

Abstract
Kaposi's sarcoma-associated herpesvirus ORF30–33 locus encodes four genes with unknown functions. We performed transcriptional mapping of these genes. Northern-hybridization, 5′- and 3′-rapid amplification of cDNA ends, and DNA sequencing identified four transcripts of 3.7, 3.6, 2.7, and 1.4 kb, none of which has alternative splicing. While all transcripts have the same termination site, their start sites vary. All transcripts are not expressed or only weakly expressed in latent cells but can be chemically induced. The 3.7 and 3.6 kb transcripts contain all four genes and are sensitive to cycloheximide (CH) but resistant to phosphonoacetic acid (PAA), indicating that they are early lytic transcripts. The 2.7 kb transcript contains ORF32 and ORF33 genes while the 1.4 kb transcript contains the ORF33 gene. Both transcripts are sensitive to CH and PAA, indicating that they are late lytic transcripts. Furthermore, we identified four promoters with functional TATA boxes, none of which is directly transactivated by RTA. Examination of the 5′ untranslated region of ORF31 failed to identify any functional internal ribosome entry sites. These results define the transcriptional patterns of the ORF30–33 locus, which should help the delineation of its function.
Keywords: Kaposi's sarcoma-associated herpesvirus (KSHV), ORF30/31/32/33, Transcript mapping, Rapid amplification of cDNA ends (RACE), Polycistronic/bicistronic transcript, Promoter
Introduction
Kaposi's sarcoma-associated herpesvirus (KSHV) is etiologically associated with Kaposi's sarcoma (KS), primary effusion lymphoma (PEL) and a subset of multicentric Castleman's disease (MCD) [1]. KSHV is a gammaherpesvirus with a genome of ~ 170 kb, consisting of terminal repeats and a 140.5 kb long unique-coding region (LUR) that encodes more than 90 genes and 12 pre-microRNAs [1, 2]. The ORF30–33 gene locus is located between ORF29b and ORF29a in KSHV genome [3]. All genes in this locus are conserved among gammaherpesviruses [4]. Recent studies in murine gammaherpesvirus-68 (MHV-68) have revealed their roles in the late stages of viral replication [5–7]. Genetic knockout of ORF30 from the MHV-68 genome led to decreased expression of viral late genes and productive infection but did not affect viral DNA replication [7]. MHV-68 ORF31 is expressed in both cytoplasm and nucleus, and its expression level peaks at 24 h post-infection [6]. Both siRNA knock-down and genetic knockout of ORF31 blocked the expression of late lytic genes and reduced viral yield [6]. MHV-68 ORF33 was defined as a viral late gene [5]. Genetic knockout of ORF33 from the MHV-68 genome had no effect on viral DNA replication, early and late gene expression, viral DNA packaging or capsid assembly but significantly affected virion morphogenesis and egress, and reduced infectious viral yield [5]. While the function of KSHV ORF30–33 locus remains unknown, KSHV ORF33 was identified as a tegument protein and shown to interact with another capsid-associated tegument protein ORF64, suggesting a possible role in virion assembly [8, 9].
In order to better define the functions of the KSHV ORF30–33 locus, it is necessary to characterize the expression patterns of the encoded transcripts. A previous study showed that transcripts from the ORF30–33 locus were not detectable in uninduced KSHV-infected PEL cells but were induced following treatment with 12-O-tetradecanoylphorbol-13-acetate (TPA), suggesting that they are viral lytic genes [10]. However, a large DNA probe of 6 kb encompassing ORF29b to ORF35 was used in this study. The structure and expression kinetics of these transcripts also remain undefined. Interestingly, the coding frames of these genes are overlapped [3], pointing to the possible presence of polycistronic transcripts. In the present study, we have characterized the mRNA transcription patterns and expression kinetics of the ORF30–33 gene cluster. We have identified two polycistronic transcripts, one bicistronic transcript and one monocistronic transcript for this locus, and mapped their 5′- and 3′-ends. Four respective functional promoters were also identified for these four transcripts while no functional internal ribosome entry site (IRES) that regulates the translation of these genes was identified in the polycistronic transcripts.
Materials and methods
Cell culture
KSHV-negative Bjab cells and KSHV-positive BCBL-1 cells were cultured in RPMI-1640 medium supplemented with 15% fetal bovine serum and 100 μg/ml of gentamicin. To induce lytic replication, cells were treated with 20 ng/ml of TPA for different lengths of time. To inhibit the expression of early and late lytic genes, TPA induction was carried out for 12 h in the presence of CH at 100 μg/ml. To inhibit the expression of late lytic genes, TPA induction was carried out for 36 or 48 h in the presence of 300 μg/ml PAA.
Northern-blot analysis
Total RNA was extracted from BCBL-1 cells or Bjab cells using Trizol reagent (Invitrogen, Carlsbad, CA). After DNase I digestion, 10 μg of RNA from each sample were separated in 1% formaldehyde-agarose gels and transferred to a Hybond-N membrane as described before [11]. Membranes were UV cross-linked before hybridization. Templates with minimal T7 RNA polymerase promoter sequence for riboprobe synthesis were prepared from cDNA using a PCR-based method. Strand-specific 32P-labeled riboprobes were prepared with the MAXIcript T7 kit (Ambion, Applied Biosystems, Austin, TX). Hybridization was carried out as previously described with some modifications [11]. In brief, membranes were pre-hybridized at 65°C for 2 h in Church's buffer containing 0.5 M NaPO4 at pH 7.2, 1% BSA, 1 mM EDTA, 7% SDS, 100 μg/ml yeast tRNA, and 100 μg/ml salmon sperm DNA followed by hybridization with 32P-labelled riboprobes at 65°C overnight. Membranes were washed twice with 2X SSC containing 0.1% SDS and once with 0.1X SSC containing 0.1% SDS at room temperature, each for 20 min. Following exposure to a screen, the signals were detected with a Typhoon Image Scanner (GE Healthcare, Piscataway, NJ).
5′- and 3′-rapid amplification of cDNA ends (RACE)
RACE was performed to determine the 5′- and 3′-ends of the transcripts. The 5′-RACE was carried out with a First Choice RLM RACE kit according to the instructions of the vendor (Ambion). In brief, 10 μg of total RNA was treated with DNase I and then with calf intestine alkaline phosphatase and tobacco acid pyrophosphatase. A RNA adapter was added to the 5′-end of mRNA. After reverse transcription, nested-PCR was carried out. For 3′-RACE, a modified oligo (dT)18NN with a multiple restriction sites in the 5′-end was used for reverse transcription. A specific primer anchoring to the downstream sequence of each viral ORFs and a universal primer matching to the multiple restriction sites of oligo(dT) were used for the amplification of the 3′ untranslated region (3′UTR). PCR products from the 5′- and 3′-RACE were run on agarose gels. Visible specific bands were isolated and cloned into the pcDNA3.1 vector (Invitrogen), which were then subjected to DNA sequencing.
Identification of functional promoters controlling the transcription of the ORF30–33 locus
Putative promoters and transcription factor binding motifs upstream of transcripts of the ORF30–33 locus were analyzed using MatInspector program [12]. Putative promoter regions were PCR-amplified from KSHV genomic DNA and inserted into the promoterless pGL4.16 vector (Promega, Madison, WI). Deletion of the putative TATA box was carried out using overlapping-PCR based method. In brief, a forward cloning primer and a reverse deletion primer were used to generate an upstream fragment while a forward deletion primer and a reverse cloning primer were used to generate a downstream fragment (Table 1). The PCR fragments were purified and used as templates to generate the full-length promoter DNA fragments with the TATA box sequence deletions. The resulting promoter reporter constructs with or without the TATA box were confirmed by DNA sequencing. Reporter assay was performed by transient transfection of 100 ng of DNA of a promoter reporter construct together with 50 ng of pSV-β-galactosidase vector DNA into 293T cells grown to 60–70% confluency in 24-well plates. Transfection was carried out with lipofectamine 2000 reagent (Invitrogen). The transfection rate reached ~90% based on the results of cotransfection with a green fluorescence protein (GFP) construct pEGFP-C1 (Clontech, Mountain View, CA). To examine the transactivation of a promoter by KSHV replication and transcription activator (RTA) encoded by ORF50, the reporter construct was cotransfected with a RTA expression vector at 100 ng. At 48 h post-transfection, cells were lysed and the supernatants were collected for luciferase assay and β-galactosidase assay as described by the manufacturer (Promega).
Table 1.
Primers for RACE, riboprobes, cloning and mutagenesis
| Primer | Sequence |
|---|---|
| Reverse transcription | 5′-CTGCAGACGCGTGCTAGCTCGAGATCTTTTTTTTTTTTTTTTTTNN-3′ |
| 5′-RACE | |
| 5′-RACE outer-F | 5′-GCTGATGGCGATGAATGAACACTG-3′ |
| ORF30 outer-R | 5′-GCAGGCAGAGTCTTTCTGTTTTGTGACA-3′ |
| ORF31 outer-R | 5′-ACAGGCAAAACACGCCGTGGAACTGACA-3′ |
| ORF32 outer-R | 5′-ACGGTTCCCGGAGCCTCGAGCGCCATGT-3′ |
| ORF33 outer-R | 5′-AATGTAGGGAAATCTCAGTCCGGATGCT-3′ |
| 5′-RACE inner-F | 5′-TTTGCGGCCGCGAACACTGCGTTTGCTGGCTTTGATG-3′ |
| ORF30 inner-R | 5′-TTTGATATCAGAGTCAGCGAGACCGAGGCCGTCGAGT-3′ |
| ORF31 inner-R | 5′-TCTGATATCCCACACTCTTCATTTCGCACCGGTGTCT-3′ |
| ORF32 inner-R | 5′-TTTGATATCTGTGGTGAGTGGCGCGGTCTCCGTCTGA-3′ |
| ORF33 inner-R | 5′-CCCGATATCTGTCCAAAGTAGATGAGCGCCATACCAT-3′ |
| 3′-RACE | |
| ORF30-F | 5′-TTTGAGCTCCTCGGTCTCGCTGACTCTCTATGTC-3′ |
| ORF31-F | 5′-TCTGGTACCTGCACGTTATCGTCTCCATCTATTC-3′ |
| ORF32-F | 5′-TCTGGTACCGTGTTACCTGGGGGTTTTGCTATTA-3′ |
| ORF33-F | 5′-TCTACCGGTGTCTTCTGTTCGATCCCATTGTCCA-3′ |
| 3′-RACE universal R | 5′-TAGACGCGTGCTAGCTCGAGATCTT-3′ |
| Riboprobe | |
| ORF30-F | 5′-ATGGGTGAGCCAGTGGATCCTGGA-3′ |
| ORF30-T7-R | 5′-TAATACGACTCACTATAGGGATCATTTCGCACCGGTGTCTAG-3′ |
| ORF31-F | 5′-ATGTCACAAAACAGAAAGACTCTG-3′ |
| ORF31-T7-R | 5′-TAATACGACTCACTATAGGGATCGTTGATAGCATGCGCATCCATT-3′ |
| ORF32-F | 5′-ATGGATGCGCATGCTATCAACGAA-3′ |
| ORF32-T7-R | 5′-TAATACGACTCACTATAGGGAAGCCATAGCGGCCTCGAATGAACA-3′ |
| ORF33-F | 5′-ATGGCTAGCCGGAGGCGCAAACTT-3′ |
| ORF33-T7-R | 5′-TAATACGACTCACTATAGGGATCAGACATTCGTAAGAGGACCCAG-3′ |
| GAPDH-F | 5′-ATGGGGAAGGTGAAGGTCGGAGTC-3′ |
| GAPDH-T7-R | 5′-TAATACGACTCACTATAGGGAGGTCTTACTCCTTGGAGGCCATGT-3′ |
| Primers for promoter analysis | |
| P30/31a-F | 5′-TCTGGTACCGGAGTTCAGAGCGTAGATGAATGTCT-3′ |
| P30/31a-R | 5′-TTTCTCGAGACGATCGGCACACACGCCCCCTTTTATTCCCAAGA-3′ |
| P30/31b-F | 5′-TTTGGTACCATGAAACCCAGAATAGCCGGCAGTGCAT-3′ |
| P30/31b-R | 5′-TTTCTCGAGACGATCGGCACACACGCCCCCTTTTATTCCCAAGA-3′ |
| P32-F | 5′-TTTCTCGAGAGCGGCTAACAGTAAAGGAGAGGAGGCGAA-3′ |
| P32-R | 5′-CTCAAGCTTTTTGTCCGCACCCGCTTGGCAATGCAGTCGT-3 |
| P33-F | 5′-TCTGGTACCCGCATTTCCTGCCCTGGTTCTAATCT-3′ |
| P33-R | 5′-CTCAAGCTTAGCGGCCTCGAATGAACACCAGATC-3′ |
| P30/31a-D-F | 5′-GTCTGACGAGTTCACGGATGATAAGCTTGGCGTCTTTCTGAAGCATG-3′ |
| P30/31a-D-R | 5′-CATGCAACAGAAAGACGCCAAGCTTATCATCCGTGAACTCGTCAGAC-3′ |
| P30/31b-D-F | 5′-ATTCGCCTCGTCTACGTAGAGCAGGAAGGTCTGTCCCCGAATGCTCT-3′ |
| P30/31b-D-R | 5′-AGAGCATTCGGGGACAGACCTTCCTGCTCTACGTAGACGAGGCGAAT-3′ |
| P32D-F | 5′-AACGGTGTACAACAGTATGCTATGCACAAAGAATAAAAAGTACGA-3′ |
| P32D-R | 3′-TCGTACTTTTTATTCTTTGTGCATAGCATACTGTTGTACACCGTT-3′ |
| P33D-F | 5′-TGTGTTACCTGGGGGTTTTGCTAAGGCCGCTATAGGGCGTCGAAG-3′ |
| P33D-R | 5′-CTTCGACGCCCTATAGCGGCCTTAGCAAAACCCCCAGGTAACACA-3′ |
| Primers for IRES assay | |
| Renilla-F | 5′-CTCGCTAGCATGACTTCGAAAGTTTATGATCCAGA-3′ |
| Renilla-R | 5′-CTCAAGCTTAGAATTATTGTTCATTTTTGAGAACT-3′ |
| Firefly-F | 5′-CGCGAATTCATGGAAGACGCCAAAAACATAAAG-3′ |
| Firefly-R | 5′-GACTCTAGAATTACACGGCGATCT-3′ |
| IRES-EMCV-F | 5′-TATGGATCCCCCTCTCCCTCCCCCCCCCCTAACGTTACTGGCCGAA-3′ |
| IRES-EMCV-R | 5′-TCTGAATTCGGTTGTGGCCATATTATCATCGTGT-3′ |
| IRES-50416-F | 5′-CTCAAGCTTATGAAACCCAGAATAGCCGGCAGT-3′ |
| IRES-50538-F | 5′-GCGAAGCTTATAAGCGGCTAACAGTAAAGGAGA-3′ |
| IRES-50762-R | 5′-TATGAATTCAGAGAGTCAGCGAGACCGAGGCCGTC-3′ |
| IRES-50852-R | 5′-TATGAATTCTTCGCACCGGTGTCTAGGCGGCACA |
Underlines denote restriction enzyme sites. Bold nucleotides denote the T7 minimal promoter F and R denote forward and reverse primers
Analysis of IRES
The basic pcDNA-DL vector was constructed by inserting DNA fragments of the Renilla luciferase gene and firefly luciferase gene into NheI-HindIII and EcoRI-XbaI sites of pcDNA3.1(+) vector (Invitrogen), respectively. A fragment of the encephalomyocarditis virus (EMCV) IRES and different fragments of putative IRES sequences upstream of ORF31 start codon was cloned into BamHI-EcoRI and HindIII-EcoRI sites of the pcDNA-DL vector, respectively, which is between the coding frames of Renilla and firefly luciferase genes. The resulting plasmid was subjected to DNA sequencing. The activity of IRES reporter was determined in 293T cells and BCBL-1 cells. Transfection of 293T cells was performed as described above. Transfection of BCBL-1 cells was carried out by electroporation using a BTX Electroporation System Electro Cell Manipulator 630 (Harvard Apparatus, Holliston, MA). BCBL-1 cells at 1 × 106 were re-suspended in 400 μl of electroporation buffer with 2 μg reporter plasmid DNA and transferred to a 0.4 cm cuvette. Cells were electroporated at 975 μF and 220 V. The transfection rate reached ~30% based on the results of cotransfection with pEGFP-C1. Transfected cells were collected and lysed at 24 h post-transfection. A Dual-Luciferase Reporter Assay System was used to assess the IRES activity following manufacturer's instruction (Promega).
Results
The ORF30-33 locus encodes multiple transcripts expressed at the late stage of viral replication
To examine the expression patterns of the ORF30-33 locus, we performed Northern-blotting with total RNA samples from BCBL-1 cells treated with TPA for various lengths of time. We selected strand-specific riboprobes based on the genomic positions of these genes (Fig. 1a). All probes detected a band around 3.7 kb, which was occasionally resolved as two bands of 3.6 and 3.7 kb (Fig. 1b). The ORF32 probe detected a 2.7 kb band while the ORF33 detected the 2.7 kb band and a 1.4 kb band in addition to the 3.7 kb band. In uninduced latent cells, the 2.7 kb transcript was not detectable while the 3.7 and 1.4 kb transcripts were weakly expressed. The expression of all the transcripts was increased following TPA induction. Such increased expression was observed at 12 h post-induction (hpi) for the 3.7 and 1.4 kb transcripts and at 24 hpi for the 2.7 kb transcript (Fig. 1b). The highest expression levels of all four transcripts were detected at 72 hpi, the latest time point examined. These results indicate that the transcripts from the ORF30-33 locus are viral lytic transcripts.
Fig. 1.

Identification of transcripts from the ORF30-33 locus. a Genomic organization of the ORF30-33 locus, probe positions, and transcripts identified by Northern hybridization. b Northern-blot detection of transcripts from the ORF30-33 locus in uninduced and TPA-induced BCBL-1 cells. Top panels show four Northern blots of total RNA from BCBL-1 cells with different riboprobes. RNA from KSHV-negative Bjab cells was used to confirm the specificity of riboprobes. Bottom panels show ethidium bromide staining of 18S rRNA to insure the even loading of RNA in different samples
Mapping of the 5′- and 3′-ends of the transcripts encoded by the ORF30-33 locus
To identify the transcription start sites of the ORF30-33 locus transcripts, we performed 5′-RACE analysis. PCR reactions were carried out using a primer complementary to the 5′-end of the cDNA adaptor sequence, and different primers within the coding frames of ORF30-33 (Fig. 2a). The PCR products were resolved on agarose gels, and all the visible specific bands were excised and sequenced (Fig. 2b). All sequences were aligned with known KSHV genomic sequence (GeneBank No.U75698) and the start sites of the transcripts were identified. Consistent with the results of Northern-blot analysis, we identified transcriptional start sites at KSHV genomic positions of 50,416, 50,538, 51,378, and 52,737, which correspond to the 3.7, 3.6, 2.7, and 1.4 kb transcripts, respectively. These results indicate that the weak 3.6 kb band that overlapped with the 3.7 kb band described in Fig. 1 is likely to represent a true transcript albeit it is difficult to separate it from the 3.7 kb transcript.
Fig. 2.
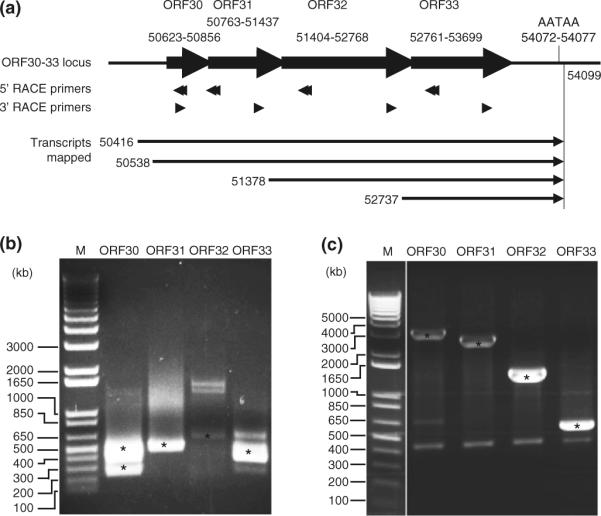
Mapping of the 5′- and 3′-ends of transcripts from the ORF30-33 locus. a Primers used for RACE, and transcripts and their 5′- and 3′-positions identified by RACE analyses. KSHV genomic positions were based on GeneBank No.U75698. b Agarose gel analysis of 5′-RACE products. c Agarose gel analysis of 3′-RACE products. Specific KSHV bands were identified by DNA sequencing and marked with star symbols
We further mapped the 3′-end of these transcripts. As shown in Fig. 2a, all the transcripts end at genomic position 54,099. A transcriptional termination site AATAA was identified at position 54,072–54,077. Thus, the ORF30-33 locus encodes four transcripts of 3,684, 3,562, 2,722, and 1,363 bp. To further confirm the presence of these transcripts, we performed PCR amplification using a primer complementary to the 5′-end of the cDNA adaptor sequence and a primer from the mapped 3′-end of the transcripts. Individual bands were isolated following resolution in gels, and subjected to DNA sequencing. As expected, sequences of the all the products matched with predicted KSHV sequences and positions (data not shown). No splicing event was identified for any of these transcripts.
Classification of the transcripts encoded by the ORF30-33 locus
The expression kinetics of the transcripts encoded by the ORF30-33 locus indicates that they are lytic transcripts (Fig. 1b). To further determine the specific class(s) of the ORF30-33 locus transcripts, we induced BCBL-1 cells with TPA in the presence of CH or PAA. CH blocks de novo protein synthesis, resulting in expression inhibition of viral early and late lytic genes but not immediately-early lytic genes. PAA blocks herpesviral DNA polymerase, resulting in expression inhibition of viral late lytic genes but not immediately-early and early lytic genes. As shown in Fig. 3, the 3.7/3.6 kb band was abolished by CH but not by PAA, indicating they are early lytic transcripts. The 2.7 and 1.4 kb bands were sensitive to both CH and PAA, indicating they are late lytic transcripts. As controls, we examined the expression of a 1.7 kb KSHV early lytic transcript encoding ORF-K9 and a 0.9 kb KSHV late lytic transcript encoding ORF65 [11, 13]. As expected, the ORF-K9 transcript was sensitive to CH but resistant to PAA while the ORF65 transcript was sensitive to both CH and PAA. Interestingly, the ORF65 probe also detected three other larger transcripts, which were likely transcribed from the ORF65-68 locus (unpublished observations). Recent genome-wide analysis of KSHV transcriptome has revealed the presence of both sense and antisense long noncoding transcripts in the ORF65 locus [14]. However, these noncoding transcripts are likely different from those detected in this study.
Fig. 3.

Determination of the classes of transcripts from the ORF30-33 locus. Northern-blot analysis with total RNA from uninduced BCBL-1 cells and BCBL-1 cells induced with TPA with and without the presence of CH or PAA
Characterization of promoters in the ORF30-33 locus
To determine the promoters controlling the expression of transcripts from the ORF30-33 locus, sequences upstream and downstream of the four mapped transcription start sites were analyzed. Putative TATA boxes were identified for all four transcriptional start sites, designated as T1–4 (Fig. 4), suggesting that each transcript might be controlled by its own promoter. Potential binding sites for several known transcription factors were identified in these putative promoters. Of interests are binding sites for AP-1, CREB, RBP-J, OCT-1, SP-1 and NF-κB, which are known to regulate the expression of KSHV genes. To test the activities of these putative promoters, we generated four promoter reporter constructs designated P1–4, which contain 818, 123, 838, and 1,326 bp upstream sequences, respectively, and 206, 82, 25, and 22 bp downstream sequences, respectively, of the four transcriptional start sites. The upstream sequence for P2 (transcriptional start site 50,538) is relatively short starting immediately after the first upstream TATA box (transcriptional start site 50,416). P1 contains the upstream sequence for transcriptional start sites 50,416 and 50,538, thus its activity should reflect both promoters. As shown in Fig. 5b, strong basal activities were observed for P1, P3, and P4 with luciferase intensities 14.7-, 11.1-, and 46.5-fold higher than the promoterless control vector, respectively. In contrast, weak promoter activity was detected for P2 with luciferase intensity 3.7-fold higher than the promoterless control vector. While the result suggests that additional upstream sequence is required for the optimal activity of this promoter, they are also consistent with the relatively weak expression of the 3.6 kb transcript compared to that of the 3.7 bp transcript (Figs. 1, 3).
Fig. 4.

Analysis of putative promoters in the ORF30-33 locus. 5′ flanking sequences upstream the start codons of all genes were scanned by MatInspector (Genomatix). Transcription start sites, putative transcription factors binding motifs, and translation start codons are underlined. Arrows indicate transcription orientation. a Promoters P1 and P2 in the upstream sequences of ORF30 and ORF31. b Promoter P3 in the upstream sequence of ORF32. c Promoter P4 in the upstream sequence of ORF33. The four transcriptional start sites were labeled as TSS while the four putative TATA boxes were labeled as T1–4
Fig. 5.

Determination of promoter activity by reporter assay. a Schematic representation of different promoter reporter constructs used for promoter mapping of the ORF30-33 locus in KSHV genome. Positions for the cloned promoters are also shown and labeled as P1–4, respectively. b Basal activity of promoters from the ORF30-33 locus. The relative activity of vector control pGL4 is shown as “1”. c Deletion of TATA boxes from the promoters in the ORF30-33 locus reduced promoter activities. d RTA failed to activate the promoter reporters from the ORF30-33 locus. vIL6 promoter, which is highly responsive to RTA, was used as a positive control in the reporter assay. Results were presented as mean and error bars from three independent experiments, each with triplicates
To determine whether the putative TATA boxes are functional, we deleted the TATA box sequences from the wild-type promoter reporter constructs P3 and P4 to generate their respective deletion constructs P3D and P4D. Deletion of TATA boxes from P3 and P4 reduced the reporter activities by 78.9% and 76.3%, respectively (Fig. 5c), indicating that the transcription of the 2.7 and 1.4 kb transcripts is TATA box-dependent. Because of the presence of two TATA boxes in the P1 reporter and the weak activity of P2 reporter, we used P1 as the template to examine the functions of these two TATA boxes. We deleted the first and second TATA boxes to generate constructs P1D-1 and P1D-2, respectively. Deletion of first TATA reduced the reporter activity by 61.2% while deletion of the second TATA box reduced the reporter activity by 68.4% (Fig. 5c). Thus, all predicated TATA boxes are functional and contribute to the transcription of the ORF30-33 locus.
RTA transactivates the expression of several KSHV lytic genes. Our results showed that all the transcripts from the ORF30-33 locus were lytic and were induced by TPA. We examined whether any of the promoters in this locus could be activated by RTA. The activities of the ORF30-33 locus promoter constructs P1–4 were activated by RTA by 2.7-, 2.5-, 4.2-, and 3.2-fold, respectively. In contrast, the reporter activity of the RTA-responsive vIL6 (ORF-K2) promoter was activated by 55.7-fold. These results indicated that none of the promoters in the ORF30-33 locus was directly responsive to RTA. Thus, their activities are likely regulated by other viral/cellular factors during KSHV lytic replication.
No IRES is found in the 5′UTR of ORF31
While both the 3.6 and 3.7 kb transcripts contain ORF30 and ORF31, no independent transcription start site was identified for ORF31, suggesting a 5′ cap-independent translation mechanism for this gene. A functional IRES controlling the translation of KSHV ORF71 has previously been identified in the ORF72 coding frame [15]. We used Bicistronic Luciferase Reporter System to explore the existence of an IRES upstream of ORF31. In this system, translation of upstream Renilla luciferase occurs via 5′ cap-dependent mechanism, whereas translation of downstream firefly luciferase relies on the IRES sequence between two luciferase genes [16]. There are two putative translation start codons for ORF31 at 50,762 and 50,852, respectively, (GeneBank No.U75698 and No.AF148805) [3, 17]. Bicistronic luciferase reporters containing the putative IRES sequences inserted between the two reporter genes were constructed (Fig. 6a). pcDNA-DL1 and pCDNA-DL2 contain the 5′UTR sequences of both 3.7 and 3.6 kb transcripts before the first putative ORF31 translation start codon while pcDNA-DL3 and pCDNA-DL4 contain the 5′UTR sequences before the second putative translation start codon. A construct pcDNA-DL-EMCV containing the well-defined EMCV IRES was included in the assay. In 293T cells, the firefly luciferase activity of pcDNA-DLEMCV increased 10.1-fold over that of the pcDNA-DL vector as expected (Fig. 6b). However, the firefly luciferase activities of pcDNA-DL1, pcDNA-DL2, pcDNA-DL3, and pcDNA-DL4 only increased 1.45-, 1.39-, 1.60-, and 1.36-fold, respectively. Similar results were obtained in BCBL-1 cells (Fig. 6c).
Fig. 6.

No IRES is found in the 5′UTR of ORF31. a Schematic illustration of the basic bicistronic Renilla/firefly luciferase reporter vector (pcDNA-DL), and reporter constructs with DNA fragments from the 5′UTR of ORF31 (pcDNA-DL1, pcDNA-DL2, pcDNA-DL3 and pcDNA-DL4), and the well-defined IRES from EMCV (pcDNA-DL-EMCV). For constructs pcDNA-DL1 and pcDNA-DL3, the firefly luciferase gene has a frame shift if the translation is initiated from the ORF30 start codon. b Firefly luciferase reporter activities of the bicistronic constructs, which represent IRES activities, examined in 293T cells. c Firefly luciferase reporter activities of the bicistronic constructs, which represent IRES activities, examined in BCBL-1 cells. The constructs were transfected into 293T or BCBL-1 cells. Dual-luciferase assay was performed 24 h post-transfection. The relative firefly luciferase activities representing IRES activities were normalized to the respective Renilla luciferase activities. Results were presented as mean and error bars from three independent experiments, each with triplicates
It has been reported that unknown splicing and cryptic promoter, which could generate a monocistronic transcript to encode the downstream reporter, might cause false-positive IRES activity [18, 19]. Our transcript mapping results did not reveal any splicing events in the ORF30–33 locus. However, the pcDNA-DL1 and pcDNA-DL3 contained a weak promoter and the ORF30 start codon (Fig. 6a). Further sequence analysis of the pcDNA-DL1 and pcDNA-DL3 revealed a translational reading-frame shift in the firefly luciferase gene if the translation is initiated from the ORF30 start codon, which would result in a nonfunctional firefly luciferase product by introducing two stop codons TAA and TGA in the coding sequence of firefly luciferase at positions 20 and 90 bp, respectively (Fig. 6a). To confirm that the weak P2 promoter did not interfere with the IRES assay, we inserted the same putative IRES fragments from pcDNA-DL1 and pcDNA-DL3 into the promoterless pGL4.16 vector between the KpnI and BglII sites to generate reporters with the same reading-frame shift, which retained the two stop codons TAA and TGA in the coding sequence of firefly luciferase at positions 20 and 90 bp, respectively. As expected, these two reporter constructs had luciferase activities similar to that of pGL4.16 vector in 293T cells (data not shown). Thus, the weak P2 promoter did not interfere with the detection of any IRES activities. Taken together, these results did not reveal the presence of any IRES in the 5′UTR of ORF31.
Discussion
Several loci in the KSHV genome encode for complex overlapping transcripts consisting of polycistronic, bicistronic, and monocistronic transcripts. Examples are the ORF71–73, ORF50/K8/K8.1, ORF34–37, and ORF58–62 loci [20–27]. In this study, we have identified another locus, the ORF30–33 locus in the KHSV genome that encodes multiple overlapping transcripts. In general, viral genes encoded by the same polycistronic or bicistronic transcript often have similar or related functions. For example, all three genes in the ORF71–73 locus are involved in regulation of KSHV latency while genes in the ORF50/K8/K8.1 locus are involved in KSHV lytic replication [28–31]. Studies of MHV-68 ORF30, ORF31, and ORF33 have so far revealed their involvements in the late events of viral lytic replication [5–7]. Thus, polycistronic and bicistronic transcripts appear to provide an economic way, and perhaps a more precise mechanism to regulate the expression of genes that share similar, sequential or related functions. Interestingly, while transcripts of these complex loci often share the same transcriptional termination poly (A) signals, they frequently utilize alternative promoters for the individual transcripts, which could provide specific regulation of individual genes. This feature is observed for the ORF71–73 and ORF50/K8/K8.1 loci [20–23], and further illustrated for the ORF30–33 locus in this study. Furthermore, RNA alternative splicing such as the cases for the ORF71–73 and ORF50/K8/K8.1 loci offers an additional level of regulation for the expression of individual genes. We have identified four transcripts, each with distinct transcriptional start sites in the ORF30–33 locus. No alternative splicing was found for any of these transcripts. The 3.7 and 3.6 kb transcripts encode for all four viral genes; the 2.7 kb transcript encodes for both ORF32 and ORF33 genes; and the 1.4 kb transcript encodes for the ORF33 gene.
A previous study detected three transcripts of 8.0, 6.0, and 3.0 kb in the ORF30–33 region. However, a large DNA probe of about 6 kb encompassing ORF29b to ORF35 was used in this study [23]. Whether these transcripts contain one or several ORFs from this locus remain unclear. The structure and expression kinetics of these transcripts also have not been mapped. While Northern-blot, RACE and DNA sequencing analyses have identified four specific products of 3.7, 3.6, 2.7, and 1.4 kb from the ORF30–33 locus, we have also observed an additional band in the range of 5 to 9 kb in the Northern blots (Fig. 1b). Both RACE and DNA sequencing analyses have so far failed to confirm these bands as KSHV specific. Interestingly, these bands were inducible by TPA, and sensitive to CH and PAA. Recent genome-wide KSHV transcript mapping has identified long lytic transcripts from both strands of the genome [14]. One of these transcripts is ~7 kb in the region of ORF26 to ORF33. Whether these transcripts are transcribed from the ORF30–33 locus remain to be further confirmed.
Four functional promoters were identified in the ORF30–33 locus, each with a functional TATA box. The transcription of the 3.7, 2.7, and 1.4 kb transcripts are likely controlled by promoters P1, P3, and P4, respectively. The weak 3.6 bp transcript can be regulated by promoters P1 and P2. The use of alternative promoters for gene transcription is common in human genes [32–35]. Several KSHV genes such as vIL6 and ORF57 utilize the same strategy for transcriptional regulation [35, 36]. However, unlike vIL6 and ORF57, we only observed a polycistronic transcript and no monocistronic transcript. Furthermore, only weak activity was detected for promoter P2 in the reporter assay. Whether the activity of promoter P2 and the expression of the 3.6 bp transcript could be enhanced in some specific condition, and the viral/cellular factor(s) that might be required for their optimal expression require further investigations. In particular, transcription factors such as AP-1, CREB, SP-1, and RBP-J that have putative binding sites in this region could be involved.
RTA has been shown to transactivate a number of KSHV genes by direct binding to the RTA-responsive element (RRE) or indirect binding to other elements in partners with cellular factors in the promoters. We failed to detect any RTA-responsive promoters in the ORF30–33 locus in reporter assays. Thus, it is unlikely that a RRE is present in these promoters. These results are consistent with the results of genome-wide screening of RTA binding sites in the KSHV genome [37]. It is likely that other factors are required for the expression of these genes during KSHV replication.
Two known mechanisms regulate gene translation in eukaryotes and viruses including 5′ cap-dependent translation and cap-independent translation. Examples for cap-independent translation include IRES and ribosome leaky scanning mediated translation [38, 39]. Results from our transcript mapping experiments indicated that ORF31 has no independent 5′ cap, and thus is unlikely translated by the 5′ cap-dependent mechanism. Further examination also failed to identify any functional IRES in the 5′UTR of ORF31. Therefore, it is likely that an alternative leaky scanning translation is involved in the translation of ORF31, which requires further experimental confirmation.
Acknowledgments
We thank Dr. Jiguo Chen at Mississippi State University for providing the p3Flg vector and members of Dr. Gao's laboratory for technical assistance and helpful discussions. This study was supported by grants from National Institute of Health (CA096 512, CA124332, and CA132637) to S-J Gao.
References
- 1.Greene W, Kuhne K, Ye F, Chen J, Zhou F, Lei X, Gao SJ. Cancer Treat. Res. 2007;133:69–127. doi: 10.1007/978-0-387-46816-7_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cullen BR. Nature. 2009;457:421–425. doi: 10.1038/nature07757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. Proc. Natl. Acad. Sci. USA. 1996;93:14862–14867. doi: 10.1073/pnas.93.25.14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alba MM, Lee D, Pearl FM, Shepherd AJ, Martin N, Orengo CA, Kellam P. Nucleic Acids Res. 2001;29:133–136. doi: 10.1093/nar/29.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guo H, Wang L, Peng L, Zhou ZH, Deng H. J. Virol. 2009;83:10582–10595. doi: 10.1128/JVI.00497-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jia Q, Wu TT, Liao HI, Chernishof V, Sun R. J. Virol. 2004;78:6610–6620. doi: 10.1128/JVI.78.12.6610-6620.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu TT, Park T, Kim H, Tran T, Tong L, Martinez-Guzman D, Reyes N, Deng H, Sun R. J. Virol. 2009;83:2265–2273. doi: 10.1128/JVI.01785-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rozen R, Sathish N, Li Y, Yuan Y. J. Virol. 2008;82:4742–4750. doi: 10.1128/JVI.02745-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu FX, Chong JM, Wu L, Yuan Y. J. Virol. 2005;79:800–811. doi: 10.1128/JVI.79.2.800-811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. J. Virol. 1998;72:1005–1012. doi: 10.1128/jvi.72.2.1005-1012.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang XP, Zhang YJ, Deng JH, Pan HY, Zhou FC, Montalvo EA, Gao SJ. Oncogene. 2001;20:523–530. doi: 10.1038/sj.onc.1204115. [DOI] [PubMed] [Google Scholar]
- 12.Quandt K, Frech K, Karas H, Wingender E, Werner T. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun R, Lin SF, Staskus K, Gradoville L, Grogan E, Haase A, Miller G. J. Virol. 1999;73:2232–2242. doi: 10.1128/jvi.73.3.2232-2242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandriani S, Xu Y, Ganem D. J. Virol. 2010;84:7934–7942. doi: 10.1128/JVI.00645-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grundhoff A, Ganem D. J. Virol. 2001;75:1857–1863. doi: 10.1128/JVI.75.4.1857-1863.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collier AJ, Tang S, Elliott RM. J. Gen. Virol. 1998;79(Pt 10):2359–2366. doi: 10.1099/0022-1317-79-10-2359. [DOI] [PubMed] [Google Scholar]
- 17.Glenn M, Rainbow L, Aurade F, Davison A, Schulz TF. J. Virol. 1999;73:6953–6963. doi: 10.1128/jvi.73.8.6953-6963.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baranick BT, Lemp NA, Nagashima J, Hiraoka K, Kasahara N, Logg CR. Proc. Natl. Acad. Sci. 2008;105:4733–4738. doi: 10.1073/pnas.0710650105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z, Dong Z, Han B, Yang Y, Liu Y, Zhang JT. Nucleic Acids Res. 2005;33:3763–3771. doi: 10.1093/nar/gki680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. J. Virol. 1998;72:8309–8315. doi: 10.1128/jvi.72.10.8309-8315.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu FX, Cusano T, Yuan Y. J. Virol. 1999;73:5556–5567. doi: 10.1128/jvi.73.7.5556-5567.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talbot SJ, Weiss RA, Kellam P, Boshoff C. Virology. 1999;257:84–94. doi: 10.1006/viro.1999.9672. [DOI] [PubMed] [Google Scholar]
- 23.Sarid R, Wiezorek JS, Moore PS, Chang Y. J. Virol. 1999;73:1438–1446. doi: 10.1128/jvi.73.2.1438-1446.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saveliev A, Zhu F, Yuan Y. Virology. 2002;299:301–314. doi: 10.1006/viro.2002.1561. [DOI] [PubMed] [Google Scholar]
- 25.Majerciak V, Yamanegi K, Zheng ZM. J. Virol. 2006;80:11968–11981. doi: 10.1128/JVI.01394-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Haque M, Wang V, Davis DA, Zheng ZM, Yarchoan R. J. Virol. 2006;80:7037–7051. doi: 10.1128/JVI.00553-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Izumiya Y, Izumiya C, Van Geelen A, Wang DH, Lam KS, Luciw PA, Kung HJ. J. Virol. 2007;81:1072–1082. doi: 10.1128/JVI.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lukac DM, Renne R, Kirshner JR, Ganem D. Virology. 1998;252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- 29.Li Q, Zhou F, Ye F, Gao SJ. Virology. 2008;379:234–244. doi: 10.1016/j.virol.2008.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye FC, Zhou FC, Xie JP, Kang T, Greene W, Kuhne K, Lei XF, Li QH, Gao SJ. J. Virol. 2008;82:4235–4249. doi: 10.1128/JVI.02370-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye FC, Zhou FC, Yoo SM, Xie JP, Browning PJ, Gao SJ. J. Virol. 2004;78:11121–11129. doi: 10.1128/JVI.78.20.11121-11129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baek D, Davis C, Ewing B, Gordon D, Green P. Genome Res. 2007;17:145–155. doi: 10.1101/gr.5872707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, Forrest AR, Alkema WB, Tan SL, Plessy C, Kodzius R, Ravasi T, Kasukawa T, Fukuda S, Kanamori-Katayama M, Kitazume Y, Kawaji H, Kai C, Nakamura M, Konno H, Nakano K, Mottagui-Tabar S, Arner P, Chesi A, Gustincich S, Persichetti F, Suzuki H, Grimmond SM, Wells CA, Orlando V, Wahlestedt C, Liu ET, Harbers M, Kawai J, Bajic VB, Hume DA, Hayashizaki Y. Nat. Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- 34.Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K, Wakaguri H, Ishii S, Sugiyama T, Saito K, Isono Y, Irie R, Kushida N, Yoneyama T, Otsuka R, Kanda K, Yokoi T, Kondo H, Wagatsuma M, Murakawa K, Ishida S, Ishibashi T, Takahashi-Fujii A, Tanase T, Nagai K, Kikuchi H, Nakai K, Isogai T, Sugano S. Genome Res. 2006;16:55–65. doi: 10.1101/gr.4039406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsuritani K, Irie T, Yamashita R, Sakakibara Y, Wakaguri H, Kanai A, Mizushima-Sugano J, Sugano S, Nakai K, Suzuki Y. Genome Res. 2007;17:1005–1014. doi: 10.1101/gr.6030107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan W, Wang S, Liu S, Wood C. Arch. Virol. 2001;146:403–413. doi: 10.1007/s007050170185. [DOI] [PubMed] [Google Scholar]
- 37.Chen J, Ye F, Xie J, Kuhne K, Gao SJ. Virology. 2009;386:290–302. doi: 10.1016/j.virol.2009.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gale M, Jr., Tan SL, Katze MG. MMBR. 2000;64:239–280. doi: 10.1128/mmbr.64.2.239-280.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malys N, McCarthy JE. CMLS. 2011;68:991–1003. doi: 10.1007/s00018-010-0588-z. [DOI] [PMC free article] [PubMed] [Google Scholar]