

Abstract
MDMX is a hetero dimeric partner of MDM2 and a critical regulator of p53. MDMX level is generally elevated in tumors with wild type p53 and contributes to p53 inactivation. MDMX degradation is controlled in part by MDM2-mediated ubiquitination. Here we show that MDMX turnover is highly responsive to changes in MDM2 level in non-transformed cells, but not in tumor cells. We found that loss of ARF expression, which occurs in most tumors with wild type p53, significantly reduces MDMX sensitivity to MDM2. Restoration of ARF expression in tumor cells enables MDM2 to degrade MDMX in a dose-dependent fashion. ARF binds to MDM2 and stimulates a second-site interaction between the central region of MDM2 and MDMX, thus increases MDMX-MDM2 binding and MDMX ubiquitination. These results reveal an important abnormality in the p53 regulatory pathway as a consequence of ARF deficiency. Loss of ARF during tumor development not only prevents p53 stabilization by proliferative stress, but also causes accumulation of MDMX that compromises p53 activity. This phenomenon may reduce the clinical efficacy of MDM2-specific inhibitors by preventing MDMX down regulation.
Keywords: MDM2, MDMX, ARF, p53, ubiquitination, Nutlin
Introduction
Regulation of the p53 tumor suppressor requires the cooperation of MDM2 and MDMX oncoproteins. MDM2 and MDMX are homologs that share amino acid sequence similarity in several domains (Shvarts et al 1997). MDM2 is an ubiquitin E3 ligase that promotes p53 ubiquitination and degradation. Although MDMX has very low E3 ligase activity, it forms heterodimer with MDM2 through the C terminal RING domain and is believed to stimulate MDM2 activity under physiological conditions (Badciong and Haas 2002, Gu et al 2002, Linares et al 2003). In addition to regulating p53 ubiquitination, MDM2 interacts with several transcriptional regulators (p300, CBP, YY1, KAP1) and chromatin modifying enzymes (HDAC1, SUV39H1, EHMT1, G9a). The recruitment of these factors may directly regulate p53 transcriptional activity at promoters. Recent studies showed that MDM2 also induces p53 conformational change and inhibits p53 DNA binding (Cross et al 2011, Sasaki et al 2007). Mouse models showed that the expression and E3 ligase activity of MDM2 are critical for preventing p53 hyper activation and ensuring the survival of mouse embryos (Itahana et al 2007, Jones et al 1995, Montes de Oca Luna et al 1995).
The biochemical activities of MDM2 are under tight regulation. MDM2 E3 ligase activity is regulated by phosphorylation near the C terminal RING domain, by interactions with several ribosomal proteins, and by the tumor suppressor ARF. MDM2 phosphorylation plays a critical role in p53 activation by genotoxic stress such as ionizing irradiation and other inducers of DNA strand breaks (Cheng et al 2009). Ribosomal proteins such as L5, L11 and L23 binds to the MDM2 central acidic region and zinc finger to cause p53 accumulation when ribosomal RNA transcription or ribosome assembly is disturbed (Zhang and Lu 2009). MDM2-L11 interaction is important for the suppression of Myc-induced tumorigenesis in a mouse model (Macias et al).
An important MDM2 regulator is the ARF tumor suppressor (Sherr 2006). ARF is encoded by an alternative open reading frame in the p16/INK4 locus. Oncogene activation induces ARF expression through transcriptional and post-translational mechanisms (Chen et al 2010). ARF binds to the acidic region of MDM2 and inhibits p53 ubiquitination (Midgley et al 2000). Mice without ARF expression are predisposed to tumor development, showing significant overlap with phenotypes of the p53-null mice (Kamijo et al 1997). ARF expression is lost in nearly all human tumors that retain wild type p53, suggesting that ARF inactivation and p53 mutation are alternative mechanisms, each sufficient to disable the p53 pathway during tumor development (Stott et al 1998).
Despite its important biological roles, the mechanism by which ARF activates p53 is still poorly understood. ARF binds to a central acidic region of MDM2 that is predicted to be unstructured in the absence of binding partners. ARF sequence also predicts that it is an unstructured protein (Sherr 2006). Interaction between an ARF peptide and the MDM2 acidic region causes significant secondary structure formation in vitro (Bothner et al 2001, Sivakolundu et al 2008). The acidic region of MDM2 is important for ubiquitination and degradation of p53 through unknown mechanisms (Kawai et al 2003b, Meulmeester et al 2003).
Mouse models demonstrated that MDMX is a critical regulator of p53 during embryonic development (Parant et al 2001). MDMX in adult somatic tissues is less important for p53 regulation compared to MDM2 (Grier et al 2006, Maetens et al 2007, Xiong et al 2006). Nonetheless, MDMX expression and phosphorylation by the ATM/Chk2 pathway has significant roles in p53 DNA damage response in adult mice (Terzian et al 2007, Wang et al 2009). MDMX overexpression has been detected in tumors with wild type p53 and presumably contributes to p53 inactivation (Danovi et al 2004, Laurie et al 2006, Ramos et al 2001). MDMX level is controlled by MDM2-mediated ubiquitination in a stress-dependent fashion (Kawai et al 2003a, Pan and Chen 2003). Whereas MDM2 has a very short half life (~30 min), MDMX is relatively stable in the absence of stress (half life 3–6 hr). Significant degradation of MDMX occurs after DNA damage or ribosomal stress induction. DNA damage induces phosphorylation of MDMX on several C terminal sites (S342 and S367 by Chk2, S403 by ATM), with S367 phosphorylation being most critical for promoting degradation by MDM2 (Chen et al 2005, Pereg et al 2005). During ribosomal stress, L11 binding to MDM2 is important for promoting MDMX degradation (Gilkes et al 2006). Artificially overexpressing MDMX or blocking MDMX degradation significantly dampens p53 response to stress, promoting tumor development (Wang et al 2009, Xiong et al 2010). Therefore, key signaling mechanisms that block p53 degradation by MDM2 also simultaneously enhance MDMX degradation by MDM2. These observations underscore the coordinated control of MDM2 and MDMX by stress signals, which is critical for proper p53 response.
MDM2 is a classic p53 transcriptional target, forming a negative feedback loop (Wu et al 1993). Although MDMX was initially thought to be non-responsive to p53 transactivation, recent studies identified bona fide p53 binding sites in intron 1 of the human MDMX gene and demonstrated that p53 activation can lead to moderate induction of MDMX transcription (Li et al 2010, Phillips et al 2010). However, the magnitude of MDMX mRNA induction (1.5–3 fold) is much lower compared to MDM2 (>10 fold) after p53 activation, and displayed cell type specificity. These studies showed that MDMX is a p53 target gene that may also provide negative feedback response to p53 activation.
MDM2 and MDMX N terminal domain form a hydrophobic pocket and binds an amphipathic helix of p53. Several small molecule disruptors of p53-MDM2 binding have been developed and showed promising anti-tumor activities (Shangary et al 2008, Vassilev et al 2004). Due to structural differences, current inhibitors of MDM2 have negligible effect on MDMX-p53 binding, thus are suboptimal for p53 activation in cells overexpressing MDMX (Hu et al 2006, Patton et al 2006, Shangary et al 2008, Wade et al 2006). Because MDM2-specific inhibitors can partially activate p53 which in turn induces MDM2 expression, these inhibitors should in theory promote MDMX degradation, thus bypassing the need for an MDMX inhibitor. However, several reports found that Nutlin treatment does not cause significant reduction of MDMX level in tumor cell lines, and may even increase MDMX level in certain cell lines due to p53-mediated induction of MDMX transcription (Hu et al 2006, Li et al 2010, Wade et al 2006). Interestingly, Nutlin induces significant MDMX reduction in non-transformed human cells (Wade et al 2006). These observations suggest important differences in the regulation of MDMX expression and degradation in normal and tumor cells.
In this report, we show that ARF is a key factor in regulating MDMX degradation by MDM2. Loss of ARF expression in tumor cells causes MDMX to display poor sensitivity to MDM2. Restoration of ARF expression enables MDM2 to bind and degrade MDMX in a dose-dependent fashion. ARF acts by stimulating heterodimer formation between MDM2 and MDMX, thus increasing MDMX ubiquitination. These results showed that loss of ARF expression during tumorigenesis results in a defect in MDMX degradation that further compromises p53 function.
Results
MDMX is resistant to MDM2 in tumor cells
To investigate whether normal and tumor cells display a difference in MDMX regulation by MDM2, we used Nutlin and MI-63 to treat a panel of human cells including normal human fibroblasts (HFF, WI-38) and tumor cells (U2OS, A549, 2102EP, 833KE, H1299, HCT116, and HCT116-p53−/−). Treatment of all p53 wild type cells caused strong increase in MDM2 due to p53 activation. Rise of MDM2 was correlated with significant decrease of MDMX level in non-transformed HFF and WI-38 cells, but not in tumor cell lines (Figure 1a, Supplemental Figure 1). Instead, certain tumor cell lines (HCT116, 2102EP, 833KE) showed moderate increase of MDMX level. As expected, Nutlin and MI-63 did not change MDM2 and MDMX levels in p53-null cells. RT-PCR analysis showed that MDMX mRNA was induced after p53 activation to various degrees in different cell lines, ranging from no induction (WI-38, HFF), ~30% (HCT116, U2OS), to 300–600% (2102EP, 833KE) (Figure 1b). These results indicate that increase of MDM2 is not sufficient to eliminate MDMX in tumor cells. Induction of MDMX mRNA may even lead to net increase in MDMX protein level. Furthermore, the significant down regulation of MDMX in Nutlin/MI-63-treated normal cells (WI-38, HFF) cannot be explained by changes in MDMX transcription, suggesting that regulation of MDMX turnover is different between normal and tumor cells (see Figure 3).
Figure 1. MDM2 inhibitors have different effects on MDMX level in normal and tumor cells.

(a) Indicated cell lines were treated with 8 μM Nutlin or 8 μM MI-63 for 16 hrs. MDMX level was analyzed by Western blot. WI-38 and HFF are used to represent normal cells, whereas U2OS, HCT116, A549, 833KE and 2102EP are tumor cells. (b) Cells were treated with 8 μM Nutlin, 8 μM MI-63 for 16 hrs or with 10 Gy gamma radiation for 4 hrs. MDMX mRNA level was analyzed by qRT-PCR (n=3).
Figure 3. Accelerated MDMX degradation after MDM2 induction in normal cells.

(a, b, c) Indicated cell lines were treated with 8 μM Nutlin for 16 hrs. Cells were harvested at different time points after 50 μg/ml CHX was added. MDMX degradation was analyzed by Western blot. (d) HFF and U2OS cells were infected for 24 hrs with MDM2 adenovirus at titers (MOI ranging from 45 to 135 and from 15 to 45, respectively) that produce similar levels of MDM2 and analyzed by Western blot. (e) U2OS cells were treated with 8 μM Nutlin for 16 hrs, followed by 10 Gy IR for 4 hrs, and analyzed for MDMX level.
It is note-worthy that a recent study showed that Nutlin treatment induces MDMX degradation in a significant number of tumor cell lines (including HCT116, A549) (Xia et al 2008). Our results above showed that MDMX in HCT116 and A549 were non-responsive to Nutlin or MI-63. The reason for this discrepancy is still unclear. It is possible that we used lower concentrations of Nutlin (8 μM of a racemic mixture) whereas the Xia study used 10 μM of active Nutlin enantiomer. Strong p53 activation and MDM2 induction may lead to other cellular stress that facilitates MDMX degradation.
Proteasome-mediated MDMX degradation after MDM2 induction in normal cells
To determine whether deficiency in MDMX degradation accounts for the inability of tumor cells to down regulate MDMX, we focused on HFF and U2OS as representative normal and tumor cell lines. Nutlin treatment reduced MDMX level in HFF cells in a dose-dependent fashion, but slightly induced MDMX in U2OS (Figure 2a). Inhibition of proteasome by MG132 blocked MDMX down-regulation by Nutlin in HFF cells (Figure 2b), suggesting that MDMX reduction was due to proteasome-mediated degradation. In contrast, MDMX level in U2OS and HCT116 showed little response to MG132, indicating relatively stable MDMX before and after MDM2 induction (Figure 2c).
Figure 2. Proteasome-mediated MDMX degradation after MDM2 induction in normal cells.

(a) HFF and U2OS cells were treated with Nultin at indicated concentrations for 16 hrs. MDMX level was analyzed by Western blot. (b) HFF cells were treated with 8 μM Nutlin for 16 hrs, with or without 30 μM MG132 for the last 4 hrs and analyzed by Western blot. (c) U2OS and HCT116 cells were treated with 8 μM Nutlin for 16 hrs, with or without 30 μM MG132 for the last 4 hrs and analyzed by Western blot. (d) Lentivirus-MDMX was used to stably infect HFF to increase its MDMX level. HFF and HFF-MDMX cells were treated with Nutlin for 16 hrs and MDMX level was analyzed by Western blot.
Tumor cells typically have higher MDMX expression level compared to non-transformed cells. It is possible that excess MDM2 in normal cells allows MDMX degradation, whereas excess MDMX in tumor cells saturates the capacity of MDM2. To test this possibility, lentivirus-MDMX was used to stably infect HFF to increase its MDMX level by 8-fold, >2-fold higher than tumor cells such as HCT116 and U2OS, Supplemental Figure 2a). When treated with Nutlin, HFF-MDMX cells still showed significant MDMX degradation compared to parental cells (Figure 2d), suggesting that normal cells are inherently more capable in degrading MDMX.
MDMX sensitivity to MDM2 is different between normal and tumor cells
To determine whether decrease of MDMX in Nutlin-treated HFF and WI-38 cells were due to accelerated degradation, MDMX degradation rate was analyzed by cycloheximide block. The results showed that MDMX was stable in untreated WI-38 and HFF, but became unstable after Nutlin treatment (Figure 3a, 3b). In contrast, Nutlin did not induce MDMX degradation in U2OS cells (Figure 3c). To further test the effect of MDM2 expression without using Nutlin treatment, WI-38 and U2OS cells were infected with MDM2 adenovirus. The result showed that increased MDM2 expression in HFF also led to MDMX degradation, whereas MDM2 expression in U2OS had negligible effect on MDMX level (Figure 3d).
Previous study showed that DNA damage promotes MDMX degradation without inducing MDM2 level, suggesting that phosphorylation of MDMX increases its sensitivity to MDM2 (Chen et al 2005). As expected, gamma irradiation promoted MDMX degradation in U2OS cells, despite inducing lower MDM2 level than Nutlin. Combination of irradiation and Nutlin resulted in further reduction of MDMX level (Figure 3e). Therefore, simply increasing MDM2 level in U2OS was not effective in degrading MDMX. Additional stress signal such as DNA damage was needed to trigger MDMX degradation.
DNA damage promotes MDMX degradation mainly by Chk2-mediated phosphorylation of S367. MDMX-367A mutant is resistant to DNA damage-induced degradation (Chen et al 2005). We found that stably expressed MDMX-367A was still efficiently degraded in HFF cells after Nutlin treatment (data not shown), thus ruling out a role of MDMX phosphorylation in its sensitivity to MDM2 in normal cells. Together, the results suggest that MDMX is sensitive to MDM2 in normal cells, but is relatively resistant to MDM2 in tumor cells. The results also suggest an important change in MDMX regulation during the transformation process.
ARF expression promotes MDMX degradation in tumor cells
A key difference between normal cells and tumor cells with wild type p53 is the loss of ARF expression due to deletion or silencing of the INK4 locus. To test whether ARF expression in tumor cells restores MDMX sensitivity to MDM2, a U2OS cell line expressing IPTG-inducible ARF (NARF6) was used (Stott et al 1998). U2OS cells have deletion of INK4 locus and are devoid of endogenous ARF (Stott et al 1998). Induction of high level MDM2 alone by Nutlin did not reduce MDMX level in NARF6 cells, similar to the parental U2OS. Induction of ARF by IPTG resulted in p53 activation, MDM2 induction, and moderate reduction of MDMX level. Importantly, simultaneous induction of MDM2 and ARF led to significant reduction of MDMX (Figure 4a). A cycloheximide block experiment showed that MDMX degradation was accelerated after ARF expression (Figure 4b). The effect of ARF was observed at expression levels similar to endogenous ARF in the p53-null H1299 cells (Supplemental Figure 3), suggesting that it can occur at physiological ARF levels. These results suggest that ARF cooperates with MDM2 to regulate MDMX stability.
Figure 4. ARF expression promotes MDMX degradation.

(a) NARF6 cells were treated with 100 μM IPTG for 48 hrs to induce ARF. The cells were then treated with 8 μM Nutlin for 16 hrs and analyzed by Western blot. (b) NARF6 cells were treated with IPTG and Nutlin for 24 hrs. CHX (50 μg/ml) were added and cells were harvested at indicated time points for western blot analysis of MDMX stability. (c) Wild type MEF and ARF-null MEF were treated with 8 μM Nutlin for 16 hrs. Cells were harvested at different time points after CHX was added. MDMX level was analyzed by Western blot. Two background bands in the mouse MDM2 blot are marked by *. (d) Quantitation of MDMX levels in (c) by densitometry.
Next, we tested whether endogenous ARF expression in non-transformed cells contributes to MDMX sensitivity to MDM2. First, we compared MDMX degradation in MEFs derived from wild type and ARF-null mice. The result showed that in the absence of Nutlin treatment, MDMX was more stable in ARF-null MEF than wild type MEF (Figure 4c). Furthermore Nutlin treatment led to more complete MDMX degradation in wild type MEF than ARF-null MEF (Figure 4d). In contrast, gamma irradiation reduced MDMX level in both cell lines (Supplemental Figure 2c). In the second experiment, stable knockdown of endogenous ARF in HFF cells using shRNA also led to stabilization of endogenous MDMX after Nutlin treatment (Supplemental Figure 2d). Therefore, loss of ARF expression reduces MDMX sensitivity to MDM2, suggesting that ARF is an important regulator of MDMX degradation in the absence of DNA damage signals.
MDMX degradation facilitates cell cycle arrest by ARF
To test the functional significance of MDMX degradation in ARF-mediated cell cycle arrest, modified U2OS cell lines expressing high, normal, or reduced levels of MDMX were infected with ARF adenovirus. ARF expression in U2OS inhibited cell proliferation but did not induce apoptosis (not shown). The cell cycle arrest by ARF measured by BrdU incorporation assay was significantly enhanced by knockdown of endogenous MDMX, whereas MDMX-overexpressing cells were completely resistant to ARF-induced arrest (Figure 5b). Western blot analysis showed that U2OS-MDMX cells maintained a high MDMX level after ARF expression despite partial degradation. MDMX overexpression did not prevent p53 stabilization by ARF, but blocked p21 induction by p53 (Figure 5a). Furthermore, MDMX knockdown enhanced p21 induction without increasing p53 level, suggesting that MDMX controls p53 transcriptional activity but not stability in this setting. Therefore, MDMX level has a significant impact on p53 response to ARF. Efficient p53 activation by ARF requires both p53 stabilization (inhibition of MDM2) and elimination of MDMX.
Figure 5. MDMX level has significant impact on ARF-mediated growth arrest.

(a) U2OS cells stably transfected with MDMX cDNA (U2OS-MDMX) or MDMX shRNA (U2OS-shRNA) expression plasmids were infected with adenovirus-ARF at indicated MOI and analyzed by western blot after 24 hrs. (b) Cells with MDMX overexpression or knockdown were infected with adenovirus ARF for 24 hrs. Cells were labeled with BrdU for 2 hrs (n=4) and analyzed for DNA replication rate using a BrdU detection ELISA kit.
ARF stimulates MDMX ubiquitination and degradation by MDM2
Our previous study showed that ARF expression promotes MDMX ubiquitination by MDM2 (Pan and Chen 2003). However, because ARF also simultaneously induced MDM2 stabilization, it was unclear whether it acts simply by increasing MDM2 dosage. The result in Figure 4a suggests that ARF functions through a mechanism different from MDM2 stabilization, since much higher MDM2 level induced by Nutlin failed to degrade MDMX.
To test whether ARF stimulates MDMX ubiquitination by increasing MDM2 specific activity, H1299 cells were transfected with MDMX, MDM2 and His6-ubiquitin. MDMX ubiquitination was detected by Ni-NTA pull down followed by MDMX western blot. MDM2 expression levels in the absence and presence of ARF were matched by reducing the amount of MDM2 plasmids cotransfected with ARF to compensate for stabilization. The results showed that increasing ARF level moderately above endogenous level increased the high molecular weight form of ubiquitinated MDMX, suggesting that ARF stimulates MDMX poly ubiquitination by MDM2 (Figure 6a). The moderate effect of exogenous ARF in this assay is probably due to the presence of endogenous ARF in H1299 cells, and the interference of MDMX phosphorylation from transfection stress (Okamoto et al 2005). Therefore, we performed the assay using the phosphorylation-resistant MDMX-367A mutant in ARF-deficient U2OS cells. As reported previously, poly-ubiquitination of MDMX-367A was inefficient even with high doses of MDM2. Coexpression of ARF resulted in efficient poly-ubiquitination of MDMX-367A in an MDM2 dose-dependent fashion (Figure 6b). MDMX-367A is resistant to degradation by MDM2 after DNA damage or in cotransfection assay due to resistance to phosphorylation by Chk2 (Chen et al 2005). However, cotransfection of ARF led to efficient MDMX-367A degradation by MDM2 (Figure 6c). ARF expression also stimulated the poly ubiquitination of MDMX-342A-367A-403A triple mutant without Chk2 and ATM phosphorylation sites (Supplemental Figure 4a). These results demonstrate that ARF stimulates MDMX ubiquitination through a mechanism different from the phosphorylation-dependent pathway after DNA damage.
Figure 6. ARF stimulates MDMX ubiquitination and degradation by MDM2.

(a) MDMX was co-transfected with MDM2, ARF and His6-ubiquitin into H1299 cells. Ubiquitinated MDMX was detected by Ni-NTA pull down followed by MDMX Western blot. (b) MDMX-367A was co-transfected with MDM2, ARF and His6-ubiquitin into U2OS cells. Ubiquitinated MDMX-367A was detected by Ni-NTA pull down followed by MDMX Western blot. Note the increase of poly ubiquitinated MDMX in the presence of ARF (upper bracket). (c) MDMX-367A was co-transfected with MDM2 and ARF into U2OS cells. MDMX-367A degradation was analyzed by Western blot.
ARF promotes MDMX-MDM2 binding
To determine how ARF stimulates MDMX ubiquitination, an in vitro ubiquitination reaction was performed using recombinant His6-MDM2 and in vitro translated MDMX. The effects of an ARF N terminal 20 amino acid peptide (ARF20) and a scrambled peptide with the same amino acid composition (R20) were added to the reaction to test the effect on MDMX ubiquitination. The ARF20 peptide has been shown to be functionally equivalent to full-length ARF in p53 activation in vivo, and inhibition of p53 ubiquitination in vitro (Midgley et al 2000). The result showed that the ARF20 but not R20 peptide stimulated MDMX ubiquitination (Figure 7a), indicating that the effect of ARF can be recapitulated in vitro using the ARF20 peptide.
Figure 7. ARF stimulates MDMX ubiquitination in vitro.

(a) An ARF N terminal 20 amino acid peptide (ARF20) or a scrambled peptide (R20) were incubated with recombinant His6-MDM2 and in vitro translated MDMX at 37 °C for 30 min followed by incubation with E1, E2 and ubiquitin for 1 hr. MDMX ubiquitination was detected by SDS-PAGE and autofluorography. (b) SJSA cells were treated with Nutlin for 16 hrs. MDM2 was captured on protein A beads using 4B2 antibody. The beads were used to capture in vitro translated MDMX, followed by washing and incubation with E1, E2 and ubiquitin. ARF20 and R20 peptides were added during MDMX capture, or during incubation with E1/E2/ubiquitin. MDMX binding and ubiquitination was analyzed by SDS-PAGE and autofluorography.
To identify the step in the ubiquitination reaction that was stimulated by ARF, MDM2 was immunoprecipitated from SJSA cells and the loaded beads were used to capture in vitro translated MDMX. The pre-formed MDM2-MDMX complex was then incubated with E1, E2 and ubiquitin to initiate the ubiquitination reaction. ARF20 was added during the MDMX binding step, or after binding. The results showed that ARF20 peptide significantly increased the binding of MDMX to immobilized MDM2, resulting in more MDMX-ubiquitin product in the subsequent ubiquitination reaction (Figure 7b). In contrast, if the peptide was added after MDMX capture, no increase in MDMX ubiquitination was detected. Therefore, we conclude that the ARF20 peptide stimulates MDMX ubiquitination by enhancing MDMX-MDM2 binding.
To test whether ARF also stimulates MDMX-MDM2 binding in vivo, its effect was examined in U2OS cells. ARF expression increased the co-precipitation of transfected MDMX with MDM2 (Figure 8a). Endogenous MDMX-MDM2 binding was also stimulated by ARF after IPTG induction in NARF6 cells (Figure 8b). Furthermore, in the absence of ARF, high level MDM2 expression only resulted in a small increase in MDM2-MDMX complex formation, whereas after ARF expression MDM2-MDMX binding became proportional to the dose of MDM2 (Figure 8c). These results suggest that ARF is a critical cofactor that allows MDM2 to bind and degrade MDMX efficiently in the absence of DNA damage.
Figure 8. ARF stimulates MDMX-MDM2 binding.

(a) MDMX was co-transfected with MDM2 and ARF in U2OS cells. Co-precipitation of MDM2 and MDMX was analyzed by MDM2 IP followed by MDMX Western blot. (b) NARF6 cells were treated with IPTG (to induce ARF) or Nutlin (to induce comparable level of MDM2) for 16 hrs and analyzed for endogenous MDMX-MDM2 binding by IP-Western blot. (c) MDMX was co-transfected with MDM2 and ARF into U2OS cells. MG132 was added 4 hrs before the cells were harvested. Co-precipitation of MDM2 and MDMX was analyzed by MDM2 IP followed by MDMX Western blot.
ARF stimulates a second-site interaction between MDM2 and MDMX
To determine how ARF stimulates MDM2-MDMX interaction, we first tested whether ARF interacts with MDMX. Coexpression of ARF with similar amounts of FLAG-MDM2 and FLAG-MDMX showed that only MDM2 was capable of coprecipitating with ARF (Supplemental Figure 4b), consistent with a previous report (Wang et al 2001). This suggests that ARF acts by binding MDM2 and increasing its affinity to MDMX.
MDM2-MDMX interaction is mainly mediated by the C terminal RING domains (Dang et al 2002, Linke et al 2008, Tanimura et al 1999). Unexpectedly, we found that the central region of MDM2 (MDM2-100–361) without the RING domain showed efficient binding to MDMX when co-expressed with ARF. As expected, in the absence of ARF, MDM2-100–361 did not bind MDMX (Figure 9a). Further tests showed that MDM2-100–361 was able to coprecipitate MDMX-100–361 in the presence of ARF (Figure 9b). Additional mapping experiments using epitope-tagged MDM2 and MDMX fragments showed that the MDM2 acidic domain (200–300), and the MDMX central region containing the acidic domain plus zinc finger (100–361) are important for ARF-mediated binding (Supplemental Figure 5). These results suggest that ARF stimulates a novel interaction between MDM2 and MDMX central region, independent of the well-characterized binding interface between C terminal RING domains (Figure 10). This second-site interaction further stabilizes the MDM2-MDMX heterodimer, promoting MDMX degradation.
Figure 9. ARF initiates binding between MDM2 and MDMX acidic domains.

(a) FLAG-tagged central region of MDM2 (100–361 out of 1–491) was coexpressed with MDMX and ARF in H1299 cells. MDM2-100–361 binding to full length MDMX was determined by FLAG IP-MDMX Western blot. (b) FLAG-MDM2-100–361 was coexpressed in H1299 cells with MDMX central region (100–361 out of 1–490) and ARF. Binding between MDM2 and MDMX fragments were determined by FLAG IP-MDMX Western blot.
Figure 10. A model of ARF-stimulated MDM2-MDMX heterodimer formation.

MDM2 and MDMX C terminal RING domains have intrinsic ability to heterodimerize. ARF binds specifically to MDM2 acidic region, causing conformational change and initiates binding to MDMX central domain. ARF may promote direct MDMX-MDM2 contact (left branch) or act as mediator by binding both MDM2 and MDMX (right branch). The second-site interaction between the central regions increases the stability of MDM2-MDMX complex and promotes MDMX ubiquitination.
Discussion
MDM2 is an E3 ligase capable of degrading MDMX in a signal-dependent fashion. Previous studies found that endogenous MDMX is a stable protein in tumor cells, strong insults such as IR or actinomycin D treatment are needed to trigger MDMX degradation (Chen et al 2005, Gilkes et al 2006). MDM2-specific inhibitors activate p53 and in turn induce high-level MDM2 expression. Presumably MDM2 induction should lead to degradation of MDMX, bypassing the need for an MDMX inhibitor. However, our results clearly show that MDMX in a variety of tumor cells is insensitive to changes in MDM2 level, which is different from normal cells. A previous study compared two other human fibroblast cell lines to tumor cells and also showed a difference in MDMX regulation after Nutlin treatment (Wade et al 2006). Together, these observations indicate the presence of a general phenomenon that has biological and clinical relevance.
Our findings suggest that at least two mechanisms contribute to the inability of MDM2 inhibitors to down-regulate MDMX in tumor cells. First, MDMX is more inducible by p53 at the transcriptional level in tumor cells than in normal fibroblasts, resulting in higher MDMX protein level in certain tumor cell lines after p53 activation. Furthermore, there is significant cell type-specificity in p53 sensitivity, i.e., testicular germ cell tumors (2102EP, 833KE) have more inducible MDMX than other tumor cell lines. The mechanism of this cell type-dependence is still not understood. One possibility is the chromatin status of the MDMX P2 promoter may be different in each cell type. Presumably the less differentiated epigenetic profile of germ cell tumors, or the presence of certain transcription factors may be responsible for higher p53 sensitivity.
The second, possibly more important mechanism of MDMX mis-regulation in tumors is ARF deficiency. Loss of ARF during transformation not only disrupts mitogenic stress signaling to MDM2, but also leads to a defect in MDMX degradation. ARF expression renders MDMX highly sensitive to changes in MDM2 levels in part by promoting MDM2-MDMX binding. In the absence of ARF, MDMX becomes resistant to MDM2-mediated degradation and loses response to changes in MDM2 level. The coordinated control of two key p53 regulators may be essential for ARF to activate p53 in normal cells. Interestingly, the current findings and previous studies together show that oncogenic stress, DNA damage, and ribosomal stress all inhibit MDM2 ubiquitination of p53 while stimulating MDMX ubiquitination. This common effect may be evolved to ensure efficient activation of p53 during different stress response. Hyperactive MDM2 and stabilized MDMX in ARF-deficient tumors cooperate to block p53 response to oncogenic stress, acting is an alternative to p53 mutation.
Our results suggest that ARF regulates MDMX degradation by binding specifically to MDM2 acidic region and initiating a second-site interaction between the central regions of MDM2 and MDMX. The secondary interaction cooperates with the RING-mediated hetero-dimerization to further stimulate MDMX degradation. The acidic regions of MDM2 and MDMX are intrinsically unstructured in the absence of binding partners. ARF sequence also has features of an unstructured protein. Interaction between an ARF peptide and the MDM2 acidic region causes significant secondary structure formation in vitro (Bothner et al 2001, Sivakolundu et al 2008). Therefore, our results are consistent with a model that ARF induces a change of MDM2 acidic domain conformation so that it becomes compatible for interacting with the central region of MDMX (Figure 10, left branch). Alternatively, initial ARF-MDM2 interaction may cause ARF to adopt a structure competent for MDMX binding. Thus ARF may act as a mediator to stabilize the MDM2-MDMX heterodimer (Figure 10, right branch). This scenario is favored since the two acidic domains may not directly interact due to strong negative charges, whereas ARF will provide the positive charges needed to form a stable trimeric complex.
It is well-established that ARF not only prevents p53 degradation, but counter intuitively also causes MDM2 stabilization and accumulation. Our results suggest that MDM2 accumulation induced by ARF may serve to promote MDMX degradation, i.e., ARF modulate and re-target the MDM2 E3 activity to further facilitate p53 activation. Therefore, MDM2 can exert negative and positive effects on p53 activity, depending on whether the cells are under stress. This intricate mechanism may explain the ability of ARF to function as key regulator of p53 and its obligatory loss during the development of tumors that express wild type p53.
The abnormal MDMX regulation in tumor cells has direct clinical relevance. MDM2-specific inhibitors are currently being tested in clinical trials (Millard et al 2011). These compounds are designed to target tumors with wild type p53. This population has invariably lost ARF expression due to deletion or gene silencing. P53 activation by targeting MDM2 alone in such tumors will be sub-optimal, or may be ineffective in cases with MDMX overexpression. Targeting MDMX may substantially improve the efficacy of MDM2 inhibitors.
Materials and methods
Cell lines and reagents
The following cell lines were used in the study: A549 (lung tumor), H1299 (lung tumor, p53-null), U2OS (osteosarcoma), HCT116 (colon carcinoma), 2102EP and 833KE (testicular germ cell tumor), WI-38 (human lung fibroblast) and HFF (human foreskin fibroblast), MEF (mouse embryo fibroblast). 833KE and 2102EP cells were provided by Dr. Stuart Lutzker. NARF6 (U2OS expressing IPTG-inducible p14ARF) was provided by Dr. Dawn Quelle. ARF-null MEF was provided by Dr. Yanping Zhang. Cells were maintained in DMEM medium with 10% fetal bovine serum. All MDM2 and MDMX constructs used in this study were human cDNA clones. Nutlin was purchased from Cayman Chemical. MI-63 was re-synthesized based on published structure (Ding et al 2006). Nutlin and MI-63 were used at 5–10 μM. HFF cells with stable expression of MDMX were generated by infection with MDMX lentivirus and selection of pooled cell line. Stable knockdown of ARF was performed using an ARF shRNA retrovirus (V2HS_195839) obtained from Open Biosystems. Cell proliferation was measured using the BrdU labeling and detection kit III (Roche).
Protein analysis
To detect proteins by western blot, cells were lysed in lysis buffer [50 mM Tris-HCl (pH 8.0), 5 mM EDTA, 150 mM NaCl, 0.5% NP40, 1 mM PMSF, 50 mM NaF] and centrifuged for 5 min at 10,000 g. Cell lysate (10–50 μg protein) was fractionated by SDS-PAGE and transferred to Immobilon P filters (Millipore). The filter was blocked for 1 hr with phosphate-buffered saline (PBS) containing 5% non-fat dry milk, 0.1 % Tween-20. The following monoclonal antibodies were used: 4B2 and 2A9 for full-length MDM2 IP (Chen et al 1993); 3G9 for western blot of full-length MDM2 and 4B11 for western blot of MDM2 C terminal fragment; DO-1 (Pharmingen) for p53 western blot; 8C6 antibody for MDMX western blot and IP (Li et al 2002); rabbit polyclonal MDMX antibody for western blot of MDMX deletion mutants; 2A10 for mouse MDM2 western blot; 7A8 for mouse MDMX western blot. p14ARF and p19ARF antibody were purchased from Neomarkers and Abcam, respectively. The filter was developed using Supersignal (Thermo Scientific) or ECL-plus reagent (Amersham).
Quantitative real-time PCR
Cells were treated with 8 μM Nutlin, 8 μM MI-63 for 16 hrs or 10 Gy IR for 4 hrs and mRNA was extracted with RNeasy Mini kit (QIAGEN). Approximately 3 μg mRNA was used as template for reverse transcription by using SuperScript™ III First-Strand synthesis System (Invitrogen). The products were used for real-time PCR using the following primer pairs: MDMX 5′-GCC TTG AGG AAG GAT TGG TA and 5′-TCG ACA ATC AGG GAC ATC AT; MDM2 5′-CCC TTA ATG CCA TTG AAC CT and 5′-CAT ACT GGG CAG GGC TTA TT; Actin 5′-GCT CGT CGT CGA CAA CGG CTC and 5′-CAA ACA TGA TCT GGG TCA TCT TCT C. The relative levels of target genes were determined using actin as an internal control.
In vivo ubiquitination assay
U2OS cells in 10 cm plates were transfected with 5 μg His6-ubiquitin, 5 μg MDMX or MDMX-367A, 1–2 μg MDM2 or MDM2 mutants and 1 μg p14ARF expression plasmids using calcium phosphate precipitation method. Thirty-two hr after transfection, cells from each plate were collected into two aliquots. One aliquot (10%) was used for direct western blot. The remaining cells (90%) were used for purification of His6-tagged proteins by Ni2+-NTA beads. The cell pellet was lysed in buffer A (6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris-Cl pH8.0, 5 mM imidazole, 10 mM β-mecaptoethanol) and incubated with Ni2+-NTA beads (Qiagen) for 4 hr at room temperature. The beads were washed with buffer A, B (8 M urea, 0.1 M Na2PO4/NaH2PO4, 0.01 M Tris-Cl pH8.0, 10 mM β-mecaptoethanol), C (8 M urea, 0.1 M Na2PO4/NaH2PO4, 0.01 M Tris-Cl pH6.3, 10 mM β-mecaptoethanol), and bound proteins were eluted with buffer D (200 mM imidazole, 0.15 M Tris-Cl pH6.7, 30% glycerol, 0.72M β-mercaptoethanol, 5% SDS). The eluted proteins were analyzed by western blot for the presence of conjugated MDMX.
In vitro ubiquitination assay
SJSA cells were treated with 8 μM Nutlin for 16 hrs, then treated with 30 μM MG132 for 4 hrs. MDM2 was immunoprecipitated with 4B2 antibody. The substrate MDMX was produced by in vitro translation in rabbit reticulocyte lysate using the TNT system (Promega) in the presence of 35S-methionine. Fifteen μl packed protein A beads loaded with MDM2 from a 15 cm plate of SJSA cells were washed with lysis buffer and reaction buffer (50 mM Tris pH7.5, 2.5 mM MgCl2, 15 mM KCl, 1 mM DTT, 0.01% Triton X-100, 1% glycerol), incubated with 10 μl of in vitro translated MDMX, with or without ARF20 peptide (MVRRFLVTLRIRRACGPPRV) or R20 scrambled peptide (ALVRTRFRVRRPCVMLGPIR) for 2 hr at 4°C, and washed with reaction buffer. The beads loaded with MDM2-MDMX complex were incubated with 100 ng purified human ubiqutin-activating enzyme His6-E1, 250 ng His6-Ubc5Hb ubiquitin conjugating enzyme, 5 μg ubiquitin (BIOMOL), and 20 μl reaction buffer in the presence of 4 mM ATP. The mixture was incubated at 37°C for 2 hr with shaking, boiled in SDS sample buffer and fractionated by SDS-PAGE. The gel was dried and MDMX was detected by autoradiography.
Supplementary Material
Indicated cell lines were treated with 8 μM Nutlin or 8 μM MI-63 for 16–24 hrs. Changes in MDMX and other indicated markers were analyzed by Western blot. Note that ARF protein is below the detection limit in HFF cells in the absence of oncogenic stress.
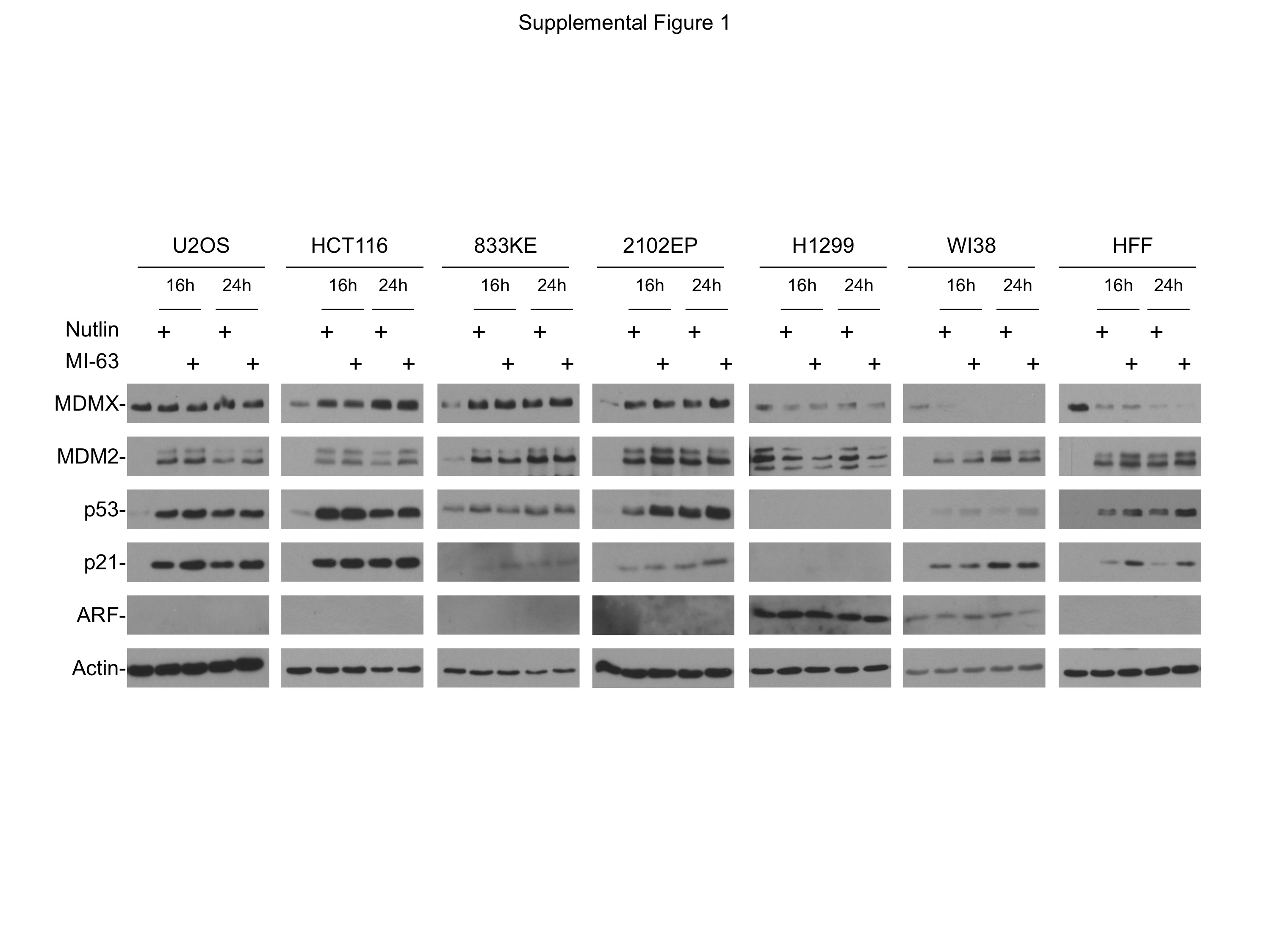{kind=link}
(a) Comparison of MDMX level in lenti-MDMX infected HFF cells to endogenous MDMX in U2OS and HCT116 by Western blot. Equal amounts of total protein were loaded for each cell line. (b) Validation of antibody specificity for detection of mouse MDMX. MEFs of indicated genetic background were analyzed using 7A8 antibody. 41.4 cells are derived from MDMX knock out mouse that expresses an N terminal truncated form of MDMX. (c) Wild type MEF and ARF-null MEF were treated with 10 Gy IR for 4 hrs. MDMX level was analyzed by Western blot using 7A8 antibody. Two background bands in the mouse MDM2 blot (2A10 antibody) are marked by *. (d) Stable knockdown of ARF leads to stabilization of MDMX. HFF cells stably infected with retrovirus expressing ARF shRNA were treated with 8 μM Nutlin for 18 hrs. MDMX stability was determined by cycloheximide block.
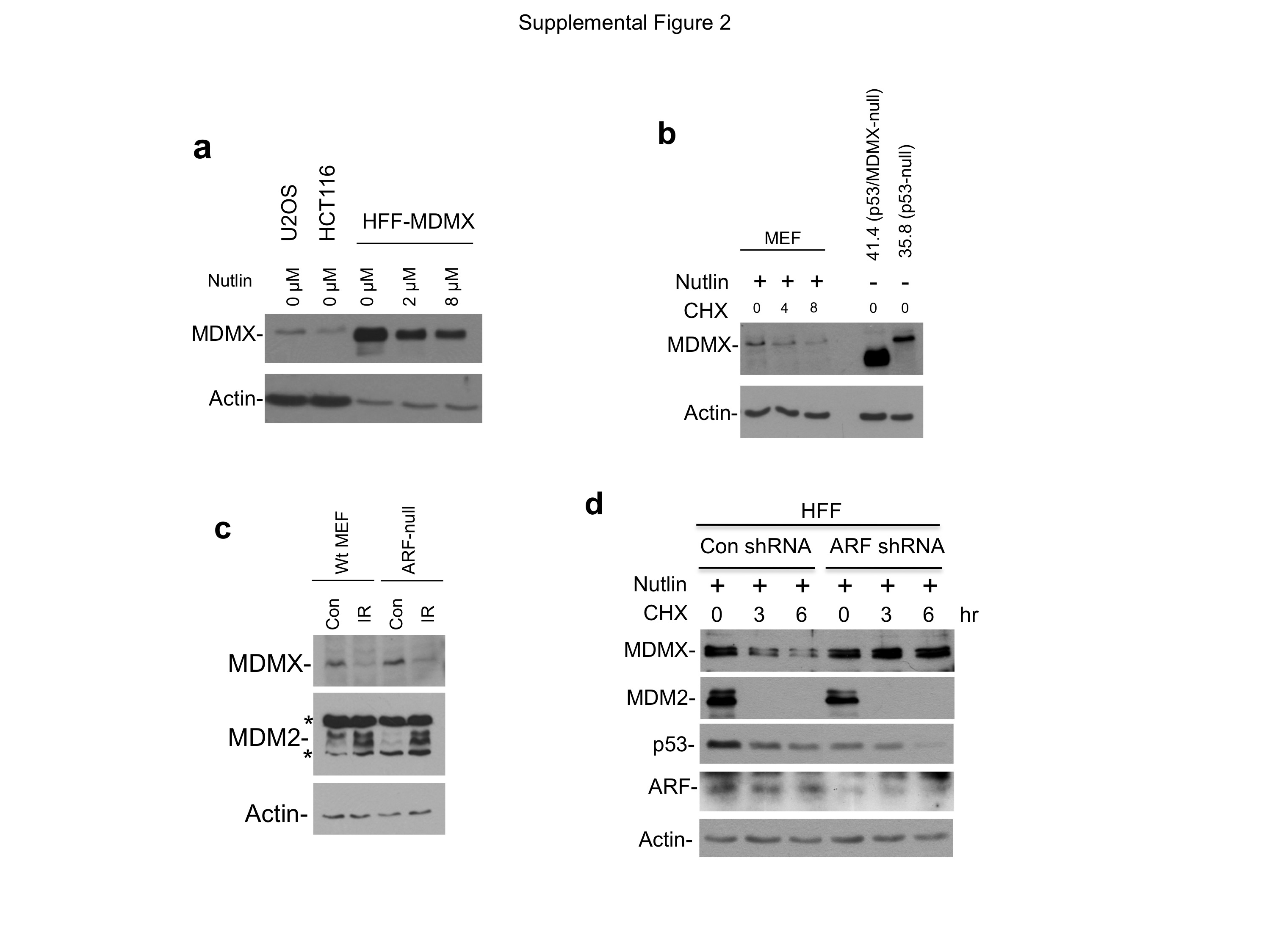{kind=link}
(a) NARF6 cells were treated with IPTG ranging from 12 μM to 50 μM for 48 hrs to induce ARF. The cells were then treated with 8 μM Nutlin for 16 hrs and analyzed by Western blot. (b) NARF6 cells were treated with IPTG for 48 hrs and analyzed by ARF Western blot in comparison to H1299.
{kind=link}
(a) MDMX-342A-367A-403A was co-transfected with MDM2, ARF and His6-ubiquitin into U2OS cells. Ubiquitinated MDMX-342A-367A-403A was detected by Ni-NTA pull down followed by MDMX Western blot. Note the increase of poly ubiquitinated MDMX in the presence of ARF (bracket). (b) ARF was co-transfected with FLAG-MDM2 or FLAG-MDMX into U2OS cells. The ability of MDM2 and MDMX to bind ARF was analyzed by FLAG IP-ARF Western blot.
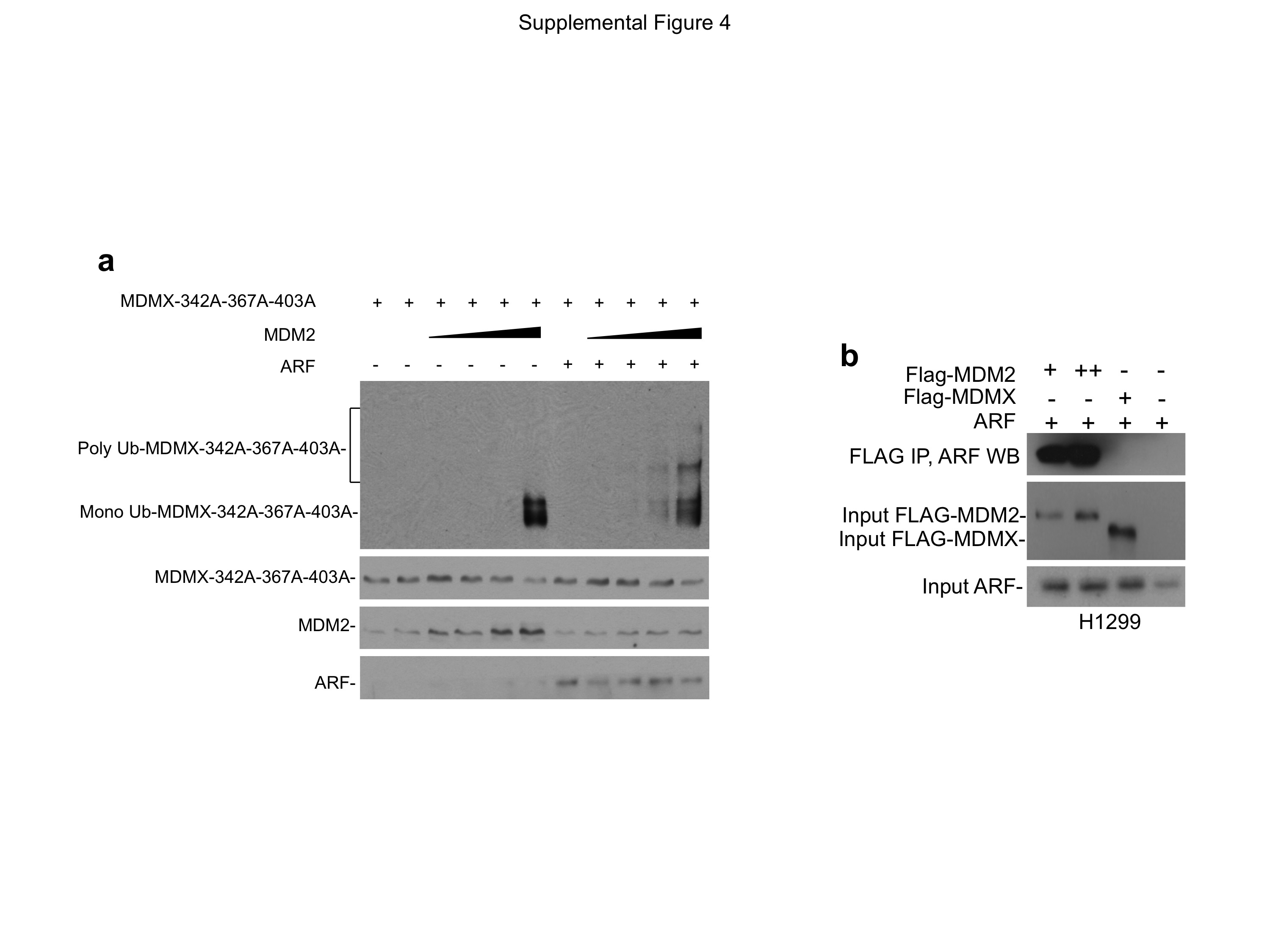{kind=link}
U2OS cells were transfected with ARF, epitope-tagged MDM2 and MDMX fragments. The ability of ARF to stimulate MDM2-MDMX binding was determined by IP-Western blot. MDM2 residue 200–300 (a) and MDMX residue 100–361 (b) are necessary for the interaction in the presence of ARF.
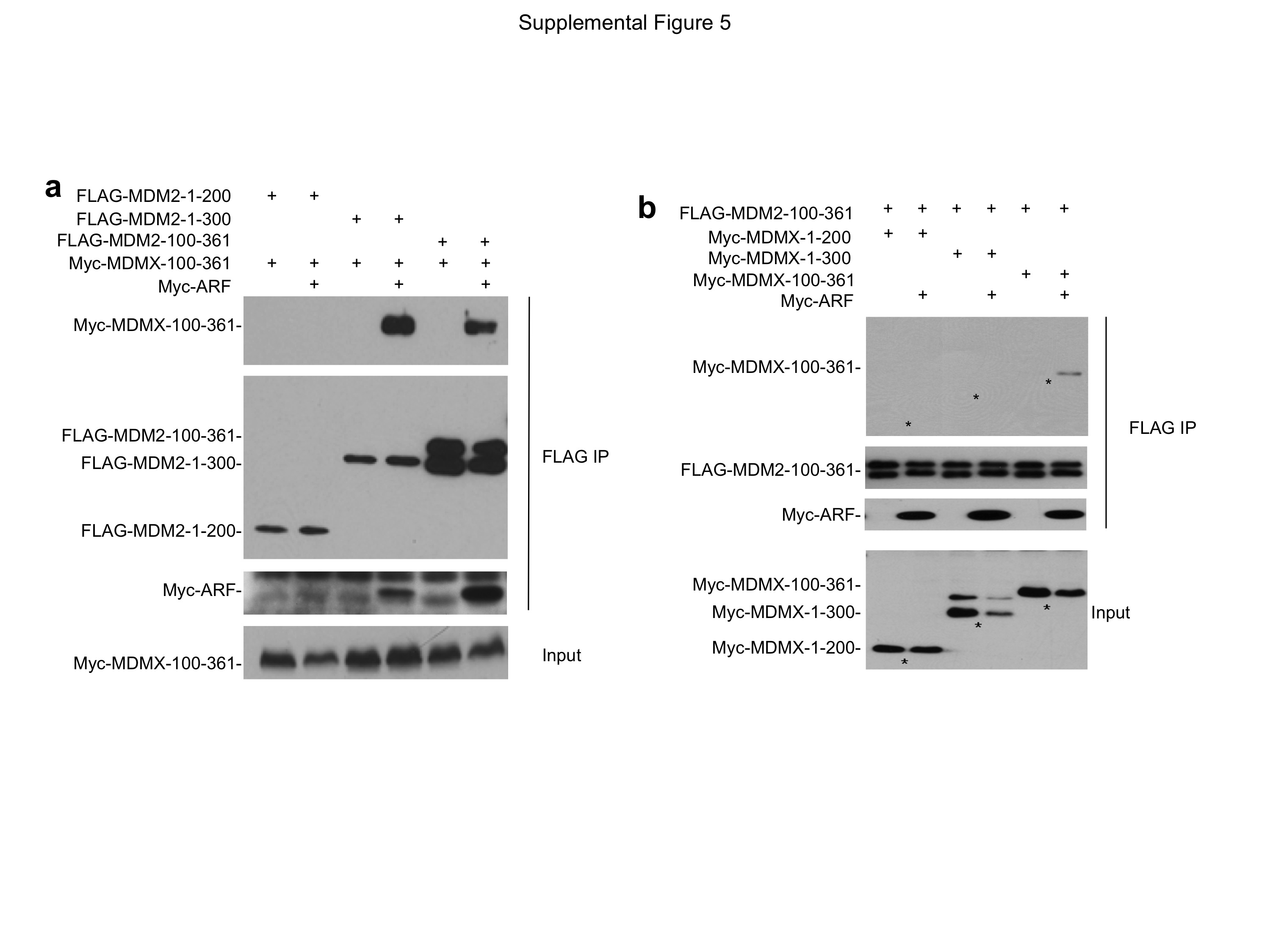{kind=link}
Acknowledgments
We thank Dr. Dawn Quelle for NARF6 cells and Dr. Yanping Zhang for ARF-null MEF. This work was supported in part by grants from the National Institutes of Health (CA141244, CA109636, CA118210).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Badciong JC, Haas AL. MdmX is a RING finger ubiquitin ligase capable of synergistically enhancing Mdm2 ubiquitination. J Biol Chem. 2002;277:49668–49675. doi: 10.1074/jbc.M208593200. [DOI] [PubMed] [Google Scholar]
- Bothner B, Lewis WS, DiGiammarino EL, Weber JD, Bothner SJ, Kriwacki RW. Defining the molecular basis of Arf and Hdm2 interactions. J Mol Biol. 2001;314:263–277. doi: 10.1006/jmbi.2001.5110. [DOI] [PubMed] [Google Scholar]
- Chen D, Shan J, Zhu WG, Qin J, Gu W. Transcription-independent ARF regulation in oncogenic stress-mediated p53 responses. Nature. 2010;464:624–627. doi: 10.1038/nature08820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Marechal V, Levine AJ. Mapping of the p53 and mdm-2 interaction domains. Mol Cell Biol. 1993;13:4107–4114. doi: 10.1128/mcb.13.7.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Gilkes DM, Pan Y, Lane WS, Chen J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. Embo J. 2005;24:3411–3422. doi: 10.1038/sj.emboj.7600812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Chen L, Li Z, Lane WS, Chen J. ATM activates p53 by regulating MDM2 oligomerization and E3 processivity. EMBO J. 2009;28:3857–3867. doi: 10.1038/emboj.2009.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross B, Chen L, Cheng Q, Li B, Yuan ZM, Chen J. Inhibition of p53 DNA Binding Function by the MDM2 Protein Acidic Domain. The Journal of biological chemistry. 2011;286:16018–16029. doi: 10.1074/jbc.M111.228981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang J, Kuo ML, Eischen CM, Stepanova L, Sherr CJ, Roussel MF. The RING domain of Mdm2 can inhibit cell proliferation. Cancer Res. 2002;62:1222–1230. [PubMed] [Google Scholar]
- Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol. 2004;24:5835–5843. doi: 10.1128/MCB.24.13.5835-5843.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, et al. Structure-based design of spiro-oxindoles as potent, specific small-molecule inhibitors of the MDM2-p53 interaction. J Med Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- Gilkes DM, Chen L, Chen J. MDMX regulation of p53 response to ribosomal stress. Embo J. 2006;25:5614–5625. doi: 10.1038/sj.emboj.7601424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grier JD, Xiong S, Elizondo-Fraire AC, Parant JM, Lozano G. Tissue-specific differences of p53 inhibition by Mdm2 and Mdm4. Mol Cell Biol. 2006;26:192–198. doi: 10.1128/MCB.26.1.192-198.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, et al. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002;277:19251–19254. doi: 10.1074/jbc.C200150200. [DOI] [PubMed] [Google Scholar]
- Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J Biol Chem. 2006;281:33030–33035. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, et al. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell. 2007;12:355–366. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Kawai H, Wiederschain D, Kitao H, Stuart J, Tsai KK, Yuan ZM. DNA damage-induced MDMX degradation is mediated by MDM2. J Biol Chem. 2003a;278:45946–45953. doi: 10.1074/jbc.M308295200. [DOI] [PubMed] [Google Scholar]
- Kawai H, Wiederschain D, Yuan ZM. Critical contribution of the MDM2 acidic domain to p53 ubiquitination. Mol Cell Biol. 2003b;23:4939–4947. doi: 10.1128/MCB.23.14.4939-4947.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie NA, Donovan SL, Shih CS, Zhang J, Mills N, Fuller C, et al. Inactivation of the p53 pathway in retinoblastoma. Nature. 2006;444:61–66. doi: 10.1038/nature05194. [DOI] [PubMed] [Google Scholar]
- Li B, Cheng Q, Li Z, Chen J. p53 inactivation by MDM2 and MDMX negative feedback loops in testicular germ cell tumors. Cell Cycle. 2010;9:1411–1420. doi: 10.4161/cc.9.7.11255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Chen L, Chen J. DNA damage induces MDMX nuclear translocation by p53-dependent and -independent mechanisms. Mol Cell Biol. 2002;22:7562–7571. doi: 10.1128/MCB.22.21.7562-7571.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci U S A. 2003;100:12009–12014. doi: 10.1073/pnas.2030930100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linke K, Mace PD, Smith CA, Vaux DL, Silke J, Day CL. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 2008;15:841–848. doi: 10.1038/sj.cdd.4402309. [DOI] [PubMed] [Google Scholar]
- Macias E, Jin A, Deisenroth C, Bhat K, Mao H, Lindstrom MS, et al. An ARF-independent c-MYC-activated tumor suppression pathway mediated by ribosomal protein-Mdm2 Interaction. Cancer Cell. 18:231–243. doi: 10.1016/j.ccr.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maetens M, Doumont G, Clercq SD, Francoz S, Froment P, Bellefroid E, et al. Distinct roles of Mdm2 and Mdm4 in red cell production. Blood. 2007;109:2630–2633. doi: 10.1182/blood-2006-03-013656. [DOI] [PubMed] [Google Scholar]
- Meulmeester E, Frenk R, Stad R, de Graaf P, Marine JC, Vousden KH, et al. Critical role for a central part of Mdm2 in the ubiquitylation of p53. Mol Cell Biol. 2003;23:4929–4938. doi: 10.1128/MCB.23.14.4929-4938.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley CA, Desterro JM, Saville MK, Howard S, Sparks A, Hay RT, et al. An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene. 2000;19:2312–2323. doi: 10.1038/sj.onc.1203593. [DOI] [PubMed] [Google Scholar]
- Millard M, Pathania D, Grande F, Xu S, Neamati N. Small-Molecule Inhibitors of p53-MDM2 Interaction: The 2006–2010 Update. Current pharmaceutical design. 2011;17:536–559. doi: 10.2174/138161211795222649. [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, Lozano G. Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature. 1995;378:203–206. doi: 10.1038/378203a0. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Kashima K, Pereg Y, Ishida M, Yamazaki S, Nota A, et al. DNA damage-induced phosphorylation of MdmX at serine 367 activates p53 by targeting MdmX for Mdm2-dependent degradation. Mol Cell Biol. 2005;25:9608–9620. doi: 10.1128/MCB.25.21.9608-9620.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Chen J. MDM2 promotes ubiquitination and degradation of MDMX. Mol Cell Biol. 2003;23:5113–5121. doi: 10.1128/MCB.23.15.5113-5121.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet. 2001;29:92–95. doi: 10.1038/ng714. [DOI] [PubMed] [Google Scholar]
- Patton JT, Mayo LD, Singhi AD, Gudkov AV, Stark GR, Jackson MW. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to Nutlin-3. Cancer Res. 2006;66:3169–3176. doi: 10.1158/0008-5472.CAN-05-3832. [DOI] [PubMed] [Google Scholar]
- Pereg Y, Shkedy D, de Graaf P, Meulmeester E, Edelson-Averbukh M, Salek M, et al. Phosphorylation of Hdmx mediates its Hdm2- and ATM-dependent degradation in response to DNA damage. Proc Natl Acad Sci U S A. 2005;102:5056–5061. doi: 10.1073/pnas.0408595102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips A, Teunisse A, Lam S, Lodder K, Darley M, Emaduddin M, et al. HDMX-L is expressed from a functional p53-responsive promoter in the first intron of the HDMX gene and participates in an autoregulatory feedback loop to control p53 activity. J Biol Chem. 2010;285:29111–29127. doi: 10.1074/jbc.M110.129726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos YF, Stad R, Attema J, Peltenburg LT, van der Eb AJ, Jochemsen AG. Aberrant expression of HDMX proteins in tumor cells correlates with wild-type p53. Cancer Res. 2001;61:1839–1842. [PubMed] [Google Scholar]
- Sasaki M, Nie L, Maki CG. MDM2 binding induces a conformational change in p53 that is opposed by heat-shock protein 90 and precedes p53 proteasomal degradation. J Biol Chem. 2007;282:14626–14634. doi: 10.1074/jbc.M610514200. [DOI] [PubMed] [Google Scholar]
- Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, et al. Temporal activation of p53 by a specific MDM2 inhibitor is selectively toxic to tumors and leads to complete tumor growth inhibition. Proc Natl Acad Sci U S A. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6:663–673. doi: 10.1038/nrc1954. [DOI] [PubMed] [Google Scholar]
- Shvarts A, Bazuine M, Dekker P, Ramos YF, Steegenga WT, Merckx G, et al. Isolation and identification of the human homolog of a new p53-binding protein, Mdmx. Genomics. 1997;43:34–42. doi: 10.1006/geno.1997.4775. [DOI] [PubMed] [Google Scholar]
- Sivakolundu SG, Nourse A, Moshiach S, Bothner B, Ashley C, Satumba J, et al. Intrinsically unstructured domains of Arf and Hdm2 form bimolecular oligomeric structures in vitro and in vivo. J Mol Biol. 2008;384:240–254. doi: 10.1016/j.jmb.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stott FJ, Bates S, James MC, McConnell BB, Starborg M, Brookes S, et al. The alternative product from the human CDKN2A locus, p14(ARF), participates in a regulatory feedback loop with p53 and MDM2. Embo J. 1998;17:5001–5014. doi: 10.1093/emboj/17.17.5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimura S, Ohtsuka S, Mitsui K, Shirouzu K, Yoshimura A, Ohtsubo M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999;447:5–9. doi: 10.1016/s0014-5793(99)00254-9. [DOI] [PubMed] [Google Scholar]
- Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, Lozano G. Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol Cell Biol. 2007;27:5479–5485. doi: 10.1128/MCB.00555-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Wade M, Wong ET, Tang M, Stommel JM, Wahl GM. Hdmx modulates the outcome of p53 activation in human tumor cells. J Biol Chem. 2006;281:33036–33044. doi: 10.1074/jbc.M605405200. [DOI] [PubMed] [Google Scholar]
- Wang X, Arooz T, Siu WY, Chiu CH, Lau A, Yamashita K, et al. MDM2 and MDMX can interact differently with ARF and members of the p53 family. FEBS Lett. 2001;490:202–208. doi: 10.1016/s0014-5793(01)02124-x. [DOI] [PubMed] [Google Scholar]
- Wang YV, Leblanc M, Wade M, Jochemsen AG, Wahl GM. Increased radioresistance and accelerated B cell lymphomas in mice with Mdmx mutations that prevent modifications by DNA-damage-activated kinases. Cancer Cell. 2009;16:33–43. doi: 10.1016/j.ccr.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Bayle JH, Olson D, Levine AJ. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Xia M, Knezevic D, Tovar C, Huang B, Heimbrook DC, Vassilev LT. Elevated MDM2 boosts the apoptotic activity of p53-MDM2 binding inhibitors by facilitating MDMX degradation. Cell Cycle. 2008;7 doi: 10.4161/cc.7.11.5929. [DOI] [PubMed] [Google Scholar]
- Xiong S, Van Pelt CS, Elizondo-Fraire AC, Liu G, Lozano G. Synergistic roles of Mdm2 and Mdm4 for p53 inhibition in central nervous system development. Proc Natl Acad Sci U S A. 2006;103:3226–3231. doi: 10.1073/pnas.0508500103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong S, Pant V, Suh YA, Van Pelt CS, Wang Y, Valentin-Vega YA, et al. Spontaneous tumorigenesis in mice overexpressing the p53-negative regulator Mdm4. Cancer Res. 2010;70:7148–7154. doi: 10.1158/0008-5472.CAN-10-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009;16:369–377. doi: 10.1016/j.ccr.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Indicated cell lines were treated with 8 μM Nutlin or 8 μM MI-63 for 16–24 hrs. Changes in MDMX and other indicated markers were analyzed by Western blot. Note that ARF protein is below the detection limit in HFF cells in the absence of oncogenic stress.
(a) Comparison of MDMX level in lenti-MDMX infected HFF cells to endogenous MDMX in U2OS and HCT116 by Western blot. Equal amounts of total protein were loaded for each cell line. (b) Validation of antibody specificity for detection of mouse MDMX. MEFs of indicated genetic background were analyzed using 7A8 antibody. 41.4 cells are derived from MDMX knock out mouse that expresses an N terminal truncated form of MDMX. (c) Wild type MEF and ARF-null MEF were treated with 10 Gy IR for 4 hrs. MDMX level was analyzed by Western blot using 7A8 antibody. Two background bands in the mouse MDM2 blot (2A10 antibody) are marked by *. (d) Stable knockdown of ARF leads to stabilization of MDMX. HFF cells stably infected with retrovirus expressing ARF shRNA were treated with 8 μM Nutlin for 18 hrs. MDMX stability was determined by cycloheximide block.
(a) NARF6 cells were treated with IPTG ranging from 12 μM to 50 μM for 48 hrs to induce ARF. The cells were then treated with 8 μM Nutlin for 16 hrs and analyzed by Western blot. (b) NARF6 cells were treated with IPTG for 48 hrs and analyzed by ARF Western blot in comparison to H1299.
(a) MDMX-342A-367A-403A was co-transfected with MDM2, ARF and His6-ubiquitin into U2OS cells. Ubiquitinated MDMX-342A-367A-403A was detected by Ni-NTA pull down followed by MDMX Western blot. Note the increase of poly ubiquitinated MDMX in the presence of ARF (bracket). (b) ARF was co-transfected with FLAG-MDM2 or FLAG-MDMX into U2OS cells. The ability of MDM2 and MDMX to bind ARF was analyzed by FLAG IP-ARF Western blot.
U2OS cells were transfected with ARF, epitope-tagged MDM2 and MDMX fragments. The ability of ARF to stimulate MDM2-MDMX binding was determined by IP-Western blot. MDM2 residue 200–300 (a) and MDMX residue 100–361 (b) are necessary for the interaction in the presence of ARF.