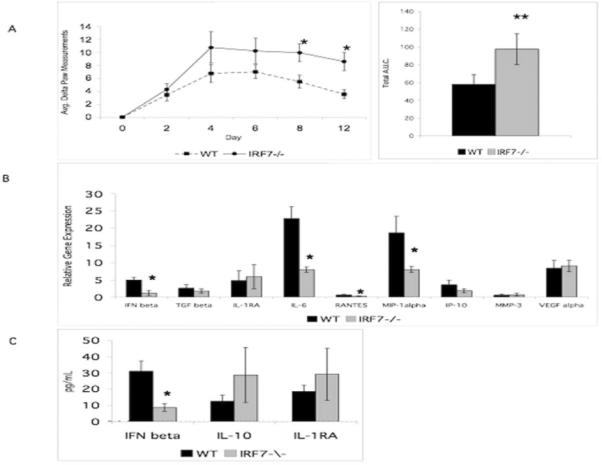
Irf7−/− mice have increased arthritis in the passive K/BxN serum transfer model. A. Irf7−/− mice developed significantly more paw swelling than WT controls day 8 and day 12 (left panel, *p<0.05). Total area under the curve (AUC) was significantly increased in Irf7−/− mice compared with WT (right panel, **p<0.05). Data are representative of 2 independent experiments. B. IFNβ was decreased in Irf7−/− synovial tissue. Relative expression (RE) of IFNβ, IL-6, RANTES, and MIP-1α was decreased (*p<0.05) compared with control. RE of TGFβ, IL-1RA, IP-10, MMP-3, and VEGFα was not significantly different. C. Serum IFNβ protein measured by ELISA was decreased in the Irf7−/− arthritic mice (*p<0.009, n=8 mice per group). Levels of IL-10 and IL-1RA were not different in sera of WT and Irf7−/− K/BxN mice.
Augmented systemic and local pro-inflammatory cytokine profiles in arthritic Irf7−/− mice
To evaluate potential mechanisms for increased arthritis in the Irf7−/− K/BxN passive transfer model, we analyzed synovial tissue extracts from ankle joints of the mice on day 4 of the arthritis model (Figure 1B). Most notably and in agreement with the clinical effect of increased arthritis, IFNβ gene expression was significantly less in IRF7 deficient synovium. Gene expression of IFNβ, RANTES, MIP1α, and IL-6 was decreased in K/BxN Irf7−/− synovial tissue compared with K/BxN WT synovium (3.9-fold, p<0.05; 2.2-fold, p<0.05; 2.4-fold, p<0.04; 1.7-fold, p<0.05 respectively). Gene expression of MMP3 and IP-10 was not significantly different in ankle Irf7−/− synovial tissue compared with WT. Of note, the significant decrease of IFNβ mRNA in the ankle tissue of the Irf7−/− mice suggested that the exacerbation of arthritis could be due to lower levels of this anti-inflammatory cytokine in mouse arthritis models. Expression of other anti-inflammatory genes, including IL-1RA and TGFβ, as well as VEGFα, was not significantly different compared with control arthritic synovial tissue.
To further investigate the possibility that IRF7 deficiency exacerbates K/BxN arthritis by decreasing type I IFN, serum levels of the anti-inflammatory cytokine IFNβ were evaluated. In agreement with decreased synovial IFNβ gene expression in arthritic Irf7−/− K/BxN mice, serum IFNβ levels were also significantly decreased in Irf7−/− mice (2.8-fold, p<0.04) compared with WT K/BxN (Figure 1C). Therefore, decreased IFNβ in serum and synovial tissue might contribute to the increase in K/BxN arthritis in Irf7−/− mice. Decreased expression of the anti-inflammatory cytokine IFNβ in both synovium and serum is supportive of the overall clinical effect of increased arthritis. Serum levels of IL-10 and IL-1RA were not significantly different in Irf7−/− mice compared with WT.
Analysis of bone damage by micro-CT image analysis
Because histologic evaluation the degree of extra-articular swelling in the tissue sections revealed no significant difference between WT and Irf7−/−, we performed micro-CT to further evaluate tissue and bone damage in the arthritic animals. We used micro-CT to determine differences in cortical and trabecular bone damage between WT and Irf7−/− arthritic serum transfer mice (Figure 2). Three-dimensional reconstruction of the calcaneus showed no significant difference in cortical bone area/total area (BA/TA) computed as percent object area for WT (79.3 ± 3.0) compared with Irf7−/− (79.5 ± 2.1). In addition, no significant difference was detected in trabecular bone volume/tissue volume (BV/TV) computed as percent bone volume in arthritic WT (20.8 ± 3.4) compared with Irf7−/− mice (21.6 ±3.4). This data supported the histological analysis that revealed no difference in bone erosions in WT compared with Irf7−/− arthritic mice.
Figure 2.

Visualization, 3D reconstruction, and quantification of serum transfer induced joint damage by microcomputed tomography. Representative images of arthritic WT and Irf7−/− mice (n=5 each) cortical and trabecular bone are shown in upper left images. Percent cortical bone area and trabecular bone volume calculations are shown in the table on the upper right. No significant difference between WT and IRF7 KO cortical bone area or trabecular bone volume was detected. The lower image demonstrates the regions of micro-CT used for cortical and trabecular bone analysis.
IFNβ treatment suppresses Irf7−/− K/BxN inflammatory arthritis
To explore the role of IFNβ in regulation of Irf7−/− K/BxN inflammatory arthritis, mice were treated with replacement recombinant IFNβ or PBS (Figure 3A). Irf7−/− mice treated with IFNβ demonstrated an average relative decrease in paw swelling similar to IFNβ treated WT mice, suggesting that the degree of arthritis inhibition from baseline by IFNβ treatment was similar and the treatment effectively decreased arthritis. The severity of arthritis by total area under the curve for Irf7−/− mice was decreased to the level of untreated WT by replacement IFNβ treatment (Figure 3A). The relative mean decrease was 47% and 36% for WT and Irf7−/− mice, respectively (p<0.05). Hence, the clinical effect of IFNβ treatment did not require IRF7 and was likely due to the direct anti-inflammatory effect of IFNβ treatment. Arthritis was significantly increased in Irf7−/− mice compared with WT mice treated with IFNβ (p<0.05).
Figure 3.

IFNβ treatment in Irf7−/− mice effectively decreased arthritis. A. Irf7−/− and WT mice were injected with serum on day 0 and 2. Mice were injected IP daily with IFNβ 1000 IU or PBS. Treatment of the Irf7−/− mice with replacement IFNβ decreased the amount of arthritis to WT levels. The inhibition of arthritis by IFNβ treatment in Irf7−/− mice was significant and IFNβ treatment also decreased arthritis in WT mice (*p<0.05). Arthritis was significantly increased in Irf7−/− mice despite IFNβ treatment (**p<0.05). B. Serum IL-1RA was increased in Irf7−/−mice treated with IFNβ compared with control treatment (*p<0.03). IL-1RA was significantly higher in Irf7−/−serum compared with WT after treatment with IFNβ (*p<0.03).
IFNβ treatment increases serum IL1-RA levels in IKKε mice (10), suggesting that IFNβ enhances the anti-inflammatory effect of IKKε deficiency through induction of IL-1RA. These results supported other studies demonstrating that the anti-inflammatory IFNβ mechanism of action requires IL-1RA (5, 21). To evaluate this potential mechanism in the Irf7−/− mice, IL-1RA ELISA was performed on serum from Irf7−/− K/BxN mice compared with control serum (Figure 3B). In contrast to control PBS, IFNβ treatment of Irf7−/− mice resulted in a significant increase in serum IL-1RA levels. Therefore, IFNβ treatment induced serum IL-1RA production in the Irf7−/− mice compared with control. This effect, as well as the direct anti-inflammatory effect of IFNβ in mouse arthritis models, might contribute to the decrease in arthritis with IFNβ treatment.
Poly (I-C) treatment decreases passive K/BxN arthritis in Irf7−/− mice
To determine the role of IRF7 in the poly (I-C) mediated inhibitory effect on serum transfer arthritis (9, 10), we performed the passive model in Irf7−/− and WT mice treated on days 0 and 2 with poly (I-C). Arthritis severity was significantly decreased in poly (I-C) treated Irf7−/− mice relative to PBS treated mice (Figure 4A). Because treatment with poly (I-C) decreased paw swelling in Irf7−/− mice compared with PBS control, other IRF or transcription factors likely play a partial role in poly (I-C) mediated inhibition of arthritis. Despite the poly (I-C) treatment, overall arthritis remained increased in the Irf7−/− mice compared with WT.
Figure 4.

Treatment with poly (I-C) decreased arthritis in the passive serum transfer model. A. Irf7−/− and WT mice were injected with serum on day 0 and 2. Treatment was performed with IP injection of poly (I-C) or PBS on day 0 and 2. Both WT and Irf7−/− poly (I-C) treatment groups had less paw swelling (*p<0.05). Treatment with poly (I-C) decreased the amount of arthritis in Irf7−/− to WT levels. Irf7−/− mice had increased arthritis compared with WT despite treatment with poly (I-C) (**p<0.05). B. Poly (I-C) mediated induction of gene expression in the synovial tissue of Irf7−/− mice demonstrated that IFNβ gene expression was restored to WT levels. C. Poly (I-C) treatment in Irf7−/− mice increases IFNβ gene expression to WT levels (*p<0.001). D. IFNβ protein by ELISA was increased in the sera of arthritic WT and Irf7−/− mice treated with poly (I-C) (*p<0.05). Baseline IFNβ in serum is decreased in Irf7−/− mice (**p<0.05).
Next, we evaluated changes in synovial tissue gene expression with poly (I-C) treatment. Figure 4B shows gene expression in ankle synovial tissue from poly (I-C) treated Irf7−/− K/BxN compared to poly (I-C) treated WT mice. Comparison of the standard serum transfer model synovial PCR (Figure 1B) with the in vivo poly (I-C) treated arthritis model PCR (Figure 4B) we noted a significant change in IFNβ gene expression. In vivo poly (I-C) treatment of Irf7−/− mice restored IFNβ gene expression (decreased WT IFNβ at baseline shown in Figure 1B), in synovial tissue to WT levels (Figure 4B), suggesting that the effect of poly (I-C) might occur via induction of IFNβ by other IRF family members. Poly (I-C) treatment did not induce a significant difference in gene expression of the anti-inflammatory cytokines IL-1RA and TGFβ in K/BxN Irf7−/− synovium. Induction of IL-6 gene expression was significantly less in poly (I-C) treated Irf7−/− synovial tissue compared with poly (I-C) treated WT; however, IL-6 is not an absolute requirement for arthritis in this serum transfer model (22). Induction of RANTES was increased in the Irf7−/− synovial tissue after in vivo poly (I-C) treatment, opposite of the clinical effect. Figure 4C summarizes and compares the different effects of poly (I-C) treatment on IFNβ synovial gene expression in Irf7−/− mice compared with WT. Poly (I-C) treatment of Irf7−/− mice resulted in 3.6-fold induction of IFNβ gene expression compared with control PBS (p<0.001), but had no significant effect on WT IFNβ gene expression in the synovium. Of note, the baseline gene expression of IFNβ is decreased in the Irf7−/− mice and poly (I-C) treatment restores mRNA to WT levels. IFNβ protein expression was also increased in the serum of Irf7−/− K/BxN mice treated with in vivo poly (I-C) compared with PBS control (Figure 4D). Serum IFNβ was significantly increased by poly (I-C) in Irf7−/− mice suggesting that serum IFNβ levels correlate with decreased arthritis in this model. Baseline serum IFNβ protein concentration was decreased in Irf7−/− mice compared with WT.
Increased induction of poly (I-C) stimulated pro-inflammatory gene expression in Irf7−/− FLS
To dissect cell specific roles of IRF7 in arthritis, we performed in vitro poly (I-C) stimulation of murine FLS lines and peritoneal macrophages derived from non-arthritic Irf7−/− mice. No significant difference in IFNβ gene induction was detected in Irf7−/− FLS. Poly (I-C) stimulation of Irf7−/− FLS resulted in increased induction of gene expression of RANTES (5.2-fold, p<0.01), MIP1α (1.8-fold, p<0.05), and MMP-3 (2.7-fold, p<0.05) compared with WT FLS (Figure 5A). Induction of IP-10 gene expression was decreased in the Irf7−/− FLS in response to poly (I-C) stimulation. Taken together, these results suggest that IRF7 might play a counter regulatory or anti-inflammatory role in poly (I-C) induced responses in murine FLS independent of IFNβ.
Figure 5.
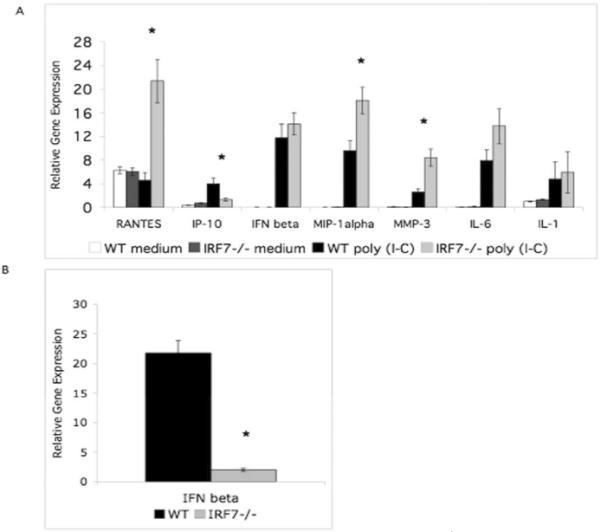
Poly (I-C) induced gene expression of murine Irf7−/− FLS and peritoneal macrophages. Mouse Irf7−/− FLS and peritoneal macrophages were stimulated with poly (I-C). QPCR analysis of cytokine gene expression was determined for RANTES, IP-10, IFNβ, MIP-1α, MMP-3, and IL-6 normalized to HPRT1. A. Poly (I-C) stimulation of Irf7−/− FLS resulted in increased induction of several pro-inflammatory cytokines compared with control (*p<0.05). B. Poly (I-C) stimulation of Irf7−/− peritoneal macrophages resulted in significantly less induction of IFNβ gene expression compared with control (*p<0.05).
Decreased induction of IFNβ gene expression in Irf7−/− macrophages
Because IRF7 did not contribute to IFNβ production in FLS, we evaluated the role of IRF7 in poly (I-C) induced IFNβ production by macrophages. To evaluate the effect of genetic deficiency of IRF7 in macrophages, we isolated cells from the peritoneal cavity and stimulated in vitro with poly (I-C) (Figure 5B). In contrast to FLS, Irf7−/− macrophages stimulated with poly (I-C) showed significantly less induction of cytokine gene expression of IFNβ compared with WT macrophages (8.8-fold decrease, p<0.01). These results are different from Irf7−/− FLS indicating a cell specific regulatory role and suggesting that IRF7 activates poly (I-C)-induced IFNβ gene expression in the hematopoietic macrophage subset. IRF7 regulates IFNβ gene expression in murine macrophages, but not in stromal FLS.
Discussion
RA is a destructive, inflammatory arthritis that involves activation of innate immunity. Although rheumatoid arthritis cannot be directly studied in mice, the passive serum transfer model is useful because of its simplicity and primary dependence on activation of innate immunity (23). This passive model of arthritis can be studied in genetically deficient mice to assess pathogenic mechanisms of innate immunity in inflammatory arthritis (24). In addition, TLR signaling through MyD88 has been implicated in the pathogenesis (25). The model requires IL-1, has modest dependence on TNFα, and pertinent to the IRF7 arthritis to be discussed, does not require IL-6 (22, 26). This model provides insights that are useful to understanding innate immune activation and effector mechanisms of IRF7 and type I IFN regulation in inflammatory arthritis.
We hypothesized that IRF7 differentially regulates synovial inflammation in a cell specific manner that can alter innate immunity and the clinical course of arthritis in vivo. To screen for a functional effect of IRF7 deficiency in arthritis, we evaluated the serum transfer model of antibody-mediated arthritis in mice. Genetic IRF7 deficiency exacerbated arthritis suggesting that IRF7 plays an overall counter-regulatory role in the later stages of serum transfer K/BxN arthritis. However, in light of the lack of a significant difference in histologic score evaluating bone erosion, cartilage erosion, and synovial inflammation, the change in paw volume might be related to vascular permeability and a difference in extra-articular swelling. The lack of effect on synovial histology despite increased paw volume suggested that IRF7 might also play a role in vascular permeability and subsequent tissue edema contributing to paw measurement differences as noted in other arthritis models (19, 20). Of note, several viruses alter vascular permeability in endothelial cells through cytokine expression as a mechanism of infection during human disease. IRF7 could contribute to regulation of cytokine expression and increased vascular permeability in this arthritis model.
Because IRF7 regulates type I IFN responses and IFNβ expression was decreased in the serum and synovium of Irf7−/− mice, we also hypothesized that the increase in arthritis in the Irf7−/− mice could be partially due to an insufficiency of IFNβ. To explore the role of IRF7 in the response to IFNβ, Irf7−/− mice were injected with replacement IFNβ concurrently with serum transfer. IFNβ treatment of Irf7−/− mice resulted in a decrease in arthritis to similar to WT PBS treated mice. Although the Irf7−/− mice were not able to mount an endogenous IFNβ response during the course of arthritis, they were able to respond adequately to the exogenous IFNβ treatment. Exogenous IFNβ treatment also enhanced serum IL-1RA production that could have also contributed to the beneficial effects of this treatment in the Irf7−/− mice, which is consistent with other reports (5, 21). More specifically, the mechanism of the beneficial effects of IFNβ in passive K/B×N arthritis are dependent on IL-1RA in this mouse model of arthritis (27).
Although IRF7 decreases serum transfer arthritis, the upstream kinase IKKε that activates IRF7 actually increases arthritis in this model. We proposed that IRF7 decreases arthritis through production of IFNβ, an anti-inflammatory cytokine. The initial type I IFN response to innate receptor activation results in IKKε/TBK1 phosphorylation of IRF3 and IRF7. This signaling induces type I IFN that bind the IFNα/β receptor and further induce IRF7 transcription and also secondary activation of IKKε (28). Therefore, an amplification loop can proceed through the IKK-related kinases, especially IKKε (29). Certain type I IFN-stimulated genes are not activated in the absence of IKKε because the IFN-stimulated gene factor 3 (ISGF3) complex does not bind to promoter of those particular genes. This pathway involves IFNβ mediated activation of IKKε, followed by IKKε phosphorylation of signal transducer and activator of transcription 1, a component of ISGF3. This might contribute to the opposite clinical effect of IRF7 and IKKε in the serum transfer arthritic mice.
TLR activation has been shown to contribute to inflammation in autoimmune diseases; however, repeated exposure to a TLR agonist can induce a hypo-responsiveness or desensitization to further TLR stimulation (30). The TLR3 agonist poly (I-C) has been previously studied in the K/B×N passive antibody-mediated arthritis model in WT and in IKKε null mice and resulted in decreased arthritis if co-administered at the onset of disease with serum (10). Serum levels of IL-1RA were increased in IKKε null IFNβ treated mice, which was proposed to contribute to the clinical decrease in arthritis. IL-1RA mediates the beneficial effect of systemic IFNβ therapy in the serum transfer model (27). Because poly (I-C) induces IRF7 in vitro, we used Irf7−/− mice to determine if the poly (I-C)-mediated decrease in serum transfer arthritis is dependent on IRF7 mediated induction of an anti-inflammatory type I IFN response. Surprisingly, poly (I-C) treatment also decreased arthritis in Irf7−/− arthritic mice to the same degree as the WT mice. The partial effect might have occurred through inducing and restoring synovial IFNβ gene expression to WT levels. However, the IFN effect might be correlative rather than causal to changes in arthritis. The functional role of synovial IFNβ is not definitive because the WT mice treated with poly (I-C) have reduced arthritis without an increase in synovial IFNβ. More importantly and consistent with the clinical response, serum IFNβ protein levels in Irf7−/− mice were also increased by poly (I-C) treatment. This indicated that other IRF or transcription factors might have contributed to poly (I-C)-mediated inhibition of arthritis in the Irf7−/− mice and that circulating IFNβ likely plays a more important role compared with synovial gene expression.
The Irf7−/− mice exhibited an accentuated clinical course in the passive serum transfer model, yet were able to respond to both the poly (I-C) and IFNβ treatments in vivo. Whole joint preparations demonstrated a reduction in the overall level of IFNβ mRNA transcription, which might reflect the cellular composition of FLS and macrophage-like synoviocytes in the swollen joints. We have studied FLS in RA and mouse models of arthritis and propose that they are important players in inflammatory arthritis. In RA, FLS display a transformed phenotype and also express cadherin-11, an adhesion molecule essential for the development of the synovium and thickening of the lining (31). Although many cell types can contribute to the vicious cycle that initiates and perpetuates inflammatory arthritis, we included FLS and macrophage data to accompany the in vivo studies. Similar to human FLS knockdown studies indicating a minimal role for IRF7 in FLS IFN production (13), no significant difference in IFNβ gene expression was detected in poly (I-C) stimulated Irf7−/− FLS. In contrast, IRF7 regulated the induction of several IFN-stimulated genes including RANTES, IP-10, MIP1α, as well as MMP3 in mouse FLS in response to poly (I-C). These results suggest that IRF7 might play a counter regulatory or anti-inflammatory role in poly (I-C)-induced responses in murine FLS independent of IFNβ.
Because the Irf7−/− FLS were able to generate IFNβ transcripts in response to in vitro poly (I-C) stimulation (Figure 5A), the lack of IFNβ expression could not be attributed to an inherent deficit produced by FLS. Hence, to further examine the reduction in IFNβ seen in the arthritic Irf7−/− synovium, we evaluated gene expression in macrophages stimulated with poly (I-C). In contrast to FLS, IRF7 deficient macrophages showed significantly less induction of IFNβ gene expression in response to poly (I-C). These results suggested a cell specific, key regulatory role for IRF7 in IFNβ production by the macrophages, but not in stromal FLS. It is uncertain whether the in vitro profile of Irf7−/− peritoneal macrophages stimulated with poly (I-C) plays any role in synovial IFNβ gene expression in the serum transfer model. The overall pro-inflammatory profile of FLS combined with the loss of macrophage anti-inflammatory IFNβ might act together to increase arthritis in the Irf7−/− K/B×N mouse model. In vitro studies in human monocytic cell lines confirm these cell specific roles for IRF in IFN-regulated gene expression (in press).
IFNβ plays a complex role and suppresses inflammation, especially in mouse models of arthritis. Mice with collagen-induced arthritis injected with fibroblasts expressing IFNβ have decreased arthritis severity and bone and cartilage destruction (5). An alternative approach in the passive serum transfer model combined IKKε deficiency with subtherapeutic IFNβ, which amplified the anti-inflammatory effects of this pathway (10). IRF7 plays a counter regulatory role in the K/B×N serum transfer model of arthritis likely through cell specific effects on IFN production and cytokine gene expression. Thus, careful dissection of the signaling pathways that regulate the type I IFN response and identification of the key regulatory transcription factors, kinases, and IFN-response genes could shed light on news ways to modulate anti-inflammatory effects of this pathway without markedly suppressing host defense in RA patients.
Acknowledgements
The authors would like to thank Dr. Gary S. Firestein and David L. Boyle for advice and assistance with data interpretation. We also thank Meghan Edgar and Lisa Ronacher for technical assistance.
Grant support: National Institute of Health K08 Career Development Award AR052800 and K08 ARRA AR052800-04S1 supplement; Arthritis Foundation.
Abbreviations
- FLS
fibroblast-like synoviocyte
- HPRT
hypoxanthine-guanine phosphoribosyltransferase
- IKK
IκB kinase
- IL-1RA
IL-1 receptor antagonist
- IP-10
IFN inducible protein 10
- IP
intraperitoneal
- IRF
IFN regulatory factor
- MMP
matrix metalloproteinase
- poly I-C
polyinosinic polycytidylic acid
- RA
rheumatoid arthritis
- RE
relative expression
- WT
wild type
Reference
- 1.Sweeney S, Firestein G. Primer: signal transduction in rheumatic disease--a clinician's guide. Nat Clin Pract Rheumatol. 2007;3(11):651–60. doi: 10.1038/ncprheum0631. [DOI] [PubMed] [Google Scholar]
- 2.Brentano F, Kyburz D, Schorr O, Gay R, Gay S. The role of Toll-like receptor signalling in the pathogenesis of arthritis. Cell Immunol. 2005;233(2):90–6. doi: 10.1016/j.cellimm.2005.04.018. [DOI] [PubMed] [Google Scholar]
- 3.Malmgaard L. Induction and regulation of IFNs during viral infections. J Interferon Cytokine Res. 2004;24:439–54. doi: 10.1089/1079990041689665. [DOI] [PubMed] [Google Scholar]
- 4.Moynagh P. TLR signalling and activation of IRFs: revisiting old friends from the NF-kappaB pathway. Trends Immunol. 2005;26(9):469–76. doi: 10.1016/j.it.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 5.van Holten J, Reedquist K, Sattonet-Roche P, Smeets TJ, Plater-Zyberk C, Vervoordeldonk MJ, et al. Treatment with recombinant interferon-beta reduces inflammation and slows cartilage destruction in the collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res Ther. 2004;6(3):R239–49. doi: 10.1186/ar1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smeets TJ, Dayer JM, Kraan MC, Versendaal J, Chicheportiche R, Breedveld FC, et al. The effects of interferon-beta treatment of synovial inflammation and expression of metalloproteinases in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43(2):270–4. doi: 10.1002/1529-0131(200002)43:2<270::AID-ANR5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 7.van Holten J, Pavelka K, Vencovsky J, Stahl H, Rozman B, Genovese M, et al. A multicentre, randomised, double blind, placebo controlled phase II study of subcutaneous interferon beta-1a in the treatment of patients with active rheumatoid arthritis. Ann Rheum Dis. 2005;64(1):64–9. doi: 10.1136/ard.2003.020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayashi T, Gray C, Chan M, Tawatao R, Ronacher L, McGargill M, et al. Prevention of autoimmune disease by induction of tolerance to Toll-like receptor 7. Proc Natl Acad Sci. 2009;106(8):2764–9. doi: 10.1073/pnas.0813037106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yarilina A, DiCarlo E, Ivashkiv L. Suppression of the effector phase of inflammatory arthritis by double-stranded RNA is mediated by type I IFNs. J Immunol. 2007;178(4):2204–11. doi: 10.4049/jimmunol.178.4.2204. [DOI] [PubMed] [Google Scholar]
- 10.Corr M, Boyle D, Ronacher L, Flores N, Firestein G. Synergistic benefit in inflammatory arthritis by targeting I kappaB kinase epsilon and interferon beta. Ann Rheum Dis. 2009;68(2):257–63. doi: 10.1136/ard.2008.095356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. Triggering the interferon antiviral response through an IKK-related pathway. Science. 2003;300(5622):1148–51. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 12.Sweeney S, Mo L, Firestein G. Antiviral gene expression in rheumatoid arthritis: role of IKK epsilon and interferon regulatory factor 3. Arthritis Rheum. 2007;56(3):743–52. doi: 10.1002/art.22421. [DOI] [PubMed] [Google Scholar]
- 13.Sweeney S, Kimbler T, Firestein G. Synoviocyte Innate Immune Responses: II. Pivotal Role of IFN Regulatory Factor 3. J Immunol. 2010;15(184):7162–8. doi: 10.4049/jimmunol.0903944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434(7034):772–7. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 15.Kouskoff V, Korganow A, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87(5):811–22. doi: 10.1016/s0092-8674(00)81989-3. 29;87(5):811–22. [DOI] [PubMed] [Google Scholar]
- 16.Korganow A, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, et al. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10(4):451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 17.Sweeney SE, Hammaker D, Boyle DL, Firestein GS. Regulation of c-Jun phosphorylation by the I kappa B kinase-epsilon complex in fibroblast-like synoviocytes. J Immunol. 2005;174(10):6424–30. doi: 10.4049/jimmunol.174.10.6424. [DOI] [PubMed] [Google Scholar]
- 18.Kim TK, Kim TH, Maniatis T. Efficient recruitment of TFIIB and CBP-RNA polymerase II holoenzyme by an interferon- enhanceosome in vitro. Proc Natl Acad Sci U S A. 1998;95(21):12191–6. doi: 10.1073/pnas.95.21.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abreu J, Dontje W, Krausz S, de Launay D, van Hennik P, van Stalborch A, et al. A Rac1 inhibitory peptide suppresses antibody production and paw swelling in the murine collagen-induced arthritis model of rheumatoid arthritis. Arthritis Res Ther. 2010;12(1):R2. doi: 10.1186/ar2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Firestein GS. `Rac'-ing upstream to treat rheumatoid arthritis. Arthritis Res Ther. 2010;12(1):109. doi: 10.1186/ar2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palmer G, Mezin F, Juge-Aubry C, Plater-Zyberk C, Gabay C, Guerne P. Interferon beta stimulates interleukin 1 receptor antagonist production in human articular chondrocytes and synovial fibroblasts. Ann Rheum Dis. 2004;63(1):43–9. doi: 10.1136/ard.2002.005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji H, Pettit A, Ohmura K, Ortiz-Lopez A, Duchatelle V, Degott C, et al. Critical roles for interleukin 1 and tumor necrosis factor alpha in antibody-induced arthritis. J Exp Med. 2002;196(1):77–85. doi: 10.1084/jem.20020439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kyburz D, Corr M. The KRN mouse model of inflammatory arthritis. Springer Semin Immunopathol. 2003;25(1):79–90. doi: 10.1007/s00281-003-0131-5. [DOI] [PubMed] [Google Scholar]
- 24.Kouskoff V, Korganow A, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87(5):811–22. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 25.Choe JY, Crain B, Wu SR, Corr M. Interleukin 1 receptor dependence of serum transferred arthritis can be circumvented by toll-like receptor 4 signaling. J Exp Med. 2003;197(4):537–42. doi: 10.1084/jem.20021850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kyburz D, Carson D, Corr M. The role of CD40 ligand and tumor necrosis factor alpha signaling in the transgenic K/BxN mouse model of rheumatoid arthritis. Arthritis Rheum. 2000;43(11):2571–7. doi: 10.1002/1529-0131(200011)43:11<2571::AID-ANR26>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 27.Corr M, Boyle D, Ronacher L, Lew B, van Baarsen L, Tak P, et al. Interleukin 1 receptor antagonist mediates the beneficial effects of systemic interferon beta in mice: implications for rheumatoid arthritis. Ann Rheum Dis. 2011;70(5):858–63. doi: 10.1136/ard.2010.141077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Borden E, Sen G, Uze G, Silverman R, Ransohoff R, Foster G, et al. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6(12):975–90. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tenoever B, Ng S, Chua M, McWhirter S, García-Sastre A, Maniatis T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315(5816):1274–8. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 30.Broad A, Jones D, Kirby J. Toll-like receptor (TLR) response tolerance: a key physiological “damage limitation” effect and an important potential opportunity for therapy. Curr Med Chem. 2006;13(21):2487–502. doi: 10.2174/092986706778201675. [DOI] [PubMed] [Google Scholar]
- 31.Lee D, Kiener H, Agarwal S, Noss E, Watts G, Chisaka O, et al. Cadherin-11 in synovial lining formation and pathology in arthritis. Science. 2007;315(5814):1006–10. doi: 10.1126/science.1137306. [DOI] [PubMed] [Google Scholar]