

Summary
Background
Heterologous expression of Factor VIII (FVIII) is about 2 to 3 orders of magnitude lower than similarly sized proteins. Bioengineering strategies aimed at different structural and biochemical attributes of FVIII have been successful in enhancing its expression levels.
Objective
Disulfide bonds are vital to the proper folding, secretion and stability of most secretory proteins. In an effort to explore additional targeted bioengineering approaches, the role of disulfide bonds in FVIII secretion and function was probed in this study.
Methods and Results
Single and paired cysteine mutants were generated by substituting with serine or glycine residues and analyzed by transient transfection into COS-1 and CHO cells. Seven of the eight disulfide bonds in FVIII were found to be indispensable for proper secretion and function. However, elimination of the disulfide bond formed by C1899 and C1903 within the conserved A3 domain improved the secretion of FVIII. The addition of the C1899G/C1903G mutations to a previously described FVIII variant, 226/N6, with high secretion efficiency increased its secretion by 2.2-fold. Finally, the addition of the A1-domain mutation, F309S in conjunction with the disulfide mutation had an additive effect resulting in a net improvement in secretion of between 35–45 fold higher than wild type FVIII in CHO cells.
Conclusion
Such combined targeted bioengineering strategies may facilitate more efficient production of recombinant FVIII toward low cost factor replacement therapy for hemophilia A.
Keywords: bioengineering, disulfide bonds, factor VIII, hemophilia A, secretion
Introduction
Coagulation Factor VIII (FVIII), a large plasma glycoprotein cofactor, performs a critical role in the intrinsic blood coagulation pathway [1]. A quantitative or qualitative deficiency of this protein results in the inherited bleeding disorder, hemophilia A, affecting 1 in 5000 males. The mainstay of treatment for this lifelong bleeding disorder is FVIII replacement, primarily with recombinantly-derived protein [2].
A major obstacle to universal implementation of low cost replacement therapy is the inherent limitation of commercial recombinant FVIII (rFVIII) production since its expression is 2 to 3 orders of magnitude lower than that of other comparably sized proteins in heterologous systems [3]. The cost of rFVIII therapy is high owing to the expense of production, purification and formulation. In addition, the requirement of specialized centralized production and distribution of rFVIII has limited its availability to developing and third world countries [4]. The high costs associated with commercial rFVIII production are for the most part attributed to several biochemical characteristics of the recombinant protein. Inefficient expression of the mRNA [3,5,6] coupled with misfolding and degradation of a sizeable portion of the primary translation product [7,8] and retention of FVIII within the endoplasmic reticulum (ER) through interaction with several ER chaperones [9–12] contribute significantly to the low expression levels of rFVIII in heterologous systems. Furthermore, a facilitated transport mechanism comprising the mannose-binding lectin LMAN1 and MCFD2 has been shown to be required for efficient transport of FVIII from the ER to the Golgi [13–17]. FVIII also requires stabilization by von Willebrand factor (VWF) upon secretion from the cell [18].
Elucidation of the structure, function and secretion pathway of FVIII has provided important insights that have aided in designing and developing bioengineered forms of rFVIII with improved secretion efficiency. Since the B-domain of FVIII was found to be dispensable for its co-factor activity [19], a B-domain-deleted (BDD)-FVIII was developed that resulted in a 17 fold-increase in mRNA levels but registered only a modest 30% increase in protein secretion suggesting other intracellular limitations to secretion in play [20]. An A1 domain mutation F309S that was introduced into FVIII to overcome the retention in the ER through association with BiP increased secretion 3-fold and reduced ATP requirement for secretion [21]. The requirement for facilitated transport of FVIII by the LMAN1/MCFD2 complex by interacting primarily through the mannose residues on N-linked oligosaccharides within its B-domain [22] has led to the development of a bioengineered FVIII variant, 226/N6, wherein the addition of a small portion of native B-domain to BDD-FVIII led to a 5–10 fold improvement in secretion [23]. Introduction of the A1-domain mutation F309S further enhanced secretion to about 15–20 fold over BDD-FVIII.
Despite these significant advances, the huge divide between rates of expression of FVIII compared to other similarly sized proteins in heterologous systems still persists. A large proportion of current research is focused on identifying and exploring new targets for FVIII bioengineering. Some of the unexplored areas of FVIII structure from a bioengineering perspective include post-translational modifications such as asparagine (N)-linked glycosylation outside of the B-domain, O-glycosylation and disulfide bond formation among others. Disulfide bonds are generally considered vital to the structural stability of most secreted and membrane proteins and play a crucial role in the quality control and secretion of proteins from the ER [24,25]. However, in certain proteins, specific disulfide loops have been identified as dispensable to the proper folding, secretion and functionality of the protein. Additionally, elimination of these loops has contributed to increased secretion efficiency in these proteins. Examples include human lysozyme [26,27] and human chorionic gonadotropin α and β-subunit [28,29]. FVIII has eight disulfide bonds in its structure [30] (Fig. 1) but the effect of these on FVIII expression has not been characterized to date.
Figure 1. Factor VIII has eight disulfide bonds in its mature form.

The A1 and A2 domains of the heavy chain and A3 domain of the light chain contain two disulfide bonds each while the C1 and C2 domains of the light chain contain one disulfide bond each (A). The positions of the free and disulfide paired cysteines of FVIII are shown in yellow in this X-ray crystal structure representation (courtesy of Dr. B.L.Stoddard, Fred Hutchinson Cancer Research Center, Seattle, WA) (B). Seven of these disulfides are buried in the hydrophobic core whereas the disulfide bridge between C1899 and C1903 is exposed to the surface.
In this report, we have systematically analyzed the eight disulfide bonds of FVIII and characterized their role in its secretion and function. While the majority of its disulfide bonds were indispensable for the proper secretion and function of FVIII, one loop was found to be non-essential and its elimination improved FVIII secretion. A further increment in secretion was observed when this disulfide mutation was introduced into the 226/N6 variant carrying the previously described A1 domain mutation, F309S, suggesting that this might be yet another independent mechanism that limits the efficient heterologous expression of FVIII.
Materials and Methods
FVIII-deficient and normal pooled human plasma were obtained from George King Biomedical (Overland Park, KS). Activated partial thromboplastin (automated aPTT reagent) and CaCl2 were purchased from bioMerieux Inc. (Durham, NC). Dulbecco modified Eagle medium (DMEM), fetal bovine serum and other tissue culture reagents were purchased from Gibco BRL (Gaithersburg, MD). COAMATIC was purchased from DiaPharma (West Chester, OH). F8C-EIA was purchased from Affinity Biologicals Inc. (ON, Canada). FuGENE-6 transfection reagent and restriction enzymes were purchased from Roche Applied Science (Indianapolis, IN). Quikchange XL-site directed mutagenesis Kit was purchased from Stratagene (La Jolla, CA).
Plasmid Mutagenesis
The mammalian expression vector, pMT-2 containing the full length human FVIII cDNA (NM_000132), designated as WT-FVIII, was subjected to mutagenesis using the Quikchange XL-site directed mutagenesis kit to generate single and paired mutants of cysteines involved in disulfide bond formation. The cysteine residue was mutated to either glycine or serine in each case. All mutant plasmids were verified by restriction enzyme digestion and DNA sequence analysis. The C1899G/C1903G double mutant within 226/N6 [23] was similarly generated and designated as 226/N6-DM. The 226/N6-DM-F309S construct was generated by subcloning the SpeI-KpnI fragment of plasmid Phe309Ser [21] into the 226/N6-DM plasmid as described previously.
Transient cell transfection and analysis
The wild type and mutant plasmid constructs were transfected into Chinese hamster ovary (CHO) cells using FuGENE-6 transfection reagent as per manufacturer’s guidelines. Transfections into COS-1 cells were carried out by the diethylaminoethanol (DEAE) - dextran method [31]. Conditioned medium was harvested at 60–70 hours post-transfection.
Stable Expression of 226/N6 disulfide mutants in CHO cells
226/N6, 226/N6-F309S, 226/N6-DM and 226/N6-DM-F309S cDNA inserts in pMT2 were excised with XhoI and SalI and inserted into a modified mammalian expression vector pDEF38 [32] at the XhoI site. The resulting plasmids were linearized with FspI prior to electroporation into DHFR-null CHO DUKX-K1 cells using amaxa cell line Nucleofector® Kit T (Lonza) with 0.5μg plasmid into 5×106 cells for growth in MEM alpha (Invitrogen) 10% heat-inactivated FBS (Invitrogen) supplemented with ADT (1μg/ml each of Adenosine, Deoxyadenosine, Thymidine). After 48 hours, the cells from a single 10 cm dish were diluted into fifteen 10cm dishes, for selection of growth in MEM alpha 10% heat-inactivated dialyzed-FBS (Invitrogen) lacking the ADT supplement. After ten to fourteen days, individual colonies autotrophic for DHFR were picked and transferred into wells of a 96 well dish. After three more weeks to allow expansion of the colonies, the highest expressing colonies at this point were secreting FVIII at around 1 U/ml. From each FVIII variant, three expressing colonies were combined into a pool for each variant for selection of growth in 50nM MTX (methotrexate (Calbiochem)) in MEM alpha-10% FBS as had been done above (dialyzed FBS lacking ADT supplements). After allowing colonies to appear, picking 96 of them, and expanding these, six of 226/N6 and 12 of the other three variants were chosen for study of expression of FVIII variants. Stably transformed cells growing well in 50nM MTX were plated in 6 cm dishes and allowed to reach confluence after four days. The number of cells was estimated to be approximately 5×106 cells per dish. The cell culture supernatants collected after two days were analyzed using aPTT and ELISA.
In vivo plasmid expression in a hemophilia A (exon 16 −/−) mouse model by hydrodynamic tail-vein injection
In vivo expression of 226/N6-F309S and 226/N6-DM-F309S constructs was analyzed in a F8 exon 16 knock-out mouse model of hemophilia A. Plasmid DNA (100ug) was diluted in 2.0 mL Lactated Ringers and infused over 10 seconds into the tail vein [33, 34]. Peripheral blood was collected from the retro-orbital venous plexus after 24 hours and anticoagulated with 3.8% sodium citrate. Plasma was separated by centrifugation at 2000 rpm for 20 min and FVIII activity and antigen levels were analyzed by COAMATIC chromogenic assay and human FVIII-specific ELISA respectively.
Factor VIII assays
An anti-FVIII light chain sandwich enzyme-linked immunosorbent assay (ELISA) was employed to quantify FVIII antigen levels using a commercial F8C-EIA kit (Affinity Biologicals) according to the manufacturer’s recommendations. FVIII activity was measured by two different methods: (i) a 1-stage aPTT clotting assay on an MLA Electra 750 fibrinometer (Medical Laboratory Automation, Pleasantville, NY) by reconstitution of human FVIII-deficient plasma. The FVIII plasma standard was normal pooled plasma (FACT) from George King Biomedical. (ii) a 2-stage chromogenic method using the COAMATIC assay kit according to the manufacturer’s instructions. The calibration standard included with this kit is assayed according to the Fourth International WHO standard.
Statistical Analysis
Data are expressed as mean values plus or minus standard deviation. Statistical analyses were performed by a 2-sided student t test. Statistical significance was established at P < 0.05.
Results
Seven of eight disulfide bonds are indispensable for FVIII secretion and function
In order to study the role of each of the eight disulfide bonds of FVIII on its secretion and function, we generated single and paired cysteine mutants by mutating them to either serines or glycines and analyzed them by transient transfection in COS-1 and CHO cells. FVIII antigen and one-stage activity assays performed on conditioned media harvested 60–70 hours post transfection revealed that of the eight paired double cysteine mutants, seven were found to be retained intracellularly while the double mutants, C1899/1903S (Fig. 2A, C) and C1899/1903G (Fig. 2B, D) showed an improved secretion over and above the wild type FVIII levels. Similar analysis of the 16 single cysteine mutants showed that 15 of them were retained intracellularly with antigen and activity levels well below background readings. In contrast, the C1903S and C1903G mutants were secreted with antigen levels of about 70–80% of that of wild type FVIII and the proteins were fully functional (data not shown).
Figure 2. Seven of the eight disulfide bonds in FVIII are essential for the proper secretion and activity of the protein.


Paired double cysteine mutants containing cysteine to serine modifications were transiently expressed in COS-1 cells (Panel A) and CHO cells (Panels C). Double cysteine mutants consisting of cysteine to glycine changes were similarly expressed in COS-1 (Panel B) and CHO (Panel D) cells. FVIII antigen (ng/ml) (■) and 1-stage activity (mU/ml) (□) levels were measured by ELISA and aPTT, respectively and are presented here as a percentage of wild type FVIII. The assays were performed on conditioned medium 60–70 hours post-transfection. Data presented is representative of several independent transfection experiments (n ≥ 6) and the error bars where shown represent the standard deviation.
The C1899/1903S double mutant displayed on average, a 1.2 fold higher secretion than wild type FVIII in COS-1 cells while it was secreted at about 1.35 fold higher than the wild type FVIII in CHO cells. The C1899/1903G double mutant on the other hand exhibited on average, a 1.3 and 1.6 fold higher secretion in COS-1 and CHO cells respectively. These results were reproducible and consistent in at least five separate transfection experiments with two different independently isolated mutant clones. The activity of the double mutant, as witnessed from both 1-stage (135–168 mU/ml) and 2-stage assays (121–149 mU/ml) correlated with the higher antigen levels (22.8–31.1 ng/ml). A similar modest improvement in secretion was observed for the C1899/C1903 double mutants in the BDD-FVIII backbone as well (Data not shown)
C1899/1903G double mutant displays higher secretion in 226/N6 backbone
Since both C1899/1903S and C1899/1903G mutants performed similarly and with the C1899/1903G mutant displaying a slightly better secretion potential, we next tested if this disulfide mutation was capable of further enhancing secretion in 226/N6, the bioengineered variant with superior secretion efficiency. To achieve this, we recreated the C1899/1903G double mutant in 226/N6 backbone and performed transfection analysis in COS-1 and CHO cells. The activity (1-stage and 2-stage) and the antigen levels were measured in conditioned media 60–70 hours post-transfection and were compared to 226/N6-FVIII. The results showed that the disulfide mutation augmented secretion of the 226/N6 variant by about 1.6 fold in COS-1 cells (Fig. 3A) and by about 2.2 fold (p<0.05) in CHO cells (Fig. 3B). Quantitation of FVIII in cell extracts by ELISA revealed that 226/N6 and 226/N6-DM were expressed at similar levels intracellularly (data not shown) suggesting a secretion advantage for the disulfide mutant. These findings were reproducible and consistent across multiple independent transfection experiments (n≥6).
Figure 3.

Secretion advantage conferred by elimination of the C1899-C1903 disulfide bond is augmented in the 226/N6 variant. 1- and 2-stage activity and FVIII antigen levels were determined for 226/N6 (■) and 226/N6-DM (□) cDNA constructs expressed transiently in COS-1 (A) and CHO (B) cells. Assays were performed on conditioned medium 60–70 hours post-transfection as described previously. Expression of the mutant was compared to the 226/N6 control and the data is expressed as fold-226/N6. Data presented here are the mean of several independent experiments (n ≥ 6) and the error bars represent the standard deviation.
Additive effect of A1 domain mutation (F309S) on 226/N6-DM secretion
Retention of FVIII in the ER through interaction with BiP is one of the major hurdles for efficient secretion. The previously described A1 domain point mutation, F309S, resulted in about 2-fold higher expression of both the full length FVIII and 226/N6 variant in cells [23]. We therefore hypothesized that addition of this mutation would further improve secretion of 226/N6-DM. We generated the 226/N6-DM-F309S hybrid construct through a sub-cloning strategy described earlier and performed transient transfection into COS-1 and CHO cells. Conditioned media harvested 60–70 hours post-transfection were analyzed for expression by activity (1-stage and 2-stage) and antigen assays. Results were expressed relative to 226/N6 transfected in parallel. The 226/N6-DM-F309S construct was secreted at an even higher level resulting in a net increase of about 3.2-fold over 226/N6 in COS-1 cells (Fig. 4A) and close to 4.1-fold in CHO cells (Fig. 4B) (p<0.05). Thus, there was an additive effect of the A1-domain mutation, F309S, to the secretion enhancement conferred by the disulfide mutation within the 226/N6 backbone. These results were similarly consistent and reproducible across several independent transfection experiments both in COS-1 and CHO cells.
Figure 4. Additive effect of the A1 domain mutation F309S and the C1899/1903G disulfide mutation on secretion of 226/N6.
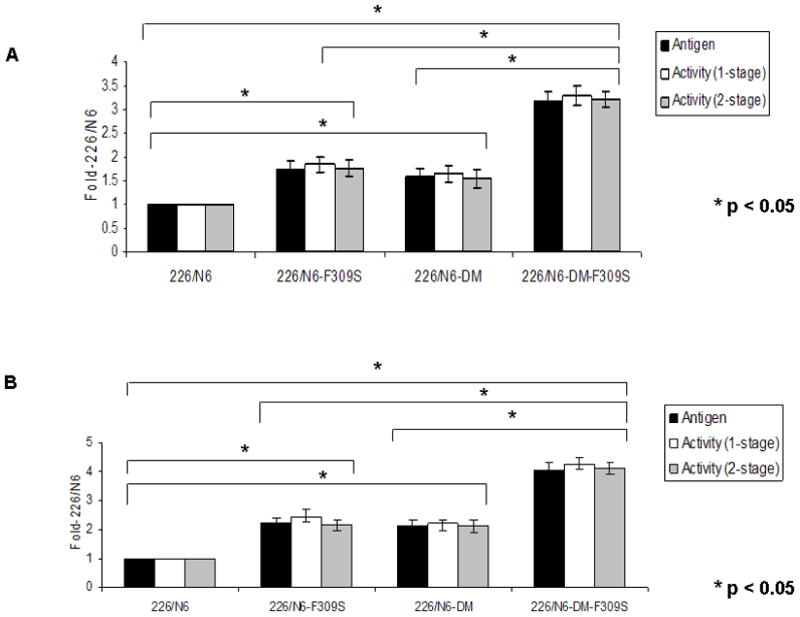
cDNA constructs of 226/N6, 226/N6-F309S, 226/N6-DM and 226/N6-DM-F309S were transiently expressed in COS-1 (A) and CHO (B) cells. FVIII antigen levels (■), 1-stage activity (□) and 2-stage activity (■) were measured from conditioned medium 60–70 hours post-transfection. Expression levels of the constructs were compared to 226/N6 control and the data presented as fold-226/N6. Data shown is representative of several independent experiments (n ≥ 6) and the error bars represent the standard deviation. *P < 0.05
Stable expression of 226/N6-DM and 226/N6-DM-F309S within CHO cells
CHO cell lines stably expressing 226/N6-DM and 236/N6-DM-F309S were prepared as described under Methods. Comparison of the high expressing clones of 226/N6, 226/N6-DM, 226/N6-F309S and 226/N6-DM-F309S revealed that the secretion advantage conferred by the C1899/1903G disulfide mutation was preserved in the stably expressing CHO cell line as well. Average FVIII antigen levels of 6.3 μg/ml, 11.9 μg/ml, 12.9 μg/ml and 23.8 μg/ml were obtained for 226/N6, 226/N6-DM, 226/N6-F309S and 226/N6-DM-F309S respectively. The average FVIII activity levels, as measured by an aPTT assay, for these four constructs were 15.7 U/ml, 31.3 U/ml, 32.3 U/ml and 54.8 U/ml, respectively. On average, the 226/N6-DM variant demonstrated a 1.8 – 2.0 fold higher secretion over 226/N6 and the 226/N6-DM-F309S variant exhibited a 1.7 – 1.85 fold improvement in secretion over 226/N6-F309S (Fig. 5).
Figure 5. Stable expression of 226/N6-DM and 226/N6-DM-F309S within CHO cells.

CHO cells stably expressing 226/N6, 226/N6-DM, 226/N6-F309S and 226/N6-DM-F309S were prepared as described under Methods. FVIII antigen [Panel A (■)] and activity levels [Panel B (□)] were measured by ELISA and aPTT assays respectively. Data presented is the mean from several of the highest expressing clones and the error bars represent the standard deviation. *P < 0.05
In vivo expression of 226/N6-DM-F309S
In order to test whether the secretion improvement observed for the disulfide mutant in COS-1 and CHO cells would also extend to an in vivo heterologous expression system, we induced transient expression of 226/N6-F309S and 226/N6-DM-F309S constructs in the liver of hemophilia A (F8−/−) using a hydrodynamic tail-vein injection of plasmid DNA as described earlier. A FVIII specific ELISA showed an average antigen level of 1.05 μg/ml for 226/N6-F309S and 1.8 μg/ml for 226/N6-DM-F309S (Fig. 6A). The FVIII activity levels measured by the COAMATIC chromogenic assay were, on average, 7.8 U/ml for 226/N6-F309S and 13.2 U/ml for 226/N6-DM-F309S (Fig. 6B). These values suggested ~ 1.7 fold higher expression levels of 226/N6-DM-F309S over 226/N6-F309S indicating that the secretion enhancement of the disulfide mutant was reproducible in vivo as well. The specific activity of 226/N6-F309S (7428.57 U/mg) was similar to 226/N6-DM-F309S (7333.33 U/mg).
Figure 6. C1899/1903G disulfide mutation confers secretion advantage in vivo in a hemophilia A mouse model.

FVIII antigen (Panel A) and activity (Panel B) in the plasma of hemophilia A mice were measured by an ELISA and a COAMATIC chromogenic assay respectively, following transient expression of 226/N6-F309S and 226/N6-DM-F309S constructs by hydrodynamic tail vein injection as described under Methods. Data presented (○) represent multiple independent expression studies (N ≥ 5) with the mean indicated by a bar. *P < 0.05
Bioengineering results in synergistic improvement in secretion of FVIII
Incorporation of the disulfide mutation in previously bioengineered FVIII variants with higher expression efficiency further boosts secretion with synergistic effect. Data shown in Fig. 7 was obtained from comparing expression levels of 226/N6-DM and 226/N6-DM-F309S constructs to wild type FVIII from multiple transfection experiments in CHO cells. Incorporation of these mutations within 226/N6 led to a cumulative effect resulting overall in a 35–45 fold higher secretion than wild type FVIII in CHO cells, suggesting that such combined targeted bioengineering strategies may facilitate more efficient production of rFVIII towards low-cost replacement therapy.
Figure 7. Bioengineering strategies lead to synergistic improvements in FVIII secretion efficiency.

Data obtained by comparing expression levels of 226/N6-DM and 226/N6-DM-F309S constructs to full length wild type FVIII (WT-FVIII) from a series of several independent transient transfection experiments in CHO cells. Data is presented as fold-WT-FVIII. Error bars represent the standard deviation. *P < 0.05
Discussion
With the exception of the report by McMullen et al [30] identifying the locations of disulfide bonds and free cysteines, there haven’t been any other studies that have focused on the role of disulfide bonds within FVIII. This report is the first to systematically analyze the role of each individual disulfide bond in the secretion and function of FVIII. While disulfide bridges are generally indispensable for the proper folding, assembly, secretion and function of most disulfide containing proteins, there have been reports of certain exceptions as in the case of human lysozyme [26,27] and human chorionic gonadotropin alpha [29] and beta [28] subunits wherein mutation of a particular cysteine residue involved in disulfide bonding or elimination of a disulfide loop improved secretion efficiency. We therefore tested whether a similar exception may exist within a complex, multi domain protein such as FVIII.
We observed that 15 of the 16 cysteine residues involved in seven disulfide bonds were indispensable and mutation of these residues to serine or glycine resulted in complete intracellular retention of the protein. All of these cysteines are buried and form core disulfide bonds that are essential for the stability of the protein. C1899 and C1903 are the only two cysteine residues that are exposed to the surface. These findings were mostly consistent with the phenotype of patient missense mutations reported in the database of Hemophilia A Mutation, Structure, Test and Resource Site (HAMSTeRS) available on the internet (URL: http://hadb.org.uk/) as well as from newer reports in the literature. Therefore, in essence, this study also provides a mechanistic basis for the clinical phenotype of missense mutations of cysteine residues involved in FVIII disulfide bonding, reported so far.
There have been no clinical reports so far of missense mutations of either C1899 or C1903 that are causative of hemophilia A in patients. The only alteration reported with C1903 in the HAMSTeRS database is that of a nonsense mutation resulting in a truncated protein causing severe hemophilia A. This further strengthens the argument that these two cysteines and the disulfide bond they form might be dispensable to the proper folding, secretion and function of FVIII. In addition, two missense mutations of C1903, namely C1903S and C1903W have been recorded in the National Center for Biotechnology Information (NCBI) database of single nucleotide polymorphisms (SNPs) (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi?locusId=2157). Both these entries have been validated by multiple, independent submissions to the refSNP cluster and have been genotyped by the HapMap project. This is quite consistent with our observations of the C1903S and C1903G single cysteine mutants that were secreted at levels of about 70–80% of wild type FVIII in COS-1 and CHO cells. Interestingly, the C1899G single mutant was retained completely yet the double mutant 1899–1903 was found to be secreted more efficiently than the wild type FVIII. Precedents for pairing cysteines behaving differently in the context of the folding and secretion of a protein can be found in the examples of human lysozyme [35] and human chorionic gonadotropin β subunit [28]. It is likely that the free thiol at C1903 in the C1899G mutant may hinder secretion by interacting with other free thiols in FVIII.
Further insights on the enhanced secretion of the 1899–1903 double mutant can be gained from the structural comparison of FVIII and its coagulation factor homolog, FV. Despite sharing an identical domain structure and approximately 40% amino acid identity within their A and C domains, FV is secreted 10–20 fold higher than FVIII [36]. A comparison of the disulfide bonds of FVIII and FV reveals that seven of the eight disulfide bonds present in FVIII are highly conserved in FV but interestingly, FV lacks a disulfide loop in the homologous position to the C1899–C1903 disulfide bridge in FVIII [30]. We hypothesize that the elimination of this disulfide loop improves secretion of FVIII through alterations in the molecular flexibilities in the secondary or tertiary structure. This, along with other factors such as little or no binding of FV to BiP [36] might explain the huge difference in secretion of these two structurally similar proteins.
We had also hypothesized that elimination of this non-essential disulfide bond would overcome a unique mechanism limiting FVIII secretion efficiency than had previously been explored with other bioengineered FVIII variants. Thus, combining this targeted strategy with other previously described bioengineered variants with improved secretion efficiency could have an additive effect. In support of this, introduction of this disulfide mutation into 226/N6 produced a further boost in secretion efficiency. The higher secretion efficiency of 226/N6 has been attributed to its optimization of the LMAN-1/MCFD2 facilitated ER-Golgi transport through the N-linked oligosaccharides within its short B-domain spacer [23]. Additionally, the incorporation of F309S, the A1 domain mutation which has been shown earlier to improve secretion by diminishing FVIII association with BiP and reducing its ATP dependence [21], into this hybrid construct resulted in additional improvement in secretion. This secretion enhancement was consistently observed both during transient as well as stable expression in cell lines. Furthermore, the secretion advantage conferred by the disulfide mutation could also be reproduced in vivo in a hemophilia A (F8−/−) mouse model.
Overall, this study provides new insights into the role of disulfide bonds on the folding, secretion and function of FVIII. Such insights are crucial in devising novel bioengineering strategies to improve commercial rFVIII production. These results also provide impetus to target other previously unexplored structural elements of FVIII such as N-linked glycosylation and other post-translational modifications to find ways to improve secretion. However, since many of these strategies involve the introduction of missense mutations to the native FVIII sequence, such variants will need to be tested for their potential in creating neoantigenic sites on FVIII that might lead to inhibitor formation in patients.
Ward et al (37) recently reported a 29–44 fold increase in expression of FVIII codon-optimized for expression in Homo sapiens. Though the mechanism is unclear, this approach most likely targets transcriptional and translational hindrances to efficient FVIII expression. We hypothesize that targeting post-translational modifications such as disulfide bonding, as we have done in the present study, could provide opportunities for improved expression efficiency that are additive to such enhancements to transcriptional and translational regulation of FVIII expression. A combination of targeted bioengineering techniques, as described in this study, has made it possible to attain about 35–45 fold higher secretion than wild type FVIII in mammalian cells. This clearly emphasizes the importance of such bioengineering methodology in achieving the ultimate goal of maximizing FVIII yield and reducing the overall costs of manufacturing that would make FVIII therapeutics more affordable and accessible to the global hemophilia population.
Acknowledgments
This work was supported by National Institutes of Health grant HL82619 to S.W.P. and by the Goerlich Foundation. Portions of this work were supported by NIH grants HL052173, and HL057346 to R.J.K.
References
- 1.Mann KG. Biochemistry and physiology of blood coagulation. Thromb Haemost. 1999;82:165–74. [PubMed] [Google Scholar]
- 2.Rogoff EG, Guirguis HS, Lipton RA, Seremetis SV, DiMichele DM, Agnew GM, Karpatkin M, Barish RJ, Jones RL, Bianco C, Knothe BD, Lee MS. The upward spiral of drug costs: a time series analysis of drugs used in the treatment of hemophilia. Thromb Haemost. 2002;88:545–53. [PubMed] [Google Scholar]
- 3.Lynch CM, Israel DI, Kaufman RJ, Miller AD. Sequences in the coding region of clotting factor VIII act as dominant inhibitors of RNA accumulation and protein production. Hum Gene Ther. 1993;4:259–72. doi: 10.1089/hum.1993.4.3-259. [DOI] [PubMed] [Google Scholar]
- 4.Mannucci PM. Hemophilia: treatment options in the twenty-first century. J Thromb Haemost. 2003;1:1349–55. doi: 10.1046/j.1538-7836.2003.00262.x. [DOI] [PubMed] [Google Scholar]
- 5.Kaufman RJ, Wasley LC, Davies MV, Wise RJ, Israel DI, Dorner AJ. Effect of von Willebrand factor coexpression on the synthesis and secretion of factor VIII in Chinese hamster ovary cells. Mol Cell Biol. 1989;9:1233–42. doi: 10.1128/mcb.9.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hoeben RC, Fallaux FJ, Cramer SJ, van den Wollenberg DJ, van Ormondt H, Briet E, van der Eb AJ. Expression of the blood-clotting factor-VIII cDNA is repressed by a transcriptional silencer located in its coding region. Blood. 1995;85:2447–54. [PubMed] [Google Scholar]
- 7.Dorner AJ, Bole DG, Kaufman RJ. The relationship of N-linked glycosylation and heavy chain-binding protein association with the secretion of glycoproteins. J Cell Biol. 1987;105:2665–74. doi: 10.1083/jcb.105.6.2665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pipe SW, Kaufman RJ. Factor VIII C2 domain missense mutations exhibit defective trafficking of biologically functional proteins. J Biol Chem. 1996;271:25671–76. doi: 10.1074/jbc.271.41.25671. [DOI] [PubMed] [Google Scholar]
- 9.Dorner AJ, Wasley LC, Kaufman RJ. Increased synthesis of secreted proteins induces expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells. J Biol Chem. 1989;264:20602–7. [PubMed] [Google Scholar]
- 10.Dorner AJ, Wasley LC, Kaufman RJ. Overexpression of GRP78 mitigates stress induction of glucose regulated proteins and blocks secretion of selective proteins in Chinese hamster ovary cells. EMBO J. 1992;11:1563–71. doi: 10.1002/j.1460-2075.1992.tb05201.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorner AJ, Wasley LC, Kaufman RJ. Protein dissociation from GRP78 and secretion are blocked by depletion of cellular ATP levels. Proc Natl Acad Sci USA. 1990;87:7429–32. doi: 10.1073/pnas.87.19.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tagliavacca L, Wang Q, Kaufman RJ. ATP-dependent dissociation of non-disulfide-linked aggregates of coagulation factor VIII is a rate-limiting step for secretion. Biochemistry. 2000;39:1973–81. doi: 10.1021/bi991896r. [DOI] [PubMed] [Google Scholar]
- 13.Nichols WC, Seligsohn U, Zivelin A, Terry VH, Hertel CE, Wheatley MA, Moussalli MJ, Hauri HP, Ciavarella N, Kaufman RJ, Ginsburg D. Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell. 1998;93:61–70. doi: 10.1016/s0092-8674(00)81146-0. [DOI] [PubMed] [Google Scholar]
- 14.Zhang B, Cunningham MA, Nichols WC, Bernat JA, Seligsohn U, Pipe SW, McVey JH, Schulte-Overberg U, de Bosch NB, Ruiz-Saez A, White GC, Tuddenham EG, Kaufman RJ, Ginsburg D. Bleeding due to disruption of a cargo-specific ER-to-Golgi transport complex. Nat Genet. 2003;34:220–5. doi: 10.1038/ng1153. [DOI] [PubMed] [Google Scholar]
- 15.Zhang B, Kaufman RJ, Ginsburg D. LMAN1 and MCFD2 form a cargo receptor complex and interact with coagulation factor VIII in the early secretory pathway. J Biol Chem. 2005;280:25881–6. doi: 10.1074/jbc.M502160200. [DOI] [PubMed] [Google Scholar]
- 16.Zhang B, McGee B, Yamaoka JS, Guglielmone H, Downes KA, Minoldo S, Jarchum G, Peyvandi F, de Bosch NB, Ruiz-Saez A, Chatelain B, Olpinski M, Bockenstedt P, Sperl W, Kaufman RJ, Nichols WC, Tuddenham EG, Ginsburg D. Combined deficiency of factor V and factor VIII is due to mutations in either LMAN1 or MCFD2. Blood. 2006;107:1903–7. doi: 10.1182/blood-2005-09-3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baines AC, Zhang B. Receptor-mediated protein transport in the early secretory pathway. Trends Biochem Sci. 2007;32:381–8. doi: 10.1016/j.tibs.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Kaufman RJ, Wasley LC, Dorner AJ. Synthesis, processing, and secretion of recombinant human factor VIII expressed in mammalian cells. J Biol Chem. 1988;263:6352–62. [PubMed] [Google Scholar]
- 19.Toole JJ, Pittman DD, Orr EC, Murtha P, Wasley LC, Kaufman RJ. A large region (approximately equal to 95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci USA. 1986;83:5939–42. doi: 10.1073/pnas.83.16.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pittman DD, Alderman EM, Tomkinson KN, Wang JH, Giles AR, Kaufman RJ. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted factor VIII. Blood. 1993;81:2925–35. [PubMed] [Google Scholar]
- 21.Swaroop M, Moussalli M, Pipe SW, Kaufman RJ. Mutagenesis of a potential immunoglobulin-binding protein-binding site enhances secretion of coagulation factor VIII. J Biol Chem. 1997;272:24121–4. doi: 10.1074/jbc.272.39.24121. [DOI] [PubMed] [Google Scholar]
- 22.Moussalli M, Pipe SW, Hauri HP, Nichols WC, Ginsburg D, Kaufman RJ. Mannose-dependent endoplasmic reticulum (ER)-Golgi intermediate compartment-53-mediated ER to Golgi trafficking of coagulation factors V and VIII. J Biol Chem. 1999;274:32539–42. doi: 10.1074/jbc.274.46.32539. [DOI] [PubMed] [Google Scholar]
- 23.Miao HZ, Sirachainan N, Palmer L, Kucab P, Cunningham MA, Kaufman RJ, Pipe SW. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103:3412–19. doi: 10.1182/blood-2003-10-3591. [DOI] [PubMed] [Google Scholar]
- 24.Fassio A, Sitia R. Formation, isomerisation and reduction of disulphide bonds during protein quality control in the endoplasmic reticulum. Histochem Cell Biol. 2002;117:151–7. doi: 10.1007/s00418-001-0364-0. [DOI] [PubMed] [Google Scholar]
- 25.Van Anken E, Braakman I. Versatility of the endoplasmic reticulum protein folding factory. Crit Rev Biochem Mol Biol. 2005;40:191–228. doi: 10.1080/10409230591008161. [DOI] [PubMed] [Google Scholar]
- 26.Taniyama Y, Yamamoto Y, Nakao M, Kikuchi M, Ikehara M. Role of disulfide bonds in folding and secretion of human lysozyme in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1988;152:962–7. doi: 10.1016/s0006-291x(88)80377-2. [DOI] [PubMed] [Google Scholar]
- 27.Inaka K, Taniyama Y, Kikuchi M, Morikawa K, Matsushima M. Role of disulfide bonds in folding and secretion of human lysozyme in Saccharomyces cerevisiae. J Biol Chem. 1991;266:12599–603. [PubMed] [Google Scholar]
- 28.Suganuma N, Matzuk MM, Boime I. Elimination of disulfide bonds affects assembly and secretion of the human chorionic gonadotropin beta subunit. J Biol Chem. 1989;264:19302–7. [PubMed] [Google Scholar]
- 29.Furuhashi M, Ando H, Bielinska M, Pixley MR, Shikone T, Hsueh AJ, Boime I. Mutagenesis of cysteine residues in the human gonadotropin alpha subunit. Roles of individual disulfide bonds in secretion, assembly, and biologic activity. J Biol Chem. 1994;269:25543–8. [PubMed] [Google Scholar]
- 30.McMullen BA, Fujikawa K, Davie EA, Hedner U, Ezban M. Locations of disulfide bonds and free cysteines in the heavy and light chains of recombinant human factor VIII (antihemophilic factor A) Prot Sci. 1995;4:740–6. doi: 10.1002/pro.5560040413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pittman DD, Kaufman RJ. Site-directed mutagenesis and expression of coagulation factors VIII and V in mammalian cells. Methods Enzymol. 1993;222:236–60. doi: 10.1016/0076-6879(93)22017-a. [DOI] [PubMed] [Google Scholar]
- 32.Running Deer J, Allison DS. High-Level Expression of Proteins in Mammalian Cells Using Transcription Regulatory Sequences from the Chinese Hamster EF-1α Gene. Biotechnol Prog. 2004;20:880–9. doi: 10.1021/bp034383r. [DOI] [PubMed] [Google Scholar]
- 33.Liu F, Song Y, Liu D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999;6:1258–66. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 34.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735–7. doi: 10.1089/10430349950017734. [DOI] [PubMed] [Google Scholar]
- 35.Taniyama Y, Yamamoto Y, Kuroki R, Kikuchi M. Evidence for difference in the roles of two cysteine residues involved in disulfide bond formation in the folding of human lysozyme. J Biol Chem. 1990;265:7570–5. [PubMed] [Google Scholar]
- 36.Pittman DD, Tomkinson KN, Kaufman RJ. Post-translational requirements for functional factor V and factor VIII secretion in mammalian cells. J Biol Chem. 1994;269:17329–37. [PubMed] [Google Scholar]
- 37.Ward NJ, Buckley SM, Waddington SN, VandenDriessche T, Chuah MK, Nathwani AC, McIntosh J, Tuddenham EG, Kinnon C, Thrasher AJ, McVey JH. Codon optimization of human factor VIII cDNAs leads to high-level expression. Blood. 2011;117:798–807. doi: 10.1182/blood-2010-05-282707. [DOI] [PubMed] [Google Scholar]