

Abstract
The Polycomb transcription repressor BMI1 is highly expressed in human neuroblastomas and is required for the clonogenic self-renewal and tumorigenicity of human neuroblastoma cell lines. The molecular basis of BMI1 action in neuroblastoma cells is not well understood. Here we report that BMI1 has a critical role in stabilizing cyclin E1 by repressing the expression of FBXW7, a substrate-recognition subunit of the SCF E3 ubiquitin ligase that targets cyclin E1 for degradation. BMI1 binds to the FBXW7 locus in vivo and represses its mRNA expression. Overexpression of cyclin E1 or abrogation of FBXW7 induction rescues the cell-death phenotype of BMI1 knockdown. Moreover, MYCN, an oncoprotein in the pathogenesis of high-risk neuroblastomas, is able to counteract the death-inducing effect of BMI1 knockdown by activating CCNE1 transcription. We further show that high cyclin E1 expression is associated with Stage 4 neuroblastomas and poor prognosis in patients. These findings suggest a molecular mechanism for the oncogenic activity of BMI1 and MYCN in neuroblastoma pathogenesis and progression by maintaining cyclin E1 levels.
Keywords: BMI1; cyclin E1; FBXW7, MYCN; Neuroblastoma
Introduction
BMI1 is a member of the Polycomb Group family of transcriptional repressors that was originally identified as an oncogenic partner of c-Myc in murine lymphomagenesis (Haupt et al 1991, van Lohuizen et al 1991). It is a component of the Polycomb repressive complex 1 required for transcriptional repression of genes important in development and differentiation (Sauvageau and Sauvageau 2010, Schwartz and Pirrotta 2007, Valk-Lingbeek et al 2004). BMI1 has diverse biological functions as demonstrated by phenotypic analysis of Bmi1-/- mice. These mice display defects in axial skeleton patterning, hematopoiesis, and neural development (van der Lugt et al 1994). Later studies have revealed a critical role of BMI1 in the self-renewal of normal and cancer stem cells (Iwama et al 2004, Lessard and Sauvageau 2003, Molofsky et al 2003, Park et al 2003).
A key downstream target of BMI1 is the Ink4a-ARF locus that encodes two tumor suppressors, p16Ink4a and p14ARF (p19ARF in the mouse) (Jacobs et al 1999a, Park et al 2004). p16Ink4a inhibits the cyclin D-CDK4/6 kinase responsible for phosphorylation of pRb during the cell cycle. The resulting hypophosphorylated pRb binds E2F and represses its transcriptional activation of the genes that promote S-phase entry, leading to cell cycle arrest and senescence. p14ARF inhibits MDM2, which targets p53 for ubiquitin-dependent degradation, leading to accumulation of p53 and transcriptional activation of its target genes that promote cell cycle arrest, senescence, and apoptosis (Lowe and Sherr 2003). Importantly, inactivation of the Ink4a-ARF locus or individual p16Ink4a and p19ARF partially rescues the self-renewal and frequency of stem cells in the central and peripheral nervous systems in Bmi1-/- mice (Bruggeman et al 2005, Molofsky et al 2003, Molofsky et al 2005), demonstrating that repression of the Ink4a-ARF locus is critical for Bmi1 to maintain stem cells. However, the partial rescue of Bmi1-/- phenotype by ablation of p16Ink4a and p19ARF also suggests the involvement of additional target genes for the biological functions of BMI1.
BMI1 is highly expressed in human neuroblastomas and neuroblastoma cell lines (Cui et al 2006, Cui et al 2007, Nowak et al 2006, Ochiai et al 2010). Neuroblastoma is a common childhood malignant tumor of the sympathetic nervous system that arises in paravertebral sympathetic ganglia and the adrenal medulla (Brodeur 2003). Both tissues originate from neural crest cells, a transient, highly migratory population of multipotent stem cells that require BMI1 for their self-renewal (Molofsky et al 2003, Molofsky et al 2005). We have recently demonstrated an essential role of BMI1 in the maintenance of the clonogenic self-renewal and tumorigenicity of human neuroblastoma cell lines (Cui et al 2006, Cui et al 2007). Moreover, we have shown that Bmi-1 cooperates with MYCN in transformation of avian neural crest cells by inhibiting the pro-apoptotic activity of MYCN (Cui et al 2007). Amplification of the oncogene MYCN, which occurs in approximately 22% of neuroblastoma cases, is strongly associated with highly malignant behavior and poor prognosis (Brodeur 2003, Maris and Matthay 1999, Schwab 2004).
In this report, we present evidence for cyclin E1 as a common downstream target of BMI1 and MYCN in promoting the survival of neuroblastoma cells. MYCN transcriptionally activates the expression of cyclin E1, whereas BMI1 represses the transcription of FBXW7 to block cyclin E1 degradation. These findings provide a molecular mechanism for maintaining high cyclin E1 expression, which is associated with poor outcome and disease progression in neuroblastoma patients.
Results
Individual neuroblastoma cells display differential sensitivities to BMI1 knockdown
To investigate the molecular basis of BMI1 action in neuroblastoma cells, we used an RNAi-based approach for knockdown of BMI1 expression in BE(2)-C cells, a human neuroblastoma cell line enriched for cells capable of clonogenic self-renewal in a BMI1-dependent manner (Cui et al 2006). We tested four retroviral constructs expressing shRNA sequences against different regions of the human BMI1 gene, and two of them (BMI1sh-48576 and-48580) were highly effective in knockdown of BMI1 expression (Supplementary Figure 1a) and had a similar inhibitory effect on tumor cell clonogenicity (Supplementary Figure 1b). These findings confirmed our previous observation of a critical role of BMI1 in maintaining the clonogenicity of neuroblastoma cells (Cui et al 2006, Cui et al 2007) and demonstrated the functional specificity of these BMI1 shRNA sequences.
As BMI1 is critical for maintaining the clonogenicity of neuroblastoma cells (Cui et al 2006, Cui et al 2007), constitutive knockdown of BMI1 may select a minor population of BE(2)-C cells that either fail to express BMI1 shRNA or acquire additional genetic or epigenetic mutations to bypass the requirement of BMI1. To minimize this possibility, we generated a “Tet-Off” system for inducible expression of the BMI1sh-48576 sequence in BE(2)-C cells and conducted our investigation either with single-cell-derived clones or with a pool of these clones. We examined 20 single-cell-derived clones that showed marked downregulation of BMI1 protein levels in the absence of doxycycline. Approximately two-third of these clones (n=14) were relatively resistant to BMI1 knockdown (referred to below as BMI1-resistant clones). As represented by clone-7 and -12 cells, BMI1 knockdown had no apparent effect on their proliferation in culture (Figure 1a and 1b), but significantly reduced their ability to grow in soft agar, as judged by both the number and size of colonies (Figure 1c). By contrast, cells of the remaining 6 clones (numbers 2, 9, 11, 15, 20, and 22) were extremely sensitive to BMI1 knockdown (referred to below as BMI1-sensitive clones), showing a marked decrease in clonogenicity (Figure 1d-e). Following BMI1 knockdown, these cells progressively rounded up and detached from the culture matrix (Figure 1f-g). The cell death phenotype induced by the BMI1sh-48576 sequence, which targets the 3’ UTR of human BMI1 mRNA (Supplementary Figure 2a), could be effectively rescued by retroviral expression of the BMI1 coding sequence (Supplementary Figure 2b and 2c), demonstrating that the effect is a result of BMI1 silencing. Together, these findings reveal differential sensitivities of individual BE(2)-C cells to the reduction in BMI1 levels.
Figure 1.

Individual BE(2)-C neuroblastoma cells display differential sensitivities to BMI1 knockdown. a, Immunoblot analysis of BMI1 levels in clone-7 and -12 cells cultured in the presence or absence of doxycycline (Doxy) for 3 days. BMI1 levels were quantified against α-tubulin and are presented as the fraction of the BMI1 level in the presence of Doxy. b, Phase contrast imaging of clone-7 and -12 cells cultured in the presence or absence of Doxy for 10 days. Scale bars, 100 μm. c, Soft agar clonogenic assay of clone-7 and -12 cells in the presence or absence of Doxy. Numbers of total colonies and of the colonies that were larger than 0.5 mm were determined (error bars, s.d., n=3). d, Immunoblot analysis of BMI1 levels in BMI1-sensitive clones cultured in the presence or absence of Doxy for 3 days. BMI1 levels were quantified against α-tubulin and are presented as the fraction of the BMI1 level in the presence of Doxy. e, Soft agar clonogenic assay of parental and luciferase shRNA-expressing BE(2)-C cells, and BMI1-sensitive clones in the presence or absence of Doxy. Numbers of total colonies were determined (error bars, s.d., n=3). All quantitative data from soft agar assays were analyzed using two-tailed Student's t-test with the p values indicated. f, Phase contrast imaging of BMI1-sensitive clones cultured in the presence or absence of Doxy for 3 days. g, Phase contrast imaging of the clone-2 cells cultured in the absence of Doxy for 0-6 days. Scale bars (f-g), 100 μm.
BMI1 knockdown induces non-apoptotic cell death independently of p16Ink4a and p14ARF
We next focused our investigation on the cell death-inducing effect of BMI1 knockdown. It has been shown previously that BMI1 can promote cell survival by suppressing apoptosis (Jacobs et al 1999b, Yamashita et al 2008). However, the majority of the dying cells following BMI1 knockdown displayed no morphologic features of apoptosis, such as cell shrinkage and membrane blebbing (Figure 1f). Propidium iodide flow cytometry analysis of BMI1-sensitive clone-2 and -9 cells following BMI1 knockdown revealed no significant numbers of cells with sub-G1 DNA content (Supplementary Figure 3a), a characteristic of apoptotic cells (Riccardi and Nicoletti 2006). We also observed no significant levels of membrane annexin V staining during BMI1 knockdown-induced cell death (Supplementary Figure 3b). Annexin V binds externalized phosphatidylserine on the plasma membrane, an early event in apoptosis (Martin et al 1995). Finally, treatment of clone-2 and -9 cells with the cell permeable pan-caspase inhibitor zVAD-FMK failed to prevent BMI1 knockdown-induced cell death although it was highly effective in protecting cells from Fas-mediated apoptosis (Supplementary Figure 3c). Together, these data indicate that BMI1 knockdown induces predominantly non-apoptotic cell death in a subset of neuroblastoma cells.
The isolation of neuroblastoma cell clones that were dependent on BMI1 for survival provided a highly sensitive functional assay system for the identification of relevant BMI1 downstream target genes. We first examined p16Ink4a and p14ARF, two well-documented target genes of BMI1 (Sauvageau and Sauvageau 2010, Valk-Lingbeek et al 2004). BMI1 knockdown had no significant effect on p16Ink4a protein levels but did result in a marked increase in the protein levels of p14ARF (Supplementary Figure 4a). However, overexpression of p14ARF in both clone-2 and -9 cells (in the absence of BMI1 knockdown) had no effect on their survival and proliferation in soft agar and in culture (Supplementary Figure 4b-d). Together, these results indicate that BMI1 knockdown induces non-apoptotic cell death in a manner independent of p16Ink4a and p14ARF.
BMI1 suppresses non-apoptotic cell death by maintaining cyclin E1 levels
The observation that BMI1 knockdown led to an accumulation of S-phase cells (Supplementary Figure 3a) prompted us to examine the effect BMI1 knockdown on the expression of major components of the cell cycle machinery in BMI1-sensitive cells. Immunoblotting analysis revealed that BMI1 knockdown consistently resulted in a rapid and marked decrease in cyclin E1 levels, but had no or modest effect on the levels of other cyclins and CDKs (Figure 2a and Supplementary Figure 5a). Knockdown of cyclin E1 by shRNA recapitulated the cell-death phenotype of BMI1 knockdown (Figure 2b and 2c). Conversely, overexpression of cyclin E1 abrogated the cell death-inducing effect of BMI1 knockdown (Figure 2d and 2e and Supplementary Figure 5b). These data demonstrate that maintaining cyclin E1 levels is a major mechanism for the pro-survival activity of BMI1.
Figure 2.

BMI1 suppresses cell death by stabilizing cyclin E1. a, Immunoblot analysis of cyclins and CDKs in pooled BMI1-sensitive clones following BMI1 knockdown. α-tubulin levels are shown as loading control. b, Immunoblot analysis of cyclin E1 levels in BE(2)-C cells either uninfected (parental) or infected with retroviruses expressing shRNA sequences against GFP (GFPsh) or different regions of CCNE1 (CCNE1sh-80 and -81). Cyclin E1 levels were quantified against α-tubulin and are presented as the fraction of the cyclin E1 level in parental cells. c, Crystal violet staining of BE(2)-C cells expressing either GFPsh or CCNE1sh-80. d, Immunoblot analysis of BMI1 and cyclin E1 levels in pooled BMI1-sensitive clones infected with retroviruses expressing either GFP or Myc-cyclin E1 and cultured in the presence or absence of Doxy for 3 days. α-tubulin levels are shown as loading control. e, Phase contrast imaging of pooled BMI1-sensitive clones expressing either GFP or Myc-cyclin E1 and cultured in the presence or absence of Doxy for 6 days. Scale bars, 100 μm. f, qRT-PCR analysis of CCND1 and CCNE1 mRNA levels in pooled BMI1-sensitive cells cultured in the presence or absence of Doxy for 3 days (error bars, s.d., n=3). g, Quantification of cyclin E1 half-life in pooled BMI1-sensitive clones cultured in the presence or absence of Doxy for 3 days. Samples were collected at various time points following addition of cycloheximide (CHX) for immunoblot analysis. Cyclin E1 levels were quantified against α-tubulin and are presented as the fraction of the initial levels at time zero (error bars, s.d., n=4). h, In vivo ubiquitination assay of pooled BMI1-sensitive cells cultured in the presence or absence of Doxy for 3 days and co-transfected with Flag-ubiquitin and Myc-cyclin E1 expression plasmids. Polyubiquitinated cyclin E1 was detected by immunoprecipitation of Myccyclin E1, followed by immunoblotting for Flag-ubiquitin.
Quantitative RT-PCR revealed that BMI1 knockdown had no effect on cyclin E1 mRNA levels (Figure 2f), but led to a marked decrease (3.2-6.7 fold) in its protein half-life (Figure 2g and Supplementary Figure 5c). Consistent with this result was our observation that BMI1 knockdown resulted in a marked increase in the level of cyclin E1 polyubiquitination (Figure 2h). Together, these data indicate that BMI1 maintains cyclin E1 levels by blocking its degradation.
BMI1 stabilizes cyclin E1 by repressing the expression of FBXW7
We next examined the molecular mechanism by which BMI1 maintains cyclin E1 levels. Two E3 ubiquitin ligases, the SKP/CUL/F-box (SCF) protein complex and the anaphase-promoting complex/cyclosome, are responsible for ubiquitination of cyclins. The specificity is conferred by substrate-recognition subunits of these complexes, including SKP2 (cyclins A, D, and E), FBXW7 (cyclin E), CDC20 (cyclins A and B), and CDH1/FZR1 (cyclins A and B) (Nakayama and Nakayama 2006). BMI1 knockdown significantly increased the mRNA level of FBXW7, but had no effect on the mRNA level of SKP2 (Figure 3a). In addition, chromatin immunoprecipitation (ChIP) and qPCR assays revealed a locus-wide association of BMI1 with the FBXW7 gene including the proximal promoter region (-1 kb to the transcription start site, TSS) and all FBXW7 introns (Figure 3b). Thus, FBXW7 is a direct target gene of BMI1. Moreover, abrogation of FBXW7 induction by shRNA restored cyclin E1 levels and rescued the cell-death phenotype of BMI1 knockdown (Figure 3c-e and Supplementary Figure 6). Collectively, these results indicate that BMI1 suppresses cell death by directly repressing FBXW7 expression to maintain cyclin E1 levels in neuroblastoma cells.
Figure 3.

BMI1 represses FBXW7 expression. a, qRT-PCR analysis of FBXW7 and SKP2 mRNA levels in pooled BMI1-sensitive cells cultured in the presence or absence of Doxy for 3 days (error bars, s.d., n=3). b, ChIP-qPCR analysis showing association of BMI1 with the FBXW7 locus in pooled BMI1-sensitive clones in the presence of Doxy (error bars, s.d., n=3). The association of BMI1 with HOXA2, a known BMI1 target gene, is shown as positive control. BMI1 does not bind to the CCNB1 promoter, which is shown as negative control. c, qRT-PCR analysis of FBXW7 mRNA levels in pooled BMI-sensitive cells either uninfected or infected with lentiviruses expressing shRNA against GFP or different regions of FBXW7 and cultured in the presence or absence of Doxy for 3 days (error bars, s.d., n=3). d, Immunoblot analysis of cyclins A2 and E1 levels in the cells described in (c). Cyclin E1 levels were quantified against α-tubulin and are presented as the fraction of the cyclin E1 level in the presence of Doxy. e, Crystal violet staining of pooled BMI-sensitive cells expressing GFP-shRNA, FBXW7-shRNA-55 or -56 and cultured in the presence or absence of Doxy for 6 days.
MYCN counteracts the effect of BMI1 knockdown by activating cyclin E1 transcription
It was recently reported that FBXW7 can ubiquitinate MYCN, thereby targeting it for degradation (Otto et al 2009). This prompted us to investigate whether FBXW7 induction, as a result of BMI1 knockdown, could lead to downregulation of MYCN in BE(2)-C cells, which carry MYCN amplification and express significant levels of MYCN protein (Cui et al 2007). Surprisingly, immunoblot analysis revealed that all BMI1-sensitive clones expressed no detectable levels of MYCN, even in the absence of BMI1 knockdown (Figure 4a, clone-2 and -9, and 4d, pooled clones). This was in contrast to parental, Tet repressor-expressing, and BMI1-resistant BE(2)-C cells, which all expressed high levels of MYCN (Figure 4a). BMI1 knockdown in the resistant clone-7 and -12 cells was able to upregulate FBXW7 mRNA levels but had no significant effect on MYCN and cyclin E1 protein levels (Figure 4b and 4c). Also interesting was the observation that when pooled BMI1-sensitive cells were cultured under the condition of BMI1 knockdown for an extended period of time, a minor population of cells survived and eventually became proliferative again. These cells showed re-expression of MYCN and cyclin E1 (Figure 4d), indicating a selective pressure for upregulation of MYCN and cyclin E1 when the BMI1 level is low. In addition, overexpression of MYCN in BMI1-sensitive cells effectively prevented cyclin E1 downregulation and cell death induced by BMI1 knockdown (Figure 4e and 4f and Supplementary Figure 7). These observations demonstrate that MYCN confers resistance to BMI1 knockdown and provide a molecular mechanism for the differential sensitivities of individual BE(2)-C cells to BMI1 knockdown. This conclusion was further supported by studies with additional human neuroblastoma cell lines. SK-N-DZ and SK-N-F1 cells expressed high and no detectable levels of MYCN, respectively (Supplementary Figure 8a). BMI1 knockdown was able to upregulate FBXW7 mRNA levels in both cell lines (Supplementary Figure 8b). However, only SK-N-F1 cells were sensitive to BMI1 knockdown, showing significant cyclin E1 downregulation and cell death (Supplementary Figure 8a and 8c).
Figure 4.

MYCN counteracts the effect of BMI1 knockdown. a, Immunoblot analysis of MYCN and BMI1 levels in parental and Tet repressor (Tet-Off)-expressing BE(2)-C cells, and in individual BE(2)-C clones with inducible expression of BMI1 shRNA. The cells were cultured in the presence of Doxy. α-tubulin levels are shown as loading control. b, Immunoblot analysis of BMI1, MYCN and cyclin E1 levels in BMI1-resistant clone-7 and -12 cells cultured in the presence or absence of Doxy for 4 days. β-actin levels are shown as loading control. c, qRT-PCR analysis of FBXW7 mRNA levels in clone-7 and -12 cells cultured in the presence or absence of Doxy for 3 days (error bars, s.d., n=3). d, Immunoblot analysis of BMI1, MYCN and cyclin E1 levels in pooled BMI1-sensitive cells cultured in the presence or absence of Doxy for 5 and 35 days, respectively. β-actin levels are shown as loading control. e, Immunoblot analysis of BMI1, MYCN and cyclin E1 levels in pooled BMI1-sensitive cells infected with retroviruses expressing either GFP or MYCN and cultured in the presence or absence of Doxy for 4 days. β-actin levels are shown as loading control. f, Phase contrast imaging of pooled BMI1-sensitive cells expressing either GFP or MYCN and cultured in the presence or absence of Doxy for 6 days. Scale bars, 100 μm.
It is well documented that MYC proteins can transcriptionally activate cyclin E1 expression, either directly or indirectly (Jansen-Durr et al 1993, Perez-Roger et al 1997, Steiner et al 1995). Consistent with these earlier findings, overexpression of MYCN in SHEP1 neuroblastoma cells, which express very low levels of endogenous MYCN (Cui et al 2005), upregulated cyclin E1 at both the mRNA and protein levels (Figure 5a and 5b). Also, primary human neuroblastomas with MYCN amplification expressed significantly higher levels of cyclin E1 mRNA than neuroblastomas without MYCN amplification (Figure 5c). Together, these findings suggest that MYCN counteracts the effect of BMI1 knockdown by activating CCNE1 transcription.
Figure 5.

MYCN activates CCNE1 transcription. a, qRT-PCR analysis of cyclin mRNA levels in SHEP1 cells infected with retroviruses expressing either GFP or MYCN (error bars, s.d., n=3). Two-tailed Student's t-test analysis revealed only significant upregulation of CCNE1 mRNA. b, Immunoblot analysis of cyclin E1 and MYCN levels in SHEP1 cells expressing either GFP or MYCN. Levels of cyclin E1 were quantified against β-actin with the cyclin E1 level in GFP-expressing SHEP1 cells being designated as 1.0. c, Scatter dot plots of CCNE1 expression levels in primary neuroblastomas with or without MYCN amplification. The data were analyzed using two-tailed Student's t-test with p values indicated.
High cyclin E1 expression is prognostic for poor outcome and disease progression in neuroblastoma patients
As both BMI1 and MYCN target cyclin E1 to promote the survival of neuroblastoma cells, we investigated the possibility of cyclin E1 as a prognostic marker for disease progression and clinical outcomes in neuroblastoma patients. Kaplan-Meier analysis of progression-free survival for the Oberthuer dataset, which includes a cohort of 251 neuroblastoma patients (Oberthuer et al 2006), showed that high cyclin E1 expression was associated with poor outcome, whereas low HOXC9 expression was associated with good prognosis (Figure 6a). We confirmed that high cyclin E1 expression is prognostic for unfavorable outcome with the Khan dataset (Figure 6b), which includes a cohort of 56 neuroblastoma patients (Chen et al 2008, Wei et al 2004). Importantly, in both datasets, only Stage 4 tumors displayed significantly higher levels of cyclin E1 expression (Figure 6c and 6d), demonstrating a positive correlation between cyclin E1 expression levels and tumor progression. Together, our analysis of two independent cohorts of neuroblastoma patients indicates that high cyclin E1 expression is prognostic for poor outcome and disease progression.
Figure 6.

High CCNE1 expression is associated with Stage 4 neuroblastomas and poor outcome in neuroblastoma patients. a-b, Kaplan-Meier analysis of progression-free survival for Oberthuer (a) and Khan (b) datasets with log-rank test p values indicated. The CCNE1 expression cutoff value 0 (a) or 0.031 (b) was determined by the online Oncogenomics algorithm, which separated the patients into high and low CCNE1 expression groups. c-d, Scatter dot plot of CCNE1 expression levels in stage (ST) 1-4S (c) or 1-4 (d) tumors. Data were analyzed using two-tailed Student`s t-test with p values indicated (ST1 vs ST2, 3, 4 or 4S).
Discussion
BMI1 is highly expressed in human neuroblastomas and neuroblastoma cell lines and is essential for the clonogenic self-renewal and tumorigenicity of neuroblastoma cells (Cui et al 2006, Cui et al 2007, Nowak et al 2006), suggesting a critical role of BMI1 in neuroblastoma development. By examining the sensitivity of single-cell-derived clones to BMI1 knockdown, our study has revealed a pro-survival activity of BMI1 in suppression of non-apoptotic cell death in neuroblastoma cells. This action of BMI1 depends on its ability to maintain cyclin E1 levels by directly repressing the expression of FBXW7, which targets cyclin E1 for ubiquitination and degradation. We further show that MYCN, an oncoprotein critical in the pathogenesis of high-risk neuroblastomas (Brodeur 2003, Maris and Matthay 1999, Schwab 2004), is able to counteract the effect of BMI1 knockdown by activating cyclin E1 transcription. Thus, BMI1 and MYCN maintain cyclin E1 levels in neuroblastoma cells through distinct mechanisms: MYCN transcriptionally activates the expression of cyclin E1, whereas BMI1 represses the transcription of FBXW7 to block cyclin E1 degradation (Figure 7). In this regard, it is interesting to note that a recent study has identified BMI1 as a direct target gene of MYCN (Ochiai et al 2010), suggesting a mechanism for reinforcing and maintaining cyclin E1 levels in neuroblastoma cells. These findings identify cyclin E1 as a common target of two important oncogenic pathways in the pathogenesis of neuroblastoma.
Figure 7.
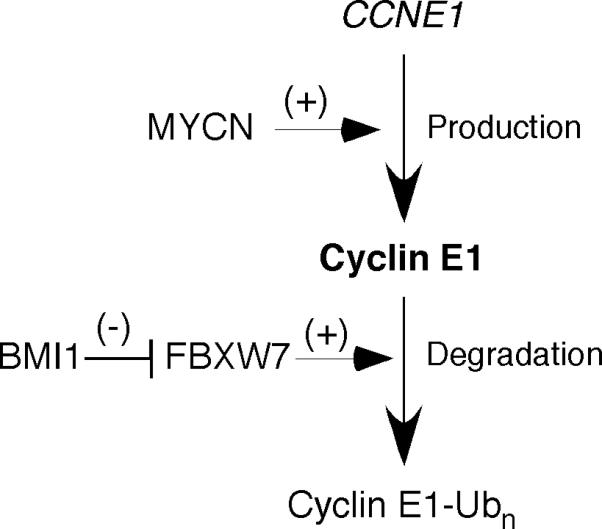
A model for the distinct mechanisms by which BMI1 and MYCN maintain cyclin E1 levels in neuroblastoma cells.
Our study has also revealed that high cyclin E1 expression is significantly associated with Stage 4 tumors and poor outcome, suggesting an important role of cyclin E1 in neuroblastoma progression. Cyclin E1 is known as a positive regulator of G1/S transition, DNA replication, and S-phase progression (Hwang and Clurman 2005, Satyanarayana and Kaldis 2009). Cyclin E1-CDK2 phosphorylates and inactivates pRb, leading to the release of E2F, which in turn promotes S-phase entry. Cyclin E1 also has a critical role in the binding of MCM proteins to replication origins and in centrosome duplication. Moreover, high cyclin E1 expression promotes genetic instability and aneuploidy (Minella et al 2002, Spruck et al 1999), which is generally thought to contribute to tumor progression by increasing genetic diversity among tumor cells (Thompson et al 2010). In this study, we have identified an essential role of cyclin E1 in the survival of neuroblastoma cells. Thus, cyclin E1 could promote neuroblastoma progression through multiple mechanisms. Together, these findings suggest that maintenance of cyclin E1 levels is likely an important mechanism for the oncogenic activity of BMI1 and MYCN in neuroblastoma pathogenesis and progression.
Previous studies have shown that BMI1 can promote cell survival by suppressing apoptosis, which is mediated, at least in part, through the ARF-MDM2-p53 pathway (Jacobs et al 1999a, Park et al 2004). Although BMI1 knockdown in BE(2)-C cells resulted in a marked increase in the protein levels of p14ARF, these cells carry mutated p53 (Kaghad et al 1997). This may be one of the reasons why BMI1 knockdown failed to induce apoptosis in BE(2)-C cells. Probably as a result, we were able to identify a novel mechanism by which BMI1 promotes cell survival by maintaining the cyclin E1 levels. The molecular basis for cyclin E1 depletion-induced non-apoptotic cell death in neuroblastoma cells remains to be determined. Notably, cyclin E1 knockdown resulted in a marked increase in the number of S-phase cells. Given the critical role of cyclin E1 in the control of DNA replication (Hwang and Clurman 2005, Satyanarayana and Kaldis 2009), we speculate that depletion of cyclin E1 may lead to an accumulation of the cells with incomplete DNA synthesis, which is known to trigger replication stress and non-apoptotic cell death (Ma et al 2011). Regardless of mechanistic details, our observations that high cyclin E1 expression is associated with Stage 4 neuroblastomas, particularly those with MYCN amplification, and is essential for the survival of neuroblastoma cells provide a strong rationale for targeting cyclin E1 as a therapeutic strategy for high-risk neuroblastomas.
FBXW7 is a general tumor suppressor and loss-of-function mutations of FBXW7 have been identified in approximately 6% of human cancers (Crusio et al 2010, Onoyama and Nakayama 2008, Welcker and Clurman 2008). In addition to cyclin E1, a large number of oncoproteins has been identified as the substrates of FBXW7, including Notch, c-MYC, c-Jun, Aurora A, GSK3-β, c-Myb, and mTor. More recently, it has been shown that FBXW7 is able to ubiquitinate MYCN and target it for degradation (Otto et al 2009), suggesting a potential tumor suppressor function for FBXW7 in neuroblastoma development. By repressing FBXW7 expression, BMI1 may stabilize these oncoproteins, and some of them might function as downstream effectors of BMI1 in promoting the clonogenic self-renewal and tumorigenicity of neuroblastoma cells. For example, inhibition of Notch signaling induces growth arrest and differentiation of neuroblastoma cells (Chang et al 2010, Ferrari-Toninelli et al 2010), suggesting a critical role of Notch signaling in the maintenance of the stem cell or progenitor state of neuroblastoma cells. In addition to neuroblastoma pathogenesis, we speculate that BMI1-mediated repression of FBXW7 may represent a common oncogenic mechanism in cancer development.
Materials and Methods
Cell culture
The human neuroblastoma cell lines BE(2)-C (CRL-2268, ATCC, Manassas, VA), SK-N-DZ (CRL-2149, ATCC) and SK-N-F1 (CRL-2141, ATCC) were cultured in DMEM and Ham's nutrient mixture F12 (1:1) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA). BE(2)-C, SK-N-DZ and SK-N-F1 cells with a Tet-Off inducible system were established as previously described (Mao et al 2011). Cells were examined and phase contrast images captured using an Axio Observer microscope and AxioVision software (Carl Zeiss MicroImaging, Thornwood, NY).
Overexpression and RNA interference
Human cDNAs for BMI1 (Cui et al 2006), MYCN (Cui et al 2005), p14ARF (Wei et al 2001), CCNE1, and FBXW7 (Open Biosystems, Huntsville, AL) were cloned into the retroviral vector pBabe-hygro. The GFP retroviral construct pBabe-hygro-GFP was used as control. Retroviral (pSM2) constructs expressing shRNA to human BMI1 (RHS4529-NM_005180) were purchased from Open Biosystems and the shRNA-coding sequences were subcloned into the retroviral shRNAmir Tet-Off vector (Silva et al 2008) for inducible expression. The luciferase shRNAmir Tet-Off construct was used as control. Retroviruses were produced in 293FT cells using the packaging plasmids pHDM-G and pMD.MLVogp. Lentiviral constructs expressing shRNA to human FBXW7 (RHS4533-NM_001013435) were purchased from Open Biosystems and to GFP (pLKO.1-GFPshRNA) from Addgene (Cambridge, MA). Lentiviruses were produced in 293FT cells using the packaging plasmids pLP1, pLP2, and pLP/VSVG (Invitrogen).
Immunoblot analysis
Immunoblotting was conducted according to standard procedures using the following primary antibodies: mouse anti-BMI1 (05-637, 1:500; Upstate, Millipore, Billerica, MA), rabbit anti-cyclin A2 (sc-751, 1:200), mouse anti-cyclin B1 (sc-245, 1:200), mouse anti-cyclin D1 (sc-20044, 1:200), mouse anti-cyclin E1 (sc-56310, 1:200), rabbit anti-cyclin E2 (ab40890, 1:2000; Abcam, Cambridge, MA), mouse anti-Flag tag (F-4042, 1:2000; Sigma-Aldrich), mouse anti-MYCN (OP13, 1:200; Calbiochem, EMD Biosciences, San Diego, CA), mouse anti-Myc tag (9E10, hybridoma supernatant, 1:10), rabbit anti-p14ARF (A300-340A, 1:500; Bethyl Laboratories, Montgomery, TX), rabbit anti-p16Ink4a (sc-468, 1:200), rabbit anti-β-actin (600-401-886, 1:2000; Rockland Immunochemicals, Gilbertsville, PA), and mouse anti-α-tubulin (B-5-1-2, 1:5000; Sigma-Aldrich). Unless indicated, all primary antibodies were purchased from Santa Cruz Biotechnology. Horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA) were used as secondary antibodies. Proteins were visualized using a SuperSignal West Pico chemiluminescence kit (Pierce, Rockford, IL) and quantified using ImageJ (version 1.42q). Films were exposed for various times for quantification of target proteins within their linear range of detection. Whenever possible, protein levels were also quantified using the Odyssey system and software (LI-COR Biosciences, Lincoln, NE).
Quantitative reverse-transcription PCR (qRT-PCR)
Cells were lysed with Trizol (Invitrogen) for total RNA purification. Reverse transcription was performed using SuperScript II Reverse Transcriptase (Invitrogen). PCR in triplicate was performed using an iQ5 real-time PCR system (Bio-Rad, Hercules, CA), a TaqMan gene expression assay kit (Applied Biosystems, Foster City, CA), and the following TaqMan probe and primers: cyclin A2 (Hs00153138_m1), cyclin B1 (Hs00259126_m1), cyclin D1 (Hs00277039_m1), cyclin E1 (Hs00233356_m1), cyclin E2 (Hs00180319_m1), FBXW7 (Hs00217794_m1), SKP2 (Hs001806634_m1), and GAPDH (Hs99999905_m1).
Fluorescence-activated cell sorting (FACS)
For cell cycle analysis, cells were fixed with 70% ethanol, incubated with ribonuclease A (Sigma-Aldrich, St. Louis, MO), and stained with 20 μg/ml propidium iodide (Invitrogen). Annexin V staining was performed using a PE Annexin V Apoptosis Detection Kit (BD Pharmingen, San Diego, CA). Samples were analyzed with a FACSCalibur system (BD Biosciences, San Jose, CA) and ModFitLT V3.2.1 software.
Protein half-life assay
Cells with Tet-Off inducible expression of BMI1 shRNA were cultured in the presence or absence of doxycycline for 3 days, followed by addition of cycloheximide (Sigma-Aldrich, St. Louis, MO) to the final concentration of 10 μg/ml. Samples were then collected at various time points for immunoblot analysis of cyclin E1 levels, which were quantified against α-tubulin using ImageJ (version 1.42q) or the Odyssey Software (LI-COR Biosciences) as described above.
In vivo ubiquitination assay
Cells with Tet-Off inducible expression of BMI1 shRNA were cultured in the presence or absence of doxycycline for 2 days and transfected with pFlag-ubiquitin and pKMyc-cyclin E1. One day after transfection, cells were pre-treated with 20 μM MG-132 for 4 h, washed with PBS and lyzed with RIPA buffer containing 20 μM MG-132 and 10 mM N-Ethylmaleimide (both from Sigma-Aldrich). Lysates were cleared by centrifugation and supernatants were collected, followed by addition of SDS to the final concentration of 1% (v/v). After boiling for 5 min to dissociate proteins, samples were diluted 1:10 with dissociation dilution buffer (50 mM HEPES, pH 7.2, 120 mM NaCl, 1% Triton-X 100, 0.5% deoxycholate, and 1 mM PMSF). Myc–cyclin E1 was immunoprecipitated with mouse anti-Myc tag (clone 4A6, Millipore, Billerica, MA) and Protein A/G Plus beads (Santa Cruz Biotechnology). Immunoprecipitates were washed and analyzed by immunoblotting as described above.
Soft agar clonogenic assay
Cells (~2000/well) were mixed with 0.3% Noble agar in growth medium and plated onto six-well plates containing a solidified bottom layer (0.6% Noble agar in growth medium). After 14 to 21 days, colonies were stained with 5 mg/ml MTT (Sigma-Aldrich), photographed and counted.
Chromatin Immunoprecipitation (ChIP)
Anti-BMI1 ChIP was performed on pooled BMI1-senitive cells cultured in the presence or absence of doxycycline for 3 days, using a monoclonal antibody to BMI1 (clone AF27, Millipore) or control mouse IgG according to the online protocol (http://jura.wi.mit.edu/young_public/hESregulation/ChIP.html). ChIP DNA in triplicate was analyzed by qPCR. Primer sequences are listed in Supplementary Table 1.
Analyses of Patient Data
Patient data used in this study were described previously (Asgharzadeh et al 2006, Oberthuer et al 2006). Gene expression datasets were obtained from the Oncogenomics Database (http://pob.abcc.ncifcrf.gov/cgi-bin/JK) (Chen et al 2008). Kaplan-Meier analysis was conducted online, and the resulting survival curves and p values (log-rank test) were downloaded. All cutoff values for separating high and low expression groups were determined by the online Oncogenomics algorithm (Chen et al 2008).
Supplementary Material
Acknowledgment
We thank Q. Du for pKMyc-cyclinE1, G. Hannon and J. Silva for shRNAmir Tet-Off, R. Mulligan for pHDM-G and pMD.MLVogp, W. Wei for pBabe-puro-p14ARF, and J. Khan for gene expression datasets from the Oncogenomics Database. This work was supported by the NIH grant CA124982 and a Georgia Cancer Coalition Distinguished Scholar Award to H.-F.D., and a grant from the National Basic Research Program of China (No. 2012cb114603) to H.C. L.M. was supported in part by a grant from the National Science Foundation of China (NSFC 81101905).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- Asgharzadeh S, Pique-Regi R, Sposto R, Wang H, Yang Y, Shimada H, et al. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. J Natl Cancer Inst. 2006;98:1193–1203. doi: 10.1093/jnci/djj330. [DOI] [PubMed] [Google Scholar]
- Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- Bruggeman SW, Valk-Lingbeek ME, van der Stoop PP, Jacobs JJ, Kieboom K, Tanger E, et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005;19:1438–1443. doi: 10.1101/gad.1299305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H-H, Lee H, Hu M-K, Tsao P-N, Juan H-F, Huang M-C, et al. Notch1 Expression Predicts an Unfavorable Prognosis and Serves as a Therapeutic Target of Patients with Neuroblastoma. Clinical Cancer Research. 2010;16:4411–4420. doi: 10.1158/1078-0432.CCR-09-3360. [DOI] [PubMed] [Google Scholar]
- Chen QR, Song YK, Wei JS, Bilke S, Asgharzadeh S, Seeger RC, et al. An integrated cross-platform prognosis study on neuroblastoma patients. Genomics. 2008;92:195–203. doi: 10.1016/j.ygeno.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crusio KM, King B, Reavie LB, Aifantis I. The ubiquitous nature of cancer: the role of the SCF(Fbw7) complex in development and transformation. Oncogene. 2010;29:4865–4873. doi: 10.1038/onc.2010.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Li T, Ding HF. Linking of N-Myc to death receptor machinery in neuroblastoma cells. J Biol Chem. 2005;280:9474–9481. doi: 10.1074/jbc.M410450200. [DOI] [PubMed] [Google Scholar]
- Cui H, Ma J, Ding J, Li T, Alam G, Ding HF. Bmi-1 regulates the differentiation and clonogenic self-renewal of I-type neuroblastoma cells in a concentration-dependent manner. J Biol Chem. 2006;281:34696–34704. doi: 10.1074/jbc.M604009200. [DOI] [PubMed] [Google Scholar]
- Cui H, Hu B, Li T, Ma J, Alam G, Gunning WT, et al. Bmi-1 is essential for the tumorigenicity of neuroblastoma cells. Am J Pathol. 2007;170:1370–1378. doi: 10.2353/ajpath.2007.060754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari-Toninelli G, Bonini SA, Uberti D, Buizza L, Bettinsoli P, Poliani PL, et al. Targeting Notch pathway induces growth inhibition and differentiation of neuroblastoma cells. Neuro-Oncology. 2010;12:1231–1243. doi: 10.1093/neuonc/noq101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65:753–763. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24:2776–2786. doi: 10.1038/sj.onc.1208613. [DOI] [PubMed] [Google Scholar]
- Iwama A, Oguro H, Negishi M, Kato Y, Morita Y, Tsukui H, et al. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 2004;21:843–851. doi: 10.1016/j.immuni.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999a;397:164–168. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Scheijen B, Voncken JW, Kieboom K, Berns A, van Lohuizen M. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999b;13:2678–2690. doi: 10.1101/gad.13.20.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen-Durr P, Meichle A, Steiner P, Pagano M, Finke K, Botz J, et al. Differential modulation of cyclin gene expression by MYC. Proc Natl Acad Sci U S A. 1993;90:3685–3689. doi: 10.1073/pnas.90.8.3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell. 1997;90:809–819. doi: 10.1016/s0092-8674(00)80540-1. [DOI] [PubMed] [Google Scholar]
- Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003;13:77–83. doi: 10.1016/s0959-437x(02)00013-8. [DOI] [PubMed] [Google Scholar]
- Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends in Molecular Medicine. 2011;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao L, Ding J, Zha Y, Yang L, McCarthy BA, King W, et al. HOXC9 Links Cell-Cycle Exit and Neuronal Differentiation and Is a Prognostic Marker in Neuroblastoma. Cancer Res. 2011;71:4314–4324. doi: 10.1158/0008-5472.CAN-11-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris JM, Matthay KK. Molecular biology of neuroblastoma. J Clin Oncol. 1999;17:2264–2279. doi: 10.1200/JCO.1999.17.7.2264. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minella AC, Swanger J, Bryant E, Welcker M, Hwang H, Clurman BE. p53 and p21 Form an Inducible Barrier that Protects Cells against Cyclin E-cdk2 Deregulation. Current biology : CB. 2002;12:1817–1827. doi: 10.1016/s0960-9822(02)01225-3. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Pardal R, Iwashita T, Park IK, Clarke MF, Morrison SJ. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 2003;425:962–967. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 2005;19:1432–1437. doi: 10.1101/gad.1299505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- Nowak K, Kerl K, Fehr D, Kramps C, Gessner C, Killmer K, et al. BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res. 2006;34:1745–1754. doi: 10.1093/nar/gkl119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberthuer A, Berthold F, Warnat P, Hero B, Kahlert Y, Spitz R, et al. Customized oligonucleotide microarray gene expression-based classification of neuroblastoma patients outperforms current clinical risk stratification. J Clin Oncol. 2006;24:5070–5078. doi: 10.1200/JCO.2006.06.1879. [DOI] [PubMed] [Google Scholar]
- Ochiai H, Takenobu H, Nakagawa A, Yamaguchi Y, Kimura M, Ohira M, et al. Bmi1 is a MYCN target gene that regulates tumorigenesis through repression of KIF1Bbeta and TSLC1 in neuroblastoma. Oncogene. 2010;29:2681–2690. doi: 10.1038/onc.2010.22. [DOI] [PubMed] [Google Scholar]
- Onoyama I, Nakayama KI. Fbxw7 in cell cycle exit and stem cell maintenance: insight from gene-targeted mice. Cell cycle (Georgetown, Tex. 2008;7:3307–3313. doi: 10.4161/cc.7.21.6931. [DOI] [PubMed] [Google Scholar]
- Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell. 2009;15:67–78. doi: 10.1016/j.ccr.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004;113:175–179. doi: 10.1172/JCI20800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Roger I, Solomon DL, Sewing A, Land H. Myc activation of cyclin E/Cdk2 kinase involves induction of cyclin E gene transcription and inhibition of p27(Kip1) binding to newly formed complexes. Oncogene. 1997;14:2373–2381. doi: 10.1038/sj.onc.1201197. [DOI] [PubMed] [Google Scholar]
- Riccardi C, Nicoletti I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nat Protoc. 2006;1:1458–1461. doi: 10.1038/nprot.2006.238. [DOI] [PubMed] [Google Scholar]
- Satyanarayana A, Kaldis P. Mammalian cell-cycle regulation: several Cdks, numerous cyclins and diverse compensatory mechanisms. Oncogene. 2009;28:2925–2939. doi: 10.1038/onc.2009.170. [DOI] [PubMed] [Google Scholar]
- Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M. MYCN in neuronal tumours. Cancer Lett. 2004;204:179–187. doi: 10.1016/S0304-3835(03)00454-3. [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet. 2007;8:9–22. doi: 10.1038/nrg1981. [DOI] [PubMed] [Google Scholar]
- Silva JM, Marran K, Parker JS, Silva J, Golding M, Schlabach MR, et al. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319:617–620. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruck CH, Won K-A, Reed SI. Deregulated cyclin E induces chromosome instability. Nature. 1999;401:297–300. doi: 10.1038/45836. [DOI] [PubMed] [Google Scholar]
- Steiner P, Philipp A, Lukas J, Godden-Kent D, Pagano M, Mittnacht S, et al. Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. Embo J. 1995;14:4814–4826. doi: 10.1002/j.1460-2075.1995.tb00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Bakhoum SF, Compton DA. Mechanisms of Chromosomal Instability. Current biology : CB. 2010;20:R285–R295. doi: 10.1016/j.cub.2010.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell. 2004;118:409–418. doi: 10.1016/j.cell.2004.08.005. [DOI] [PubMed] [Google Scholar]
- van der Lugt NM, Domen J, Linders K, van Roon M, Robanus-Maandag E, te Riele H, et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 1994;8:757–769. doi: 10.1101/gad.8.7.757. [DOI] [PubMed] [Google Scholar]
- van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell. 1991;65:737–752. doi: 10.1016/0092-8674(91)90382-9. [DOI] [PubMed] [Google Scholar]
- Wei JS, Greer BT, Westermann F, Steinberg SM, Son CG, Chen QR, et al. Prediction of clinical outcome using gene expression profiling and artificial neural networks for patients with neuroblastoma. Cancer Res. 2004;64:6883–6891. doi: 10.1158/0008-5472.CAN-04-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Hemmer RM, Sedivy JM. Role of p14(ARF) in replicative and induced senescence of human fibroblasts. Mol Cell Biol. 2001;21:6748–6757. doi: 10.1128/MCB.21.20.6748-6757.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer. 2008;8:83–93. doi: 10.1038/nrc2290. [DOI] [PubMed] [Google Scholar]
- Yamashita M, Kuwahara M, Suzuki A, Hirahara K, Shinnaksu R, Hosokawa H, et al. Bmi1 regulates memory CD4 T cell survival via repression of the Noxa gene. J Exp Med. 2008;205:1109–1120. doi: 10.1084/jem.20072000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.