

Abstract
The degree to which the level of genetic variation for gene expression is shared across multiple tissues has important implications for research investigating the role of expression on the etiology of complex human traits and diseases. In the last few years, several studies have been published reporting the extent of overlap in expression quantitative trait loci (eQTL) identified in multiple tissues or cell types. Although these studies provide important information on the regulatory control of genes across tissues, their limited power means that they can typically only explain a small proportion of genetic variation for gene expression. Here, using expression data from monozygotic twins (MZ), we investigate the genetic control of gene expression in lymphoblastoid cell lines (LCL) and whole blood (WB). We estimate the genetic correlation that represents the combined effects of all causal loci across the whole genome and is a measure of the level of common genetic control of gene expression between the two RNA sources. Our results show that, when averaged across the genome, mean levels of genetic correlation for gene expression in LCL and WB samples are close to zero. We support our results with evidence from gene expression in an independent sample of LCL, T-cells, and fibroblasts. In addition, we provide evidence that housekeeping genes, which maintain basic cellular functions, are more likely to have high genetic correlations between the RNA sources than non-housekeeping genes, implying a relationship between the transcript function and the degree to which a gene has tissue-specific genetic regulatory control.
Analyzing transcript abundance as a quantitative trait is a powerful tool used in understanding the contribution of gene expression to the etiology of many diseases (Chen et al. 2008; Emilsson et al. 2008; Cookson et al. 2009). Transcript expression levels act as an intermediate phenotype between DNA sequence variation and complex, observable phenotypes and are known to be attributable to both genetic and non-genetic factors (Monks et al. 2004; Williams et al. 2007; Cheung and Spielman 2009; Idaghdour et al. 2010). Variation influencing gene expression can manifest itself as gene expression differences between populations (Spielman et al. 2007; Storey et al. 2007; Idaghdour et al. 2010), between individuals in a population (Cheung et al. 2005; Storey et al. 2007), and in response to environmental factors, such as drug exposure (Choy et al. 2008). The genetic basis of individual and population gene expression variation has traditionally been investigated by measuring transcript abundance in a single tissue (or cell type) and the identification of quantitative trait loci correlated with gene expression variation in a single or multiple populations (Hubner et al. 2005; Dixon et al. 2007; Goring et al. 2007; Spielman et al. 2007; Stranger et al. 2007; Dimas et al. 2009; Idaghdour et al. 2010; Zeller et al. 2010).
The complexity in higher eukaryotes results in a vast range of highly specialized cell types and tissues. From a series of studies, we are beginning to understand that although some genes exhibit ubiquitous patterns of expression, others act in a highly tissue- or cell type–specific manner (Saito-Hisaminato et al. 2002; Yanai et al. 2005; Heinzen et al. 2008; Kwan et al. 2009; Jacox et al. 2010). Most attempts to use data from multiple tissues have first mapped expression QTL (eQTL) from individual tissues and then compared results among them (Petretto et al. 2006; Emilsson et al. 2008; Bullaughey et al. 2009; Dimas et al. 2009; Ding et al. 2010; Nica et al. 2011). For example, tissue specificity of eQTLs in T-cells, LCLs, and fibroblasts was determined by first mapping for eQTL against expression levels from each tissue independently, and, secondly, calculating the proportion of eQTL there were either unique to a tissue or observed in multiple tissues (Dimas et al. 2009). Dimas et al. (2009) reported that ∼70%–80% of the identified regulatory variants operate in a cell type–specific manner; however, such studies suffer in their ability to detect only eQTL with effects above a certain size as a consequence of sample size, meaning that the true degree of common regulatory variation between tissues is unknown.
Among recent work on regulatory control is interest in the location of eQTL with respect to the position of the transcript, with cis and trans used to describe near- and distant-acting regulatory variation, respectively. The exact definition of cis- and trans-acting varies considerably between studies, and so, for the sake of brevity, here we use the definition of cis-acting as “on the same chromosome” and trans-acting as “a different chromosome to the transcript location,” unless specified otherwise. It is currently unclear to what extent eQTL common between cell types and tissues operate in cis- compared with trans-, as low power to detect trans-eQTL makes comparisons difficult (Gilad et al. 2008; Ding et al. 2010). Recently, several studies mapping cis and trans eQTL have suggested that considerable proportions of regulatory variation act in trans (Price et al. 2008, 2011; Cheung et al. 2010; Montgomery et al. 2010; Pickrell et al. 2010). This should lead us to reexamine the inferences drawn from comparing cis-eQTL overlap in multiple tissues. Using an identity-by-descent (IBD) method to partition regulatory variation acting on either cis- or trans-chromosomes showed that on average only 37% of causal loci affecting regulatory variation for expression in blood and 24% in adipose tissue occurred on the cis-chromosome (Price et al. 2011). Furthermore, an estimate of common genetic control of gene expression between these two tissues revealed an average of 0.03 ± 0.006 for the 18,735 transcripts investigated, which, when partitioned into cis- and trans-chromosomes, corresponded to 0.031 ± 0.001 and −0.001 ± 0.006, respectively. Although this suggests that common genetic control acts predominantly on cis-chromosomes, mean estimates are very close to zero, implying that, on average, genes share little common regulatory variation between blood and adipose tissues.
A broader and unbiased quantification of the genetic control of expression variability between different tissues and cell types would be helpful for several reasons. First, for the majority of transcripts, a comparison of eQTL can only provide limited information on the relationship of genetic control between tissues and cell types. eQTL studies, limited by power and multiple testing corrections, do not detect variants that explain a small proportion of phenotypic variation, and the eQTL that are detected cumulatively explain typically a relatively small proportion of the heritability in gene expression (Cheung and Spielman 2009). Second, although the role of gene expression in diseases is not fully understood, the etiology of almost all complex diseases is likely to involve multiple cell types and tissues. Therefore, studying expression variability might shed light on the complexity of expression and gene pathways involved in mediating disease susceptibility. Considering that studies investigating gene expression patterns on disease profiles are often hindered by the limited availability of the relevant human tissues, instead relying on inferences drawn from analysis of available tissues or cell types, understanding the genetic control of gene expression across tissues and cell types is of upmost importance.
In a genetically informative design, narrow or broad sense heritability for gene expression can be estimated using the concept of IBD (Visscher et al. 2008). These estimates of heritability refer to the combined effects of all causal variants that segregate in the population. Similarly, the genetic correlation between gene expression levels in different tissues can be estimated. These estimates quantify the combined effects of all causal variants on the genetic covariance and are a direct measure of a genotype by tissue interaction. A large and positive genetic correlation implies that genetic variants that affect expression in one tissue also tend to affect gene expression in another tissue, in the same direction. A negative genetic correlation implies that, on average, the same variants affect both tissues but in opposite directions. A genetic correlation close to zero implies that the genetic control of gene expression in one tissue is independent of that in another tissue, when averaged over the genome. None of these cases (positive, negative, zero correlation) preclude the existence of eQTLs common to multiple tissues, because the correlations summarize the effects of causal loci across the entire genome. For example, a gene can have a genetic correlation of zero across tissues but still have one or more eQTL common to two tissues because the correlation represents the averaged effects of all causal loci, regardless of their ability to be detected by a genome scan. However, knowledge of genetic correlations is useful because it predicts the likelihood of detecting variant-expression associations across tissues and, by implication, the success of experimental designs that aim to detect eQTLs in one tissue (say, blood) to draw inference about disease association with another tissue (e.g., brain). This provides an alternative view to that offered by comparison of eQTL results, which normally represent small proportions of genetic variance and are limited by statistical power.
In this study, the global genetic control of expression levels for 9555 genes was examined from two samples of RNA sources, collected on pairs of monozygotic (MZ) twins. From each individual, gene expression profiles were generated from whole blood (WB), collected using the PAXgene tube system, and lymphoblastoid cell lines (LCL). Using expression data collected from MZ twins is a powerful means of estimating the genetic contribution of expression variation. In particular, the observed phenotypic covariance in gene expression in LCLs and WBs between MZ twins can be partitioned into variation due to within-person environmental effects and between-person genetic effects.
Results
Data pre-processing and normalization
The entire experimental design is summarized in Figure 1. Gene expression levels were generated for LCL and WB samples from each of 50 and 47 MZ twin pairs, respectively, measured using the Illumina HumanHT-12 v3.0 whole genome chip. There were a total of 47 pairs of twins for which gene expression was profiled from each of the two RNA sources. Of the 37,857 genes whose expression levels were measured on the chip, only genes significantly expressed in at least 50% of samples were carried forward, leaving a total of 9555 genes for analysis of the genetic control of expression. Analyzing the number of genes significantly expressed in each sample showed that no single sample or group of samples caused a marked decrease in the number of genes significantly expressed across all samples (Supplemental Fig. 1). Normalization of data across chips and genes demonstrated that the vast majority (98%) of the variance in the transformed expression levels was due to differences in average expression levels across genes. As expected, the across-chip expression variance was negligible due to scaling performed during the data pre-processing stages (see Methods).
Figure 1.

Diagram of study design. Gene expression levels were collected from two RNA sources, LCL and WB, from both twins within an MZ pair. From this study design, we can calculate the following correlations: rLCL and rWB (dotted arrows) are the phenotypic correlations between MZ twins within RNA sources; rbetween (dashed arrows) is the phenotypic correlation between transcript abundance in LCL from a sample of one of an MZ twin pair and the transcript abundance in WB from the sample of the co-twin. Under the assumption that there are no shared environmental effects between twins, this correlation is a function of genetic effects only: rbetween = rGHWBHLCL, rwithin (solid arrows) is the phenotypic correlation of a RNA source within a sample. As the covariance of WB and LCL transcript abundance in a sample can be due to shared genetic and/or shared environmental effects,  , where rE is the within sample correlation of environmental effects calculated as
, where rE is the within sample correlation of environmental effects calculated as 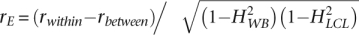 . These correlations are shown in Figure 4A–C and supporting material, respectively. This genetically informative study design provides a framework for estimating gene expression
. These correlations are shown in Figure 4A–C and supporting material, respectively. This genetically informative study design provides a framework for estimating gene expression  (dotted arrows) for each tissue, as well as genetic correlation
(dotted arrows) for each tissue, as well as genetic correlation  (dashed arrows) of a gene's expression between the two RNA sources, by partitioning variance into within and between MZ components.
(dashed arrows) of a gene's expression between the two RNA sources, by partitioning variance into within and between MZ components.
Expression variability across tissues
As a first step, we examined whether the 9555 genes had similar levels of within-sample variability. For each gene, we quantified the within-sample expression variability by calculating its coefficient of variation cv, which is the ratio of the standard deviation of sample expression level to the mean value (Kaern et al. 2005; Raser and O'Shea 2005). The standard deviation and mean expression levels were calculated from the mean of the normalized expression values of MZ twin pairs. Although other metrics are available to quantify expression variability, cv is known to be one of the most robust and unbiased measures (Kaern et al. 2005). Between LCL and WB samples, many genes exhibit divergent within-sample variability, with the LCL cv correlated with that in WB (r2 = 0.64) (Fig. 2). Although subject to sampling noise, this modest correlation of within-sample expression variability suggests that either different levels of constraint affect expression variability in both samples for most genes, or the cis- or trans-regulatory mechanisms of these genes are different.
Figure 2.

Correlation of expression variability for 9555 genes between the LCL and WB samples. The coefficient of variation (cv) was calculated from the mean normalized gene expression values of MZ pairs. Each data point represents one gene. Regression coefficient = 0.96 with SE = 0.005 and correlation coefficient = 0.64.
Effect of sex
The sex of MZ pairs was included as a covariate in the mixed model estimating the effect of genotype on expression levels (Eq. 2), although we first conducted analyses to test the significance of the effect of sex on gene expression levels from LCL and WB samples. Distributions of test statistics for difference in mean expression levels between male and female MZ twin pairs are given in Supplemental Figure 2. These are calculated such that a positive test statistic represents an increased level of expression in females compared with males. Comparison of test statistics for differential expression between sexes obtained from LCL and WB samples is shown in Figure 3. The low correlation between test statistics for the effect of sex in LCL and WB samples suggests that either the majority of genes have little common effect of sex between LCL and PCBM samples or the effect of sex has a different direction of effect between samples.
Figure 3.

Test statistics for the effect of sex on expression levels for 9555 genes in the WB and LCL samples. Each point represents a single gene. Regression coefficient = 0.13 with SE = 0.012 and correlation coefficient = 0.012.
Correlations between MZ twins within and across RNA sources
The series of normalization steps made expression phenotypes comparable across individuals and across transcripts, resulting in normally distributed expression phenotypes. Between-pair phenotypic correlations (rLCL and rWB) of transcript abundance in LCL and WB samples were calculated for each of the 9555 genes. Histograms of the phenotypic correlation coefficients are shown in Figure 4, A and B. Mean correlation coefficients for transcript abundance between MZ pairs are 0.44 and 0.34 for LCL and WB samples, respectively. Lower mean correlation coefficients of WB over LCL samples may, in part, be due to WB containing a variety of cell types, including lymphocytes, monocytes, and macrophages, compared with the single cell type of LCL. The exact effect of the cellular heterogeneity of WB on expression variability is not fully understood because each of the contributing cell types will express a unique gene expression signature relating to its function and possibly be influenced by the relative proportions of cell types (Whitney et al. 2003; Min et al. 2010). Given differences in the cellular heterogeneity between WB and LCL, we tested the effects of blood components and cell type counts on expression variability and show that all covariates tested had negligible effects on phenotypic correlations and estimates of heritability in both LCL and WB samples (Supplemental Fig. 3). A total of 14 blood biochemical traits and cell counts (listed in Supplemental Fig. 3) were investigated for their effect on expression levels. Although all of these had an insignificant effect on expression, we cannot rule out the possibility that an unknown covariate has an effect on gene expression in either one or both samples. The distribution of correlation coefficients of gene expression levels from LCL samples in one MZ twin and WB samples in the co-twin (rbetween) has a mean of 0 and a standard deviation of 0.15 (Fig. 4C). In the absence of environmental factors that are shared between twins, these phenotypic correlations between LCL and WB samples, calculated between MZ pairs, reflect genetic covariance. Correlation coefficients calculated between expression levels of LCL and WB samples from the same individuals (rwithin), which reflect both a within-person genetic and environmental covariance, are close to those calculated between MZ pairs (rbetween) (Supplemental Fig. 4), consistent with the absence of shared environmental factors. For details of the definitions of the correlations, see Figure 1.
Figure 4.

Distributions between MZ twin phenotypic correlation coefficients of transcript abundance from 9555 genes significantly expressed in all samples. (A,B) Distributions of the correlation coefficients for rLCL and rWB samples, respectively. (C) Two measures of rbetween were calculated: one between the expression value for LCL in MZ_1 and WB in MZ_2 and a second between the expression value for WB in MZ_1 and LCL in MZ_2. The figure shows the distribution of mean of the two rbetween correlations. (D) The distribution of rE. The mean and variances of the distributions are given in each part.
Estimates of heritability
Figure 4, A and B, showed a strong phenotypic correlation in gene expression between members of an MZ pair, implying underlying genetic factors. The variance in transcript abundance levels for each of the 9555 genes in both LCL and WB samples was partitioned using a linear mixed model and a least-squares analysis. Out of the 9555 genes analyzed, 377 (3.9%) and 401 (4.2%) provided a negative estimate for the genetic variance in transcript abundance in LCL and WB samples, respectively (Fig. 5). The observed distributions show considerably lower proportions of negative estimates than the 50% expected under the null hypothesis of no effect of genetic factors for all transcripts. The distribution of  above zero is close to a symmetrical decay around the mode of the distribution. Mean
above zero is close to a symmetrical decay around the mode of the distribution. Mean  is 0.38 for LCL and 0.32 for WB samples. The variance of gene
is 0.38 for LCL and 0.32 for WB samples. The variance of gene  is 0.043 for LCL and 0.034 for WB samples, both considerably greater than the expected sampling variance of 0.007 (predicted at H2 = 0.39 for all genes) and 0.009 (predicted at H2 = 0.32 for all genes) for LCL and WB, respectively (Eq. 4). The greater observed variance implies that there are real differences in heritability of gene expression across these genes.
is 0.043 for LCL and 0.034 for WB samples, both considerably greater than the expected sampling variance of 0.007 (predicted at H2 = 0.39 for all genes) and 0.009 (predicted at H2 = 0.32 for all genes) for LCL and WB, respectively (Eq. 4). The greater observed variance implies that there are real differences in heritability of gene expression across these genes.
Figure 5.

Distributions of intraclass correlations  of the 9555 genes significantly expressed in all samples (both LCL and WB). (A,B) Distributions of the correlation coefficients for the WB and LCL samples, respectively. (Light gray) Housekeeping genes (Zhu et al. 2008b) (see Results). Mean estimates of
of the 9555 genes significantly expressed in all samples (both LCL and WB). (A,B) Distributions of the correlation coefficients for the WB and LCL samples, respectively. (Light gray) Housekeeping genes (Zhu et al. 2008b) (see Results). Mean estimates of  for all (All) and housekeeping (HK) genes are shown. Distributions of genes with significant heritabilities (FDR = 0.05) are given in Supplemental Figure 5.
for all (All) and housekeeping (HK) genes are shown. Distributions of genes with significant heritabilities (FDR = 0.05) are given in Supplemental Figure 5.
The heritability estimates calculated here are from the covariance between monozygotic twins and may include both additive and non-additive genetic components, as well as shared environmental effects. Thus, they represent broad estimates and will be biased upward compared with narrow-sense heritability, which is attributable to variation in additive genetic factors (Visscher et al. 2008). However, similarities between heritabilities estimated using MZ correlations and those estimated using pedigree data (McRae et al. 2007), and clear evidence for additive genetic variation from eQTL studies (Monks et al. 2004; Goring et al. 2007; Zeller et al. 2010), lead us to make the assumption that our estimates of 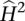 reflect mostly genetic variation. In addition, theory and data on complex traits are consistent with most genetic variation being additive (Hill et al. 2008), so MZ correlations may be a reasonable estimate of narrow sense heritability.
reflect mostly genetic variation. In addition, theory and data on complex traits are consistent with most genetic variation being additive (Hill et al. 2008), so MZ correlations may be a reasonable estimate of narrow sense heritability.
Power to detect low heritability means that many genes will not have estimates of  that are significant. Indeed, when a false discovery rate of 0.05 was used, 6184 genes in WB and 7039 in LCL were detected as heritable (Supplemental Fig. 5). Of the significantly heritable genes, the mean
that are significant. Indeed, when a false discovery rate of 0.05 was used, 6184 genes in WB and 7039 in LCL were detected as heritable (Supplemental Fig. 5). Of the significantly heritable genes, the mean 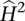 is 0.48 for LCL and 0.43 for WB samples. A total of 4721 genes are significantly heritable in both WB and LCL samples.
is 0.48 for LCL and 0.43 for WB samples. A total of 4721 genes are significantly heritable in both WB and LCL samples.
Estimates of genetic correlations
For the 4721 genes with estimates of significant  in both LCL and WB samples, we computed estimates of the genetic correlations
in both LCL and WB samples, we computed estimates of the genetic correlations  by dividing the estimates of genetic covariance by the product of the square root of the genetic standard deviations (Eq. 3). A histogram of
by dividing the estimates of genetic covariance by the product of the square root of the genetic standard deviations (Eq. 3). A histogram of  is given in Figure 6. The distribution has a mean
is given in Figure 6. The distribution has a mean  of −0.031 and a variance of 0.1 and shows a normal decline around the mean. The expected sampling variance of
of −0.031 and a variance of 0.1 and shows a normal decline around the mean. The expected sampling variance of  , calculated assuming
, calculated assuming  for both traits, is 0.039, noticeably smaller than the observed empirical variance. The empirical distribution of
for both traits, is 0.039, noticeably smaller than the observed empirical variance. The empirical distribution of  shows that there is considerable real variation in the genetic correlation of expression levels across genes, over and above sampling variation. As the average correlation is close to zero, the extent of common genetic control is observed in both positive and negative directions. Overall, the results show that a small number of genes have either highly positive or negative levels of common genetic control for gene expression levels between WB and LCL. For these genes, the high genetic correlations imply that the same causal variants affect expression in both tissues, although when the correlation is negative, the direction of the affect is reversed between tissues. Our results also show that for the majority of genes, the genetic correlation is close to zero; although this does not exclude common eQTL, it suggests that across the genome, on average, the effects of genetic variants on gene expression in the two tissues are uncorrelated.
shows that there is considerable real variation in the genetic correlation of expression levels across genes, over and above sampling variation. As the average correlation is close to zero, the extent of common genetic control is observed in both positive and negative directions. Overall, the results show that a small number of genes have either highly positive or negative levels of common genetic control for gene expression levels between WB and LCL. For these genes, the high genetic correlations imply that the same causal variants affect expression in both tissues, although when the correlation is negative, the direction of the affect is reversed between tissues. Our results also show that for the majority of genes, the genetic correlation is close to zero; although this does not exclude common eQTL, it suggests that across the genome, on average, the effects of genetic variants on gene expression in the two tissues are uncorrelated.
Figure 6.

 of the 4721 genes with significant
of the 4721 genes with significant  in both LCL and WB samples. (A) (Solid line) ± one standard deviation from the mean
in both LCL and WB samples. (A) (Solid line) ± one standard deviation from the mean 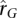 ; (dashed line) ± one standard deviation calculated from the expected sample variance given
; (dashed line) ± one standard deviation calculated from the expected sample variance given  and
and  . (B) The distribution of
. (B) The distribution of  shown in A with the number of housekeeping genes (Zhu et al. 2008b) (see Results) within each
shown in A with the number of housekeeping genes (Zhu et al. 2008b) (see Results) within each  bin shown in dark gray. The black line shows the percentage of genes within each
bin shown in dark gray. The black line shows the percentage of genes within each  bin that are housekeeping genes. We performed a Kolmogorov-Smirnov test for differences in the distributions of
bin that are housekeeping genes. We performed a Kolmogorov-Smirnov test for differences in the distributions of  for housekeeping genes (dark gray) and non-housekeeping genes (light gray); the P-value is given in the figure.
for housekeeping genes (dark gray) and non-housekeeping genes (light gray); the P-value is given in the figure.
The 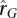 distribution shown in Figure 6 is restricted to those genes that have significant heritability estimates in both WB and LCL samples. Despite results showing a mean
distribution shown in Figure 6 is restricted to those genes that have significant heritability estimates in both WB and LCL samples. Despite results showing a mean 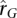 close to zero, a large number of genes have strong positive and negative estimates of genetic correlation. In Supplemental Table 1, we provide a list of the genes in the top
close to zero, a large number of genes have strong positive and negative estimates of genetic correlation. In Supplemental Table 1, we provide a list of the genes in the top  and bottom
and bottom  two percentile of the
two percentile of the  distribution (Fig. 6). High heritabilities of these genes are consistent with those reported in LCLs by Dixon et al. (2007), indicating that the genetic control is not specific to our sample population. To investigate if the
distribution (Fig. 6). High heritabilities of these genes are consistent with those reported in LCLs by Dixon et al. (2007), indicating that the genetic control is not specific to our sample population. To investigate if the  and
and 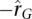 genes shared common biological functions (e.g., metabolic pathways or similar Gene Ontology functional annotation), the genes in the
genes shared common biological functions (e.g., metabolic pathways or similar Gene Ontology functional annotation), the genes in the  and
and  groups were separately subjected to GO enrichment analysis using GOEAST (Zheng and Wang 2008). The
groups were separately subjected to GO enrichment analysis using GOEAST (Zheng and Wang 2008). The  group is overrepresented by genes that are involved in GO terms relating to the MHC complex, whereas the
group is overrepresented by genes that are involved in GO terms relating to the MHC complex, whereas the  group is overrepresented by genes with GO classifications relating to intercellular components and membrane binding. A full description of the analysis and a list of the top 10 pathways listed for the
group is overrepresented by genes with GO classifications relating to intercellular components and membrane binding. A full description of the analysis and a list of the top 10 pathways listed for the  and
and  groups are given in Supplemental Table 2.
groups are given in Supplemental Table 2.
Housekeeping genes
The biological functionality of the regulatory control of a gene's expression in different tissues is typically unknown for most genes. Nevertheless, when a gene is expressed in multiple tissues, regulatory control may be common across the tissues. To investigate this, we looked at the  and
and  for genes described as housekeeping genes from a comprehensive analysis of publicly available expression profiles in 18 human tissues (Zhu et al. 2008b). In both LCL and WB samples, mean heritability of housekeeping genes is significantly (p < 1.4 × 10−73 and p < 7.8 × 10−86, respectively) greater than the mean for all genes (Fig. 5A,B). Although high heritabilities for housekeeping genes are perhaps not surprising (Butte et al. 2001; Alba and Castresana 2005), they may be due to low levels of environmental variance, rather than increases in genetic variance, relative to non-housekeeping genes. To test for this, we looked at estimates of the environmental variance for housekeeping and non-housekeeping genes and found no significant differences in the means, indicating that the higher heritabilities of housekeeping genes are due to increases in genetic variance. Compared with the
for genes described as housekeeping genes from a comprehensive analysis of publicly available expression profiles in 18 human tissues (Zhu et al. 2008b). In both LCL and WB samples, mean heritability of housekeeping genes is significantly (p < 1.4 × 10−73 and p < 7.8 × 10−86, respectively) greater than the mean for all genes (Fig. 5A,B). Although high heritabilities for housekeeping genes are perhaps not surprising (Butte et al. 2001; Alba and Castresana 2005), they may be due to low levels of environmental variance, rather than increases in genetic variance, relative to non-housekeeping genes. To test for this, we looked at estimates of the environmental variance for housekeeping and non-housekeeping genes and found no significant differences in the means, indicating that the higher heritabilities of housekeeping genes are due to increases in genetic variance. Compared with the  for all genes (Fig. 6A), housekeeping genes are overrepresented in the tails of the distribution (Fig. 6B). A Kolmogorov-Smirnov test for difference in the distributions of
for all genes (Fig. 6A), housekeeping genes are overrepresented in the tails of the distribution (Fig. 6B). A Kolmogorov-Smirnov test for difference in the distributions of  from housekeeping and non-housekeeping genes is very highly significant (p < 2.1 × 10−16).
from housekeeping and non-housekeeping genes is very highly significant (p < 2.1 × 10−16).
Correlations between multiple tissues in an independent sample
Publicly available expression data on three cell types—LCL, T-cells, and fibroblasts—in 85 unrelated individuals were downloaded from NCBI's Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) (Dimas et al. 2009). These expression data were collected as part of the GenCord project and are described in detail in Dimas et al. (2009). We calculated phenotypic correlations between normalized expression levels for each of the three pairwise combinations of cell types (Supplemental Fig. 6). The phenotypic correlation between expression in one cell type and another cell type is a combination of the genetic and environmental correlations. Thus, a distribution of the phenotypic correlation coefficients provides information on the likely distribution of the genetic correlations (Cheverud 1988). For each pairwise combination of cell types, we observe distributions with a symmetrical decay around mean values close to zero (Supplemental Fig. 6). It seems improbable that the distributions of genetic and environmental correlations are strongly skewed in opposite directions, and therefore the observed phenotypic correlation distributions imply that the genetic correlations are also likely to show a symmetrical decay around a mean close to zero.
The three pairwise combinations of phenotypic correlations for the genes listed in the  and
and  groups are given in Supplemental Table 1. These genes are characterized by high levels of shared heritable variation and high heritabilities in both LCL and WB samples. Thus, for genes that show similar
groups are given in Supplemental Table 1. These genes are characterized by high levels of shared heritable variation and high heritabilities in both LCL and WB samples. Thus, for genes that show similar  and phenotypic correlations (either positive or negative), we could infer that shared heritable expression control is consistent between the tissues in this study and Dimas et al. (2009). However, overall, there is little agreement between the phenotypic correlations from the study of Dimas et al. (2009) and our estimates of
and phenotypic correlations (either positive or negative), we could infer that shared heritable expression control is consistent between the tissues in this study and Dimas et al. (2009). However, overall, there is little agreement between the phenotypic correlations from the study of Dimas et al. (2009) and our estimates of 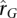 , which is likely to be due to two main factors: (1) Environmental effects, and therefore correlations, are different between the tissues and study populations. (2) Genetic correlations for transcript abundance across a pair of tissues are tissue-specific. Indeed, the correlation coefficients of the pairwise phenotypic correlations from Dimas et al. (2009) are very low (0.04–0.07), implying that even within a single study, environmental and genetic correlations are tissue-specific.
, which is likely to be due to two main factors: (1) Environmental effects, and therefore correlations, are different between the tissues and study populations. (2) Genetic correlations for transcript abundance across a pair of tissues are tissue-specific. Indeed, the correlation coefficients of the pairwise phenotypic correlations from Dimas et al. (2009) are very low (0.04–0.07), implying that even within a single study, environmental and genetic correlations are tissue-specific.
Discussion
In this study, the genetic (co)variation affecting gene expression in LCL and WB RNA sources was examined. Transcriptional regulatory networks are expected to dictate tissue specificity of regulatory effects (Ravasi et al. 2010), although the extent of this has been debated. Here, we present estimates of  (Fig. 6A) from 4721 genes, which show a symmetrical decay around a mean of −0.03 and an empirical variance approximately three times greater than would be expected by chance given this sample size. Our results show that, when averaged across the genome, mean levels of regulatory control for gene expression in LCL and WB samples due to genetic factors are close to zero. These results are supported by phenotypic correlations between gene expression in LCL, T-cells, and fibroblasts from an independent sample (Dimas et al. 2009) and are consistent with estimates of cross-tissue heritability reported by Price et al. (2011).
(Fig. 6A) from 4721 genes, which show a symmetrical decay around a mean of −0.03 and an empirical variance approximately three times greater than would be expected by chance given this sample size. Our results show that, when averaged across the genome, mean levels of regulatory control for gene expression in LCL and WB samples due to genetic factors are close to zero. These results are supported by phenotypic correlations between gene expression in LCL, T-cells, and fibroblasts from an independent sample (Dimas et al. 2009) and are consistent with estimates of cross-tissue heritability reported by Price et al. (2011).
Our results showing that, on average, the genetic correlation of regulatory variation for gene expression in LCL and WB is close to zero does not contradict published reports presenting comparisons of eQTL in multiple tissues. Estimates of  represent the combined effects of all causal loci; thus, even estimates of zero do not preclude the existence of common eQTL. Rather, the genetic covariance (CovG) is the sum of covariance for each causal loci (CovG), such that:
represent the combined effects of all causal loci; thus, even estimates of zero do not preclude the existence of common eQTL. Rather, the genetic covariance (CovG) is the sum of covariance for each causal loci (CovG), such that:
where n is total the number of causal loci. Knowledge of the genetic correlation for regulatory variation across tissues is important, particularly when trying to understand the role of gene expression in the etiology of complex diseases that affect multiple tissues and how transcriptional regulatory pathways work on a multicellular system level.
With a strong current interest in multitissue genetic control of expression (Montgomery and Dermitzakis 2011), one question is, How do our results integrate with other published studies in this field? With the exception of the genes with highly negative or positive  estimates, it is possible that the majority of genetic control for expression is likely to be controlled by different variants in the two samples with these tissue-specific loci more likely to occur in trans-regions (Dimas et al. 2009; Montgomery et al. 2010; Pickrell et al. 2010). Indeed, Price et al. (2011) recently showed that, on average, the amount of shared heritable variation for gene expression in blood and adipose tissues was lower on trans- compared with cis-chromosomes, suggesting that the fraction of common eQTL shared between tissues is inflated upward because trans associations are not tested for due to the limitations of power (Ding et al. 2010). Furthermore, the actual proportion of genes for which common cis-eQTL have been identified is very small. For example, of the genes tested in multiple tissues in recent studies, only 0.4%–0.5% of genes have significant cis-eQTL in two or more tissues (Dimas et al. 2009; Ding et al. 2010; Nica et al. 2011). A study with one of the largest sample sizes to date found that the median percentage of expression variability explained by the best eQTL SNP was 7.7% (Zeller et al. 2010). Therefore, the empirical evidence presented in these studies is that only a small proportion of genes have shared eQTL and that the proportion of phenotypic variation (and likely the proportion of heritability) explained by individual eQTL is relatively small. Here, our genetically informative design has allowed us to gain a whole genome estimate of the level of shared regulatory elements in LCL and WB RNA sources, providing an important complement to eQTL studies.
estimates, it is possible that the majority of genetic control for expression is likely to be controlled by different variants in the two samples with these tissue-specific loci more likely to occur in trans-regions (Dimas et al. 2009; Montgomery et al. 2010; Pickrell et al. 2010). Indeed, Price et al. (2011) recently showed that, on average, the amount of shared heritable variation for gene expression in blood and adipose tissues was lower on trans- compared with cis-chromosomes, suggesting that the fraction of common eQTL shared between tissues is inflated upward because trans associations are not tested for due to the limitations of power (Ding et al. 2010). Furthermore, the actual proportion of genes for which common cis-eQTL have been identified is very small. For example, of the genes tested in multiple tissues in recent studies, only 0.4%–0.5% of genes have significant cis-eQTL in two or more tissues (Dimas et al. 2009; Ding et al. 2010; Nica et al. 2011). A study with one of the largest sample sizes to date found that the median percentage of expression variability explained by the best eQTL SNP was 7.7% (Zeller et al. 2010). Therefore, the empirical evidence presented in these studies is that only a small proportion of genes have shared eQTL and that the proportion of phenotypic variation (and likely the proportion of heritability) explained by individual eQTL is relatively small. Here, our genetically informative design has allowed us to gain a whole genome estimate of the level of shared regulatory elements in LCL and WB RNA sources, providing an important complement to eQTL studies.
In this study, we observe a small fraction of genes with  , which implies that the same causal variants affect expression in LCL and WB, but in opposite directions. Studies comparing cis-eQTL in multiple tissues observe the same direction of allelic effect for common eQTL (Bullaughey et al. 2009; Ding et al. 2010; Nica et al. 2011). This discrepancy may arise if eQTL with opposing allelic effects in different tissues have either small effect sizes or occur outside of the defined cis-regions used in these studies. Unfortunately, these scenarios are currently difficult to evaluate due to low power to detect eQTL with small effect or in trans-regions.
, which implies that the same causal variants affect expression in LCL and WB, but in opposite directions. Studies comparing cis-eQTL in multiple tissues observe the same direction of allelic effect for common eQTL (Bullaughey et al. 2009; Ding et al. 2010; Nica et al. 2011). This discrepancy may arise if eQTL with opposing allelic effects in different tissues have either small effect sizes or occur outside of the defined cis-regions used in these studies. Unfortunately, these scenarios are currently difficult to evaluate due to low power to detect eQTL with small effect or in trans-regions.
Although there is still some debate about the exact definition and characteristics of housekeeping genes (Zhu et al. 2008a), it is widely accepted that they are expressed in all tissues and are required to maintain basic cellular function (Butte et al. 2001). Given this, our results showing high heritabilities and a significant overrepresentation of strong positive and negative  in housekeeping genes (Zhu et al. 2008b) support an important role in basal biological function. Compared with non-housekeeping genes, housekeeping genes are known to evolve more slowly (Zhang and Li 2004), and although coding sequences are more conserved, show less conservation in promoter, particularly distal, regions (Farre et al. 2007). As sequence conservation is likely due to increased functional constraints (Alba and Castresana 2005), the difference in selection economy in transcription and translation regions implies that although the function of housekeeping genes is under strong stabilizing selection, their regulatory control is not. Furthermore, functional similarities for genes in the
in housekeeping genes (Zhu et al. 2008b) support an important role in basal biological function. Compared with non-housekeeping genes, housekeeping genes are known to evolve more slowly (Zhang and Li 2004), and although coding sequences are more conserved, show less conservation in promoter, particularly distal, regions (Farre et al. 2007). As sequence conservation is likely due to increased functional constraints (Alba and Castresana 2005), the difference in selection economy in transcription and translation regions implies that although the function of housekeeping genes is under strong stabilizing selection, their regulatory control is not. Furthermore, functional similarities for genes in the  and in the
and in the  groups, as represented by the enrichment of GO terms, suggest that patterns of regulatory control between the tissues are influenced by the biological roles of cells (Altschuler and Wu 2010).
groups, as represented by the enrichment of GO terms, suggest that patterns of regulatory control between the tissues are influenced by the biological roles of cells (Altschuler and Wu 2010).
The MZ twin design is a powerful tool for estimating heritability and genetic correlations by partitioning variance into within- and between-MZ effects (Visscher 2004; Visscher et al. 2006; McRae et al. 2007). A limitation of this design is an inability to partition the observed covariance of the twins into components due to genetic factors and common environmental factors, leading to a possible upward bias of heritability, although this might be mitigated by the use of LCLs, which are maintained in a homogeneous environment (Cheung et al. 2003; Cheung and Spielman 2009) (see Methods). Although to date many studies have investigated the role of regulatory variation in gene expression using LCLs (Monks et al. 2004; Duan et al. 2008; Dimas et al. 2009), the application of immortalization and virus transformation steps has led to some criticism of their use, particularly in relation to understanding the role of expression in the etiology of disease (Carter et al. 2002; Çalışkan et al. 2011). Nevertheless, LCLs have been used in many studies investigating regulatory control and have shown high levels of replication across populations and samples (Li et al. 2008; Ding et al. 2010).
In summary, we have presented results on the whole genome genetic control of gene expression in two RNA sources, LCLs and WB, from a genetically informative MZ twin design. Our estimation of genetic correlations of regulatory variation for 9555 genes provides strong evidence that the average level of common genetic control in LCL and WB samples is very small. Certain genes, particularly housekeeping genes, have high negative or positive genetic correlations, pertaining to a relationship between transcript function and tissue-specific genetic control. An important implication of our results is the need to consider the degree of common genetic control between tissues, particularly when investigating the role of gene expression on the etiology of complex diseases acting in multiple cell and tissue systems. We caution against the use of gene expression measured in one tissue (e.g., blood) to draw inferences about disease association with another tissue (e.g., brain), unless knowledge of the level of common genetic control is available. Further work is clearly required to provide a deeper understanding of how causal variants that affect common and tissue-specific gene regulation function in transcription pathways across a broad range of tissues.
Methods
Monozygous twin sample
The sample consisted of 50 pairs of MZ twins, 27 female and 23 male pairs, recruited as part of a study that focuses on genetic aspects of melanocytic naevi in Australian adolescents of European descent (McGregor et al. 1999), with all study participants provided informed consent. Zygosity was tested using an AmpFLSTR Profiler Plus PCR Amplification Kit (Applied Biosystems) and Genescan v3.7.1 software (Applied Biosystems) to confirm MZ status. Total study design consisted of 100 individuals (50 MZ pairs), and expression data from LCL and WB samples for each individual (summarized in Fig. 1).
RNA preparation
Whole blood samples were collected from MZ twin pairs and processed within 24 h of collection. Whole blood was collected directly into PAXgene tubes (QIAGEN), and a second sample was collected in an ACD (acid citrate dextrose) vacuum tube. Mononucleated cells were isolated using a Ficoll gradient and LCL established by Epstein-Barr virus transformation of lymphocytes (Neitzel 1986) and stored in liquid nitrogen. For RNA isolation, established cell lines were regrown under tightly controlled growth conditions in the same batch of RPMI 1640 media with 10% FCS and antibiotics, to limit the cell culture effects on RNA preparation. Total RNA was extracted from samples using QIAGEN RNeasy Midi-Kits (QIAGEN), when cells were in log-phase growth. Total RNA was extracted from PAXgene tubes using the WB gene RNA purification kit (QIAGEN). RNA from all samples was run on an Agilent Bioanalyzer to assess quality, and to estimate RNA concentrations, RNA was converted to cDNA, amplified, and purified using the Ambion Illumina TotalPrep RNA Amplification Kit (Ambion).
Gene expression quantification
Expression profiles were generated by hybridizing 750 ng of cRNA to Illumina HumanHT-12 v3.0 BeadChips according to Illumina whole-genome gene expression direct hybridization assay guide (Illumina Inc.). Briefly, 500 ng of total RNA was used to generate biotinylated cRNA, which was fragmented and hybridized to an Illumina whole genome expression chip, HumanHT-12 v3.0, for 18 h at 58°C. BeadChips were then washed and stained and subsequently scanned to obtain fluorescence intensities. More than half (54%) of the samples were scanned using an Illumina Bead Array Reader (BAR), and the remaining (46%) of samples were scanned using an Illumina iScan when the BAR was unavailable. A Latin square design was used to randomize the samples on the chips and chip positions.
Data processing and normalization
Relative expression values were generated for each transcript using Illumina Genome Studio software (Illumina Inc.). To minimize the influence of overall signal levels, which may reflect RNA quantity and quality rather than a true biological difference between individuals, the following standardization procedures were used. Background noise detected from negative control beads was subtracted from raw expression values for each transcript. Data were then filtered for gene transcripts that were present in at least 50% of samples at p < 0.05 according to the global-error threshold calculated by Genome Studio's cross-gene error model. Using a 50% threshold is a compromise between removing genes with low proportions of sample expression and maintaining a large number of genes analyzed. Filtering based on this criterion removes genes that are either not expressed or only expressed in a low number of samples. Estimates of heritability and genetic correlation for genes not expressed are essentially meaningless because variance of measured fluorescence intensities will be due entirely to experimental variance rather than containing a genetic component. An important consequence of only including genes detected as expressed in this proportion of samples is the removal of all Y-chromosome transcripts.
To prevent the introduction of bias between the LCL and WB samples, the raw microarray data from both tissue samples were quantile normalized together. Adjusted expression levels for each transcript were transformed using a quantile transformation (Bolstad et al. 2003; Smyth and Speed 2003) to achieve a stabilized variance distribution across average expression levels. Further normalization was performed to allow expression levels to be compared across chips and genes. This was achieved fitting a linear mixed model:
where yijklm is the log-transformed expression level for individual i on chip j. The variable μ represents the mean expression level across all individuals, and Cj, Dk, Pl, and Rm are random effects removing variation in the data due to chip j, date of scanning k, chip position l, and scanner m, respectively. ɛijklm is the residual. The between-chip variance is expected to be small due to the scaling that was performed during the pre-processing of the data. The residuals from this model were used in all further analyses.
Estimating heritability and genetic correlations
Linear mixed models were used to assess the effect of sex and genotype on the normalized gene expression levels using the following model:
where y_adjijk is the normalized transcript values for individual i in MZ pair j, μ represents the mean normalized transcript levels across all individuals, Gj is the random effect of MZ pair j, Sk is the fixed effect of sex, and ɛijk is the residual. For each of the 9555 genes, Equation 2 was applied to the normalized transcript values from LCL and WB samples separately. Variance components were estimated using least squares. The intraclass correlation for each gene was calculated as:
where subscripts follow those used in Equation 2. This is simply the proportion of the variance in the data explained by the MZ pair and in the absence of common environmental effects is a measure of the broad sense heritability of a gene's expression level. Using least squares to estimate the variance components can lead to some 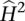 with values below zero. In a linear model framework, where errors are uncorrelated and have equal variances, least-squares analysis provides the best unbiased estimator. For balanced data, results from least squares and restricted maximum likelihood (REML) are identical, unless least-squares estimates are negative, because REML estimates of variance components are usually constrained to be non-negative. The performance of the least-squares estimator was evaluated by comparison with
with values below zero. In a linear model framework, where errors are uncorrelated and have equal variances, least-squares analysis provides the best unbiased estimator. For balanced data, results from least squares and restricted maximum likelihood (REML) are identical, unless least-squares estimates are negative, because REML estimates of variance components are usually constrained to be non-negative. The performance of the least-squares estimator was evaluated by comparison with  estimates, calculated from Equation 2 solved using REML (Gilmour et al. 1995, 2007). For least-squares
estimates, calculated from Equation 2 solved using REML (Gilmour et al. 1995, 2007). For least-squares  values above zero, REML values are very similar (Supplemental Fig. 7), as expected.
values above zero, REML values are very similar (Supplemental Fig. 7), as expected.
By treating transcript abundance, measured in LCL and WB samples as separate phenotypic traits, we are able to calculate genetic correlation coefficient (rG) for expression in each gene, which provides information on the extent of genetic influence on expression levels common to both RNA sources. Given our assumptions, the population genetic correlation coefficient can be expressed as a function of the population phenotypic correlations between MZ pairs:
where rbetween is the phenotypic correlation between transcript abundance in one RNA source from a sample of one of an MZ pair and the transcript abundance in the other RNA source from the sample of the co-twin, and rLCL and rWB are the phenotypic correlations between MZ twins for LCL and WB, respectively (for details, see Fig. 1). For estimation of the genetic correlation  , we use the between- and within-MZ mean squares and mean cross-products, as detailed in Visscher (2004).
, we use the between- and within-MZ mean squares and mean cross-products, as detailed in Visscher (2004).
Sampling variances for  and
and 
Estimates of sampling variance, or expected empirical variance across transcripts, of  and
and 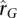 are used to quantify the empirical variance of estimates that we would expect to see given the sample size and true population values of
are used to quantify the empirical variance of estimates that we would expect to see given the sample size and true population values of  and
and  . Expected sampling variance for
. Expected sampling variance for  is calculated as:
is calculated as:
 |
where N is the sample size and H2 is the true population mean heritability. Within the classic MZ and dizygotic (DZ) twin model framework, Visscher (2004) derived a formula for estimating the sampling variance of  estimated using least-squares methods. Here we adapt this formula to calculate the sampling variance of
estimated using least-squares methods. Here we adapt this formula to calculate the sampling variance of  estimates from an MZ model. The expected sampling variance of
estimates from an MZ model. The expected sampling variance of  is, approximately:
is, approximately:
 |
where  and
and  are heritabilities for LCL and WB, respectively. To check the validity of Equations 4 and 5, results were compared with simulations. Mean square and mean cross products were sampled from a Wishart distribution under an MZ twin model, and
are heritabilities for LCL and WB, respectively. To check the validity of Equations 4 and 5, results were compared with simulations. Mean square and mean cross products were sampled from a Wishart distribution under an MZ twin model, and  and
and  were estimated using least squares. When one or both of the
were estimated using least squares. When one or both of the  were zero or negative, their values were used to calculate the empirical mean and SE of the estimates but did not contribute to an empirical estimate of
were zero or negative, their values were used to calculate the empirical mean and SE of the estimates but did not contribute to an empirical estimate of  . The performance of Equations 4 and 5 was evaluated for a range of N,
. The performance of Equations 4 and 5 was evaluated for a range of N,  , and
, and  values. One hundred thousand replicates were run for all combinations of parameters that were considered, and the empirical variance was compared with the expected sampling variance. Prediction of the sampling variance of the estimates
values. One hundred thousand replicates were run for all combinations of parameters that were considered, and the empirical variance was compared with the expected sampling variance. Prediction of the sampling variance of the estimates  and
and  was very close to the observed empirical variance across replicates (Supplemental Tables 3, 4), with asymptotically the same values at larger sample sizes.
was very close to the observed empirical variance across replicates (Supplemental Tables 3, 4), with asymptotically the same values at larger sample sizes.
Data access
Gene expression data been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE33321.
Acknowledgments
We gratefully acknowledge the participation of the individuals sampled in this work. We acknowledge funding by Australian National Health and Medical Research Council (NHMRC) grants 241944, 339462, 389927, 389875, 389891, 389892, 389938, 442915, 442981, 496739, 552485, and 552498, and Australian Research Council grants A7960034, A79906588, A79801419, DP0770096, DP0212016, and DP0343921 for building and maintaining the adolescent twin family resource through which samples were collected. We thank Anthony Caracella, Megan Campbell, Kalpana Patel, and Sara Smith for their technical assistance with the microarray hybridizations; and Alison Mackenzie, Marlene Grace, and Ann Eldridge for data collection. This research was supported by NHMRC grants 389892, 496667, and 613601. A.F.M., G.W.M., and P.M.V. are supported by the NHMRC Fellowship Scheme. E.T.D. acknowledges funds from the Louis-Jeantet Foundation. We thank Mike Goddard for helpful comments on the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.126540.111.
References
- Alba MM, Castresana J 2005. Inverse relationship between evolutionary rate and age of mammalian genes. Mol Biol Evol 22: 598–606 [DOI] [PubMed] [Google Scholar]
- Altschuler SJ, Wu LF 2010. Cellular heterogeneity: Do differences make a difference? Cell 141: 559–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193 [DOI] [PubMed] [Google Scholar]
- Bullaughey K, Chavarria CI, Coop G, Gilad Y 2009. Expression quantitative trait loci detected in cell lines are often present in primary tissues. Hum Mol Genet 18: 4296–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte AJ, Dzau VJ, Glueck SB 2001. Further defining housekeeping, or “maintenance,” genes. Focus on “A compendium of gene expression in normal human tissues”. Physiol Genomics 7: 95–96 [DOI] [PubMed] [Google Scholar]
- Çalışkan M, Cusanovich DA, Ober C, Gilad Y 2011. The effects of EBV transformation on gene expression levels and methylation profiles. Hum Mol Genet 20: 1643–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter KL, Cahir-McFarland E, Kieff E 2002. Epstein-Barr virus-induced changes in B-lymphocyte gene expression. J Virol 76: 10427–10436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YQ, Zhu J, Lum PY, Yang X, Pinto S, MacNeil DJ, Zhang CS, Lamb J, Edwards S, Sieberts SK, et al. 2008. Variations in DNA elucidate molecular networks that cause disease. Nature 452: 429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Spielman RS 2009. Genetics of human gene expression: Mapping DNA variants that influence gene expression. Nat Rev Genet 10: 595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS 2003. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet 33: 422–425 [DOI] [PubMed] [Google Scholar]
- Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT 2005. Mapping determinants of human gene expression by regional and genome-wide association. Nature 437: 1365–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung VG, Nayak RR, Wang IXR, Elwyn S, Cousins SM, Morley M, Spielman RS 2010. Polymorphic cis- and trans-regulation of human gene expression. PLoS Biol 8: e1000480 doi: 10.1371/journal.pbio.1000480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheverud JM 1988. A comparison of genetic and phenotypic correlations. Evolution 42: 958–968 [DOI] [PubMed] [Google Scholar]
- Choy E, Yelensky R, Bonakdar S, Plenge RM, Saxena R, De Jager PL, Shaw SY, Wolfish CS, Slavik JM, Cotsapas C, et al. 2008. Genetic analysis of human traits in vitro: Drug response and gene expression in lymphoblastoid cell lines. PLoS Genet 4: 16 doi: 10.1371/journal.pgen.1000287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson W, Liang L, Abecasis G, Moffatt M, Lathrop M 2009. Mapping complex disease traits with global gene expression. Nat Rev Genet 10: 184–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H, Ingle C, Beazley C, Arcelus MG, Sekowska M, et al. 2009. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 325: 1246–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Gudjonsson JE, Liang LM, Stuart PE, Li Y, Chen W, Weichenthal M, Ellinghaus E, Franke A, Cookson W, et al. 2010. Gene expression in skin and lymphoblastoid cells: refined statistical method reveals extensive overlap in cis-eQTL signals. Am J Hum Genet 87: 779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon AL, Liang L, Moffatt MF, Chen W, Heath S, Wong KCC, Taylor J, Burnett E, Gut I, Farrall M, et al. 2007. A genome-wide association study of global gene expression. Nat Genet 39: 1202–1207 [DOI] [PubMed] [Google Scholar]
- Duan S, Huang RS, Zhang W, Bleibel WK, Roe CA, Clark TA, Chen TX, Schweitzer AC, Blume JE, Cox NJ, et al. 2008. Genetic architecture of transcript-level variation in humans. Am J Hum Genet 82: 1101–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. 2008. Genetics of gene expression and its effect on disease. Nature 452: 423–428 [DOI] [PubMed] [Google Scholar]
- Farre D, Bellora N, Mularoni L, Messeguer X, Alba M 2007. Housekeeping genes tend to show reduced upstream sequence conservation. Genome Biol 8: R140 doi: 10.1186/gb-2007-8-7-r140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad Y, Rifkin SA, Pritchard JK 2008. Revealing the architecture of gene regulation: The promise of eQTL studies. Trends Genet 24: 408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmour AR, Thompson R, Cullis BR 1995. Average information REML: An efficient algorithm for variance parameter estimation in linear mixed models. Biometrics 51: 1440–1450 [Google Scholar]
- Gilmour AR, Gogel BJ, Cullis BR, Thompson R 2007. ASReml User Guide. http://www.animalgenome.org/bioinfo/resources/manuals/ASReml/UserGuide.pdf
- Goring HHH, Curran JE, Johnson MP, Dyer TD, Charlesworth J, Cole SA, Jowett JBM, Abraham LJ, Rainwater DL, Comuzzie AG, et al. 2007. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nat Genet 39: 1208–1216 [DOI] [PubMed] [Google Scholar]
- Heinzen EL, Ge DL, Cronin KD, Maia JM, Shianna KV, Gabriel WN, Welsh-Bohmer KA, Hulette CM, Denny TN, Goldstein DB 2008. Tissue-specific genetic control of splicing: Implications for the study of complex traits. PLoS Biol 6: 2869–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill WG, Goddard ME, Visscher PM 2008. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet 4: e1000008 doi: 10.1371/journal.pgen.1000008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubner N, Wallace CA, Zimdahl H, Petretto E, Schulz H, Maciver F, Mueller M, Hummel O, Monti J, Zidek V, et al. 2005. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nat Genet 37: 243–253 [DOI] [PubMed] [Google Scholar]
- Idaghdour Y, Czika W, Shianna KV, Lee SH, Visscher PM, Martin HC, Miclaus K, Jadallah SJ, Goldstein DB, Wolfinger RD, et al. 2010. Geographical genomics of human leukocyte gene expression variation in southern Morocco. Nat Genet 42: 62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacox E, Gotea V, Ovcharenko I, Elnitski L 2010. Tissue-specific and ubiquitous expression patterns from alternative promoters of human genes. PLoS ONE 5: 15 doi: 10.1371/journal.pone.0012274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaern M, Elston TC, Blake WJ, Collins JJ 2005. Stochasticity in gene expression: From theories to phenotypes. Nat Rev Genet 6: 451–464 [DOI] [PubMed] [Google Scholar]
- Kwan T, Grundberg E, Koka V, Ge B, Lam KCL, Dias C, Kindmark A, Mallmin H, Ljunggren O, Rivadeneira F, et al. 2009. Tissue effect on genetic control of transcript isoform variation. PLoS Genet 5: e1000608 doi: 10.1371/journal.pgen.1000608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JZ, Absher DM, Tang H, Southwick AM, Casto AM, Ramachandran S, Cann HM, Barsh GS, Feldman M, Cavalli-Sforza LL, et al. 2008. Worldwide human relationships inferred from genome-wide patterns of variation. Science 319: 1100–1104 [DOI] [PubMed] [Google Scholar]
- McGregor B, Pfitzner J, Zhu G, Grace M, Eldridge A, Pearson J, Mayne C, Aitken JF, Green AC, Martin NG 1999. Genetic and environmental contributions to size, color, shape, and other characteristics of melanocytic naevi in a sample of adolescent twins. Genet Epidemiol 16: 40–53 [DOI] [PubMed] [Google Scholar]
- McRae AF, Matigian NA, Vadlamudi L, Mulley JC, Mowry B, Martin NG, Berkovic SF, Hayward NK, Visscher PM 2007. Replicated effects of sex and genotype on gene expression in human lymphoblastoid cell lines. Hum Mol Genet 16: 364–373 [DOI] [PubMed] [Google Scholar]
- Min JL, Barrett A, Watts T, Pettersson FH, Lockstone HE, Lindgren CM, Taylor JM, Allen M, Zondervan KT, McCarthy MI 2010. Variability of gene expression profiles in human blood and lymphoblastoid cell lines. BMC Genomics 11: 96 doi: 10.1186/1471-2164-11-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks SA, Leonardson A, Zhu H, Cundiff P, Pietrusiak P, Edwards S, Phillips JW, Sachs A, Schadt EE 2004. Genetic inheritance of gene expression in human cell lines. Am J Hum Genet 75: 1094–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SB, Dermitzakis ET 2011. From expression QTLs to personalized transcriptomics. Nat Rev Genet 12: 277–282 [DOI] [PubMed] [Google Scholar]
- Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET 2010. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464: 773–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neitzel H 1986. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum Genet 73: 320–326 [DOI] [PubMed] [Google Scholar]
- Nica AC, Parts L, Glass D, Nisbet J, Barrett A, Sekowska M, Travers M, Potter S, Grundberg E, Small K, et al. 2011. The architecture of gene regulatory variation across multiple human tissues: The MuTHER Study. PLoS Genet 7: e1002003 doi: 10.1371/journal.pgen.1002003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petretto E, Mangion J, Dickens NJ, Cook SA, Kumaran MK, Lu H, Fischer J, Maatz H, Kren V, Pravenec M, et al. 2006. Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet 2: e172 doi: 10.1371/journal.pgen.0020172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras J-B, Stephens M, Gilad Y, Pritchard JK 2010. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464: 768–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson N, Hancks DC, Myers S, Reich D, Cheung VG, Spielman RS 2008. Effects of cis and trans genetic ancestry on gene expression in African Americans. PLoS Genet 4: e1000294 doi: 10.1371/journal.pgen.1000294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Helgason A, Thorleifsson G, McCarroll SA, Kong A, Stefansson K 2011. Single-tissue and cross-tissue heritability of gene expression via identity-by-descent in related or unrelated individuals. PLoS Genet 7: e1001317 doi: 10.1371/journal.pgen.1001317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raser JM, O'Shea EK 2005. Noise in gene expression: Origins, consequences, and control. Science 309: 2010–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, et al. 2010. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 140: 744–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito-Hisaminato A, Katagiri T, Kakiuchi S, Nakamura T, Tsunoda T, Nakamura Y 2002. Genome-wide profiling of gene expression in 29 normal human tissues with a cDNA microarray. DNA Res 9: 35–45 [DOI] [PubMed] [Google Scholar]
- Smyth GK, Speed T 2003. Normalization of cDNA microarray data. Methods 31: 265–273 [DOI] [PubMed] [Google Scholar]
- Spielman RS, Bastone LA, Burdick JT, Morley M, Ewens WJ, Cheung VG 2007. Common genetic variants account for differences in gene expression among ethnic groups. Nat Genet 39: 226–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Madeoy J, Strout JL, Wurfel M, Ronald J, Akey JM 2007. Gene-expression variation within and among human populations. Am J Hum Genet 80: 502–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Nica AC, Forrest MS, Dimas A, Bird CP, Beazley C, Ingle CE, Dunning M, Flicek P, Koller D, et al. 2007. Population genomics of human gene expression. Nat Genet 39: 1217–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM 2004. Power of the classical twin design revisited. Twin Res 7: 505–512 [DOI] [PubMed] [Google Scholar]
- Visscher PM, Medland SE, Ferreira MA, Morley KI, Zhu G, Cornes BK, Montgomery GW, Martin NG 2006. Assumption-free estimation of heritability from genome-wide identity-by-descent sharing between full siblings. PLoS Genet 2: e41 doi: 10.1371/journal.pgen.0020041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Hill WG, Wray NR 2008. Heritability in the genomics era—concepts and misconceptions. Nat Rev Genet 9: 255–266 [DOI] [PubMed] [Google Scholar]
- Whitney AR, Diehn M, Popper SJ, Alizadeh AA, Boldrick JC, Relman DA, Brown PO 2003. Individuality and variation in gene expression patterns in human blood. Proc Natl Acad Sci 100: 1896–1901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RBH, Chan EKF, Cowley MJ, Little PFR 2007. The influence of genetic variation on gene expression. Genome Res 17: 1707–1716 [DOI] [PubMed] [Google Scholar]
- Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. 2005. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21: 650–659 [DOI] [PubMed] [Google Scholar]
- Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, Maouche S, Germain M, Lackner K, Rossmann H, et al. 2010. Genetics and beyond—The transcriptome of human monocytes and disease susceptibility. PLoS ONE 5: e10693 doi: 10.1371/journal.pone.0010693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LQ, Li WH 2004. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol Biol Evol 21: 236–239 [DOI] [PubMed] [Google Scholar]
- Zheng Q, Wang XJ 2008. GOEAST: A web-based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res 36: W358–W363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, He F, Hu S, Yu J 2008a. On the nature of human housekeeping genes. Trends Genet 24: 481–484 [DOI] [PubMed] [Google Scholar]
- Zhu J, He F, Song S, Wang J, Yu J 2008b. How many human genes can be defined as housekeeping with current expression data? BMC Genomics 9: 172 doi: 10.1186/1471-2164-9-172 [DOI] [PMC free article] [PubMed] [Google Scholar]