

Abstract
New sequencing technology has dramatically altered the landscape of whole-genome sequencing, allowing scientists to initiate numerous projects to decode the genomes of previously unsequenced organisms. The lowest-cost technology can generate deep coverage of most species, including mammals, in just a few days. The sequence data generated by one of these projects consist of millions or billions of short DNA sequences (reads) that range from 50 to 150 nt in length. These sequences must then be assembled de novo before most genome analyses can begin. Unfortunately, genome assembly remains a very difficult problem, made more difficult by shorter reads and unreliable long-range linking information. In this study, we evaluated several of the leading de novo assembly algorithms on four different short-read data sets, all generated by Illumina sequencers. Our results describe the relative performance of the different assemblers as well as other significant differences in assembly difficulty that appear to be inherent in the genomes themselves. Three overarching conclusions are apparent: first, that data quality, rather than the assembler itself, has a dramatic effect on the quality of an assembled genome; second, that the degree of contiguity of an assembly varies enormously among different assemblers and different genomes; and third, that the correctness of an assembly also varies widely and is not well correlated with statistics on contiguity. To enable others to replicate our results, all of our data and methods are freely available, as are all assemblers used in this study.
The rapidly falling cost of sequencing means that scientists can now attempt whole-genome shotgun (WGS) sequencing of almost any organism, including those whose genomes span billions of base pairs. Interest in genome sequencing of new species has increased rapidly, inspired by high-profile successes such as the panda genome (Li et al. 2010a), the turkey (Dalloul et al. 2010), and several human resequencing efforts (Li et al. 2010b; Schuster et al. 2010; Ju et al. 2011), most of which used reads primarily or exclusively from Illumina sequencers. The read lengths in these projects ranged from 35 to 100 bp, and depth of coverage ranged from 50-fold to 100-fold. In contrast, earlier WGS projects using Sanger sequencing, such as the mouse (Waterston et al. 2002) and dog (Lindblad-Toh et al. 2005) genomes, used read lengths of 750–800 bp and required only sevenfold to 10-fold coverage.
The much deeper coverage of short-read sequencing projects does not entirely compensate for the shorter read length. A side-by-side comparison of the best assemblies produced with short-read data shows that assemblies with longer reads have far better contiguity than the latest short-read assemblies (Gnerre et al. 2011). This illustrates that assembling large genomes from short reads remains a very challenging problem, albeit one that has seen considerable progress in just the past two years. Indeed, except for a limited number of specialists in genome assembly, very few scientists know how to optimally design a sequencing strategy and then construct an assembly, and even these experts might not agree. The GAGE (Genome Assembly Gold-standard Evaluations) study was designed to provide a snapshot of how the latest genome assemblers compare on a sample of large-scale next-generation sequencing projects. The study, which was conceived in 2010 in response to the growing use of NGS for de novo assembly and the growing number of genome assembly packages, was designed to help answer questions such as:
What will an assembly based on short reads look like?
Which assembly software will produce the best results?
What parameters should be used when running the software?
As we show below, the answers to these questions depend critically on features of the genome, the design of the sequencing experiments, and on the software used for assembly.
Our results include the full “recipe” that we used for assembling each genome with each assembler. It is important to note in this context that similarly complete instructions are not available for any of the major landmark genomes including human (Lander et al. 2001; Venter et al. 2001) and mouse (Waterston et al. 2002), nor for recently published genomes such as panda (Li et al. 2010a). Whatever the cause, this lack of complete assembly information has made it impossible for others to replicate the assemblies of major published species. In contrast, we describe all procedures and parameters and provide the complete data sets used for each assembly in our study (see the Supplemental Material). This, coupled with the use of open-source assemblers, should permit replication of any of our results, in contrast with other recent assembly evaluations such as the Assemblathon (Earl et al. 2011) in which the assembly parameters were not described.
We also note that all of the data used in our evaluations were real sequence data from high-throughput sequencing machines, unlike the Assemblathon, which used data from a simulated genome (Earl et al. 2011). Simulated data may not capture the actual patterns of errors in real data or the variability present in naturally occurring genomes.
Results
The data
We chose whole-genome shotgun data from four deep-coverage sequencing projects covering two bacteria, a bee, and the human genome (Table 1). Three of the species were previously sequenced and finished to a very high standard using conventional Sanger technology, and later resequenced using Illumina technology. Having a finished genome allowed us to evaluate the correctness of each assembler on these species. We also included one species for which the “true” assembly is unknown: the bumble bee, Bombus impatiens. This genome is typical of many de novo assembly projects today, where the goal is primarily to create a draft-quality assembly that is the first representative of that species. Correct or not, these assemblies will likely remain for many years as the only reference sequence available.
Table 1.
Details of the four next-generation sequence data sets used for the GAGE assembly comparison

As Table 1 shows, the four genomes also represent a wide range of genome sizes, from 3 million base pairs (Mb) to 250 Mb (bee) to 3000 Mb (human). For human, however, we used only a single chromosome (chromosome 14) as a representative for the complete genome. We chose to use this smaller sample, just 1/30 of the genome, because some of the assemblers in our comparison would take many weeks to assemble the complete genome, and others would fail entirely. The NGS reads for human derived from a whole-genome sequencing project; we created our data set by first mapping all reads to the genome and then extracting those mapped to chromosome 14 (see Methods).
The Staphylococcus genome has one main chromosome and a small plasmid, while the Rhodobacter genome has two chromosomes and five plasmids. Thus even the bacteria had multiple chromosomes. The read lengths (all Illumina) ranged from 37 to 124 bp.
The assemblers
We chose eight of the leading genome assemblers, each of which is able to run large, whole-genome assemblies using Illumina-only short read data:
ABySS (Simpson et al. 2009)
ALLPATHS-LG (Gnerre et al. 2011)
Bambus2 (Koren et al. 2011) (http://www.cbcb.umd.edu/software/bambus).
CABOG (Miller et al. 2008)
SGA (Simpson and Durbin 2012)
SOAPdenovo (Li et al. 2010b)
Velvet (Zerbino and Birney 2008)
All of these are open source assemblers. For each genome and each assembler, we ran multiple assemblies using different parameters until we obtained what appeared to be an optimal or near-optimal result from that assembler. We used contig and scaffold N50 sizes as the primary metric to determine the best assembly for each program, without consideration of assembly errors. This strategy mimics what is commonly practiced among groups assembling a new genome: the assembly with the largest contigs and scaffolds is usually preferred. Software versions and details of the parameters used for each assembly are given in the Supplemental Material.
Some of these assemblers use a modular design, making it possible to mix and match different modules in different programs. For example, MSR-CA has its own “super-read” module to error-correct high-coverage Illumina reads and extend them into longer reads, which it then processes with modules from CABOG. Bambus2 uses CABOG modules to build contigs and then builds scaffolds from those.
Error correction and data cleaning
One of the most important steps in any assembly, often taking much longer than the assembly itself, is the data cleaning process. WGS data are never perfect, and the various types of errors can cause different problems for different assemblers. High-quality data can produce dramatic differences in the results: for example, one assembly of the Rhodobacter sphaeroides data (using an earlier release of SOAPdenovo) had a contig N50 size of just 233 bp, but after error correction the same assembler achieved a contig N50 of 7793 bp, more than 30 times larger.
Some of the assemblers we ran have their own built-in error-correction routines, but we wanted to tease apart the effectiveness of error correction and the assembly algorithms themselves. Therefore, the first step we ran with each of the data sets was an independent error correction method. We allowed assemblers that incorporate their own error correction routines to do further corrections in addition to this pre-processing. Abyss, SOAPdenovo, Velvet, and CABOG all produced improved results using error correction provided by a separate program, while the other assemblers were most effective when using their own error correction routines.
For all data sets, we ran the Quake error corrector (Kelley et al. 2010) to detect and correct sequencing errors. Quake bases its error detection on k-mers that occur only once or twice in a data set, indicative of a base-calling error. It then tries to replace the lowest-quality base with another base in order to create a k-mer that appears to belong to the genome. For most of the data sets, we also ran the ALLPATHS-LG error corrector (Gnerre et al. 2011). Although ALLPATHS-LG is primarily an assembler, we found that use of its corrected reads in some cases led to better assemblies than those based on Quake. Therefore, we extracted the corrected reads from ALLPATHS-LG and used them as another input to all of the assembly algorithms. We ran assemblers using both sets of error-corrected reads and chose the better assembly to report.
For some data sets, additional customized pre-processing was required. For B. impatiens, the large insert libraries (3 kb and 8 kb) used an adaptor sequence as part of the library construction protocol. Both libraries had significant numbers of reads that contained adaptor sequences. These adaptors were carefully trimmed out from all reads.
The assemblies
In Tables 2–5, we present snapshots of each assembly using a few metrics: the number, N50 size, and error-corrected sizes of contigs and scaffolds. The N50 value is the size of the smallest contig (or scaffold) such that 50% of the genome is contained in contigs of size N50 or larger. Precise recipes describing how to run each of the assemblers on each of our data sets can be found in the Supplemental Material and at http://gage.cbcb.umd.edu/recipes. These include the parameters used for each assembler as well as the series of steps required to run them, for those assemblers that require multiple steps. If an assembler could not be run on a given data set, then results for that assembler are not included.
Table 2.
Assemblies of S. aureus (genome size 2,872,915)

Table 5.
Assemblies of the bumble bee, B. impatiens (estimated size 250 Mb)

Corrected assembly contiguity analysis
It is critical to note here that the statistics in Tables 2–5 can be very misleading if an assembly contains errors; e.g., when two contigs are erroneously concatenated, the resulting assembly has larger contigs, but the assembly is worse, not better. Using the alignments to the reference genomes, we reevaluated the contig sizes for the three finished genomes. For this corrected analysis, we broke contigs at every misjoin and at every indel longer than 5 bases. This produced a revised picture of what the assembly's contiguity statistics would be if every error could be identified and the assembly could be split at that point. Note that errors can be very difficult to find, and assemblies with large numbers of errors present other problems for analysis. To present a more complete picture, Tables 2–4 include the numbers of errors and corrected N50 statistics for each assembler.
Table 4.
Assemblies of human chromosome 14 (ungapped size 88,289,540)

Evaluation of assembly accuracy
We assessed the correctness of the assemblies by aligning them to a completed reference genome. Tables 6 and 7 summarize the validation results for the three genomes for which a completed reference is available: Staphylococcus aureus, R. sphaeroides, and Hs14. A few common assembly problems are readily apparent: many small “chaff” contigs, missing reference sequence, unnecessarily duplicated contigs, repeat compressions, and widespread contig “misjoin” errors. Some of these errors are specific to certain assemblers (e.g., unaligned reference bases), while others are endemic across all of them (e.g., contig misjoins).
Table 6.
Statistics showing bases that failed to align or were present in different copy numbers in the reference genomes and the assemblies of S. aureus, R. sphaeroides, and Hs14

Table 7.
Statistics on insertions, deletions, and misassembly errors in the various assemblies of S. aureus, R. sphaeroides, and Hs14

For the analysis in Table 6, a “chaff” contig is defined as a single contig <200 bp in length. In many cases, these contigs can be as small as the k-mer size used to build the de Bruijn graph (e.g., 36 bp) and are too short to support any further genomic analysis.
One of the more difficult aspects of genome assembly is the estimation of repeat copy numbers. The statistics in Table 6 summarizing duplicated and compressed reference bases illustrate performance of the various assemblers on this task. A duplicated repeat is one that appears in more copies than necessary in the assembly, and a compressed repeat is one that occurs in fewer copies. Interestingly, the duplicated repeats appear to be a preventable problem, one that many of the assemblers handle better than others.
For example, in the S. aureus assemblies, ALLPATHS-LG, Bambus2, and SGA all produce only on the order of hundreds of bases in duplications. This may be explained by the tendency of assemblers to output the fewest copies of a repeat that can be explained by the data. In contrast, compressed repeats appear to be a systematic problem with the short-reads assemblers, with all assemblers compressing a significant number of base pairs. Suppression of segmental duplications is a well-known deficiency of modern sequencing and assembly strategies (Kelley and Salzberg 2010).
Single nucleotide polymorphisms (SNPs) and short insertions and deletions (indels), shown in Table 7, also vary by assembler. The number of SNPs and indels varied by an order of magnitude, possibly as a function of the “aggressiveness” of the assembler. An important caveat regarding the human SNPs is that we did not have a true reference for the human sample, NA12878, and this individual genome contains many true SNPs when compared with the human reference genome. However, because we are using a common reference genome and read set, the relative number of SNPs between assemblers should be a valid proxy measure of their single nucleotide errors.
A more aggressive assembler (e.g., SOAPdenovo) is prone to creating more segmental indels as it strives to maximize the lengths of its contigs, while a conservative assembler (e.g., SGA) minimizes errors at the expense of contig size. Interestingly, each assembler has a unique profile of indel error types. Figure 1 shows indel profiles for indels <100 bp in the ALLPATHS-LG, CABOG, and SOAPdenovo assemblies of human chr14. These plots demonstrate that ALLPATHS-LG and CABOG share a similar error pattern, with the majority of indel errors attributed to misestimation of tandem repeat copy numbers, and a relative balance between compressions and expansions. In contrast, SOAPdenovo shows tandem copy errors with a slight bias toward compressions, in addition to an unusual number of segmental deletions (characterized in Fig. 1 by indels plotted at x > 0 and y ≈ 0; for more details, see the Supplemental Material). With short reads, tandem repeat length estimation is a notoriously difficult problem—however, many segmental deletions can be avoided with careful use of mate-pair libraries or read threading algorithms.
Figure 1.

Comparison of the indel profiles for three assemblies of human Chr14. Every indel in the assembly is defined by the two aligned segments on either side. For each indel, the x-axis displays the distance between the two adjacent segments in the reference, and the y-axis displays the distance in the query. Thus, the point x = 100, y = 0 indicates a 100-bp deletion in the assembly, relative to the reference. Deletions from the assembly lie below the line y = x, and insertions in the assembly lie above. The indels can be roughly categorized by quadrant: (top right) divergent sequence; (bottom right) segmental assembly deletion; (bottom left) tandem repeat collapse/expansion; (top left) segmental assembly insertion. No points lie on the line y = x because only indels >5 bp are displayed. For details, see the Supplemental Methods.
“Misjoin” errors are perhaps the most harmful type, in that they represent a significant structural error. A misjoin occurs when an assembler incorrectly joins two distant loci of the genome, which most often occurs within a repeat sequence. We have tallied three types of misjoins: (1) inversions, where part of a contig or scaffold is reversed with respect to the true genome; (2) relocations, or rearrangements that move a contig or scaffold within a chromosome; and (3) translocations, or rearrangements between chromosomes. For scaffolds, relocations and indels are grouped together as Reloc/Indel, where an indel error in a scaffold means that a contig (>200 bp in length) has been deleted or inserted incorrectly. These larger-scale indels are essentially relocations where a contig has been moved. (Note that interchromosomal rearrangements were not possible for our human assembly because only one chromosome was used. Table 7 reports both types of errors under the “Reloc” category, but they are broken out separately in the Supplemental Material.)
One conclusion from our analysis is that no assembler is immune from this type of serious error, and certain assemblers seem to be repeat offenders, while others are consistently more correct. Figure 2 shows a dot plot of the Rhodobacter genome as assembled into scaffolds by SOAPdenovo and Velvet. In this example, SOAPdenovo has clearly captured the correct structure of the chromosome and plasmids, and no misjoins are visible at this resolution. However, the Velvet assembly exhibits multiple inversion and relocation errors in the main chromosome. This relative performance is captured in Table 7, where ALLPATHS-LG and SOAPdenovo have the fewest scaffold misjoins (12) and Velvet has the largest (38).
Figure 2.

A dot-plot comparison of the SOAPdenovo and Velvet scaffolds of R. sphaeroides. The finished reference chromosomes are plotted on the x-axis and the assembly scaffolds on the y-axis. Dotted lines indicate scaffold or chromosome boundaries. The apparent rearrangement at the top right of the SOAPdenovo plot is an artifact of the circular reference plasmid.
Effect of multiple libraries on assembly
An important question in the design of any whole-genome sequencing experiment is that of the number and sizes of paired-end libraries to use. Creating long-range paired-end libraries can be very helpful for assembly, but the sequencing protocols are much more costly and technically more difficult. With today's technology, paired-end libraries in the 100–300-bp range are the most economical. To evaluate the effect of library variety and size on assembly, we reassembled the Rhodobacter genome using the two original libraries plus one additional library, which consisted of 100-bp reads from 210-bp fragments, downloaded from the Sequence Read Archive. The 210-bp library had approximately the same number of reads as the 180-bp library. We assembled the genome 32 times, using all combinations of two libraries and the short library along with each assembler. The results are shown in Figure 3 and Supplemental Table 2. For ease of comparison, only two statistics are reported: the number of contigs and the (uncorrected) N50 contig size.
Figure 3.

Assemblies of R. sphaeroides using four different combinations of paired-end libraries as input to the assemblers. Each run used either one library (180 bp only) or a different combination of two libraries from 180 to 3000 bp. Note that N50 values are uncorrected; see Table 3 for the true N50 sizes for the 180 bp + 3 kb combination, which are much lower in some instances; e.g., SOAPdenovo has a corrected N50 of 14.3 kb (rather than 131.7 kb) for assembly with the 180-bp and 3-kb libraries.
For five of the assemblers, the best N50 statistic was obtained with the 180-bp and 3-kb library combination; however, ABySS, SGA, and MSR-CA obtained better results using the 180-bp and 210-bp combination. The MSR-CA result was almost twice as large, suggesting that it was able to extract more contiguity information from the additional coverage provided by the second short fragment library. This result may also suggest that the 3-kb library contained artifacts that reduced its usefulness for some assemblers. We also note that the use of more than two libraries might produce superior results for some assemblers: The SOAPdenovo assembly of the giant panda genome (Li et al. 2010a) used five libraries with fragment sizes ranging from 150 bp to 10 kb.
Discussion
Comparison of assembly size and contiguity
The tables show very large differences in performance among assemblers, as well as variation in the performance of each individual assembler when applied to different genomes. Note that larger contigs are not always correct, and below we take note of some cases where misassembled contigs produced artificially large N50 values. As Table 6 shows, certain assemblers generate chaff contigs in large amounts. For Hs14, for example, SGA outputs more base pairs in chaff contigs than it does for the rest of the assembly. ABySS also has an unusually high quantity of chaff. This can be indicative of the assembler being unable to integrate short repeat structures into larger contigs, or not properly correcting erroneous bases. These problems might create numerous very short, unambiguous paths through the graph. Alternatively, the other assemblers might simply be eliminating short contigs from their output. In either case, though, this problem can easily be addressed by ignoring the chaff contigs.
Coverage of the reference genome can be measured by the percentage of reference bases aligned to any assembled contig. The best assemblers have both a low incidence of chaff and a high coverage of the reference genome. By this metric, ALLPATHS-LG and CABOG perform admirably well on Hs14 with only 0.03% of the assembly in chaff contigs, and only 2.8% and 1.7% of the chromosome (respectively) missing from the assembly. It would appear that these assemblers are able to resolve the complex repeat structure of the human genome by a combination of accurate error correction and good use of mate-pair information. Despite its performance on Hs14, however, CABOG leaves more of R. sphaeroides uncovered (7.5%) than any other assembler.
To provide a context, it is also worth considering whether some genomes are intrinsically more difficult to assemble than the others. Assembly difficulty is partly a function of repetitiveness, which also interacts with read length: In general, a repeat sequence creates a gap unless the reads fully contain (and are longer than) the repeat. Assemblers can fill in many of these gaps using paired-end information, as long as the paired-end distances are longer than the repeats. One measure of repetitiveness is K-mer uniqueness (Schatz et al. 2010), defined as the percentage of a genome that is covered by unique sequences of length K. We computed this ratio for the three known genomes in our study and compared it with the full human genome and the nematode Caenorhabditis elegans (Fig. 4). As the figure shows, the two bacteria are less repetitive than Hs14, and Hs14 is noticeably less repetitive than the full human genome.
Figure 4.

K-mer uniqueness ratio for the three genomes assembled in GAGE: the bacteria S. aureus and R. sphaeroides and human chromosome 14. The ratio is defined as the percentage of a genome that is covered by unique (i.e., non-repetitive) DNA sequences of length K. Shown for comparison are the k-mer uniqueness ratios for the full human genome and for the nematode C. elegans.
Importance of error correction
For all four genomes and for all eight assemblers used in GAGE, the best assemblies were created from reads that had been processed through extensive error correction routines. As noted above, contig sizes after error correction often increased dramatically, as much as 30-fold. This highlights the critical importance of data quality to a good assembly. For most of the assemblers, the best results came from using reads that had been corrected either by Quake or by ALLPATHS-LG (for details, see the Supplemental Material). MSR-CA and SGA produced better results using their own built-in error correction routines, but in all cases, error correction was a key part of the assembly process.
S. aureus
Table 2 shows that SOAPdenovo produced much larger contigs for S. aureus than any of the other systems, with an N50 size of 288 kb. However, after comparing it with the reference genome, we found that SOAPdenovo contained multiple assembly errors (Table 2). Breaking the assembly at these errors produced a much smaller N50 value of 63 kb. The N50 size for ALLPATHS-LG was initially 97 kb, and with many fewer assembly errors, breaking the contigs reduced the N50 value less dramatically, to 66 kb, making it the best of the assemblers on this genome. MSR-CA's corrected N50 of 48 kb placed it below SOAPdenovo, but with about half as many assembly errors (34 vs. 65), MSR-CA would appear preferable to SOAPdenovo.
ALLPATHS-LG, MSR-CA, and Bambus2 all produced very large scaffolds, with MSR-CA producing a single scaffold containing the entire main chromosome. However, this scaffold contained several inversions, and only ALLPATHS-LG and Bambus2 produced scaffolds with no major errors.
Note that CABOG was not run on S. aureus because one of the two paired-end libraries contains reads of just 37 bp, and CABOG has a minimum read length of 64 bp.
R. sphaeroides
For Rhodobacter (Table 3), Bambus2 had the smallest number of contigs and scaffolds, with relatively large N50 sizes in both categories. The largest contigs were built by SOAPdenovo (with an N50 size of 132 kb), followed by Bambus2 (93 kb) and ALLPATHS-LG (42 kb).
Table 3.
Assemblies of R. sphaeroides (genome size 4,603,060)

As with Staphylococcus, however, the errors in the assemblies made some, particularly SOAPdenovo, appear to be better than they really were. With 422 errors, SOAPdenovo was the most error-prone of all the assemblers for Rhodobacter, and after breaking contigs at these errors, its N50 size was just 14.3 kb, dropping it to fifth place for contiguity. Bambus2 had almost as many errors and dropped even further after correction, to 12.8 kb. ALLPATHS-LG's contiguity dropped the least, and after correction its contig N50 of 34.4 kb was the best, followed by MSR-CA at 19.1 kb.
ALLPATHS-LG also produced the best scaffolding results, with the main chromosome entirely spanned by a single scaffold, closely followed by MSR-CA and Bambus2. SOAPdenovo's scaffolding results were a distant fourth place, approximately five times smaller than ALLPATHS-LG. An important caveat on these results is that the Rhodobacter data set was created following the ALLPATHS-LG “recipe” for library construction, which makes it an ideal data set for that assembler.
Although the overall results were similar for the two bacterial data sets, the sizes of the contigs were generally much larger for S. aureus, and the contigs for a given assembler varied by as much as sixfold (for ABySS). This variation illustrates how one of the most important variables in predicting assembly contiguity may be the genome itself, which is an element that cannot be controlled.
Human chromosome 14
For the human chromosome data, most of the assemblers produced relatively poor results, and the differences between the best and worst assemblers were dramatic. As with Rhodobacter, the sequencing strategy and mate-pair data were designed specifically for ALLPATHS-LG, and the creators of some of the assemblers might not have anticipated or taken full advantage of this type of data (particularly the library with overlapping mates). Regardless of the reason, ALLPATHS-LG and CABOG clearly outperformed all of the other assemblers in the contiguity statistics shown in Table 4. CABOG's contigs were 30% larger than those from ALLPATHS-LG (45.3 kb vs. 36.5 kb), but both were far larger than those produced by any of the other methods, most of which built contigs in the 2–4-kb range. Even more dramatic was the exceptionally large scaffold produced by ALLPATHS-LG, which contained almost the entire chromosome in one scaffold of 81.6 Mb. The largest scaffold generated by any other assembler was one produced by Velvet, at only 4.6 Mb.
After adjusting for misassemblies (Table 4), CABOG remained slightly ahead of ALLPATHS-LG, with both dropping substantially, to 23.7 kb and 21.0 kb, respectively. They remained far ahead of the third-best assembler, SOAPdenovo, with an N50 size of just 7.4 kb. It is also important to note that all of the leading performers had thousands of assembly errors on this chromosome, which translates into tens of thousands of errors on a full human genome. Fewer errors were found in the assemblies of ABySS (704 errors) and SGA (981 errors), but their more-cautious approaches produced very small contig N50 sizes of 2.0 and 2.7 kb. Thus, despite all efforts at error correction and repeat identification, assembly of a mammalian genome from NGS data remains an extremely challenging problem.
B. impatiens
Unlike the other three genomes, the bumble bee (B. impatiens) does not have a finished reference. Based on the results above, contiguity and size statistics should be interpreted very cautiously; it is possible that assembly errors, if known, would dramatically change these values, as they did in our experiments on S. aureus above. Nonetheless, we found that SOAPdenovo generated contigs with nearly double the N50 size of CABOG, 57 kb versus 24 kb. The scaffold N50 sizes were all similar, although SOAPdenovo's were slightly larger than the others. Worth noting here is that in experiments using an earlier (2010) release of SOAPdenovo, it could only produce contigs with an N50 of 6.4 kb, indicating a substantial improvement in that assembler in its more recent version.
Most of the other assemblers could not assemble these data at all, for various reasons. ALLPATHS-LG could not be used because it requires at least one library with overlapping mate pairs, which this project did not have. The other assemblers appeared to be unable to handle the large number of reads (∼500 million), and most of them crashed, often after several days running on a 256-GB multi-core computer. This illustrates an underappreciated fact of genome assembly with current technology: For larger genomes, the choice of assemblers is often limited to those that will run without crashing.
Shared assembly errors
To address the question of whether assembly errors were common or different among all of the algorithms, we looked at the intersections of errors on the assembly of Hs14. Insofar as the errors are unique, then it might be beneficial to merge the results of multiple assemblers to produce a consensus assembly. We focused on errors >5 bp, which include the collapse or expansion of small tandem repeats as well as larger errors. As shown in Figure 5, Bambus2, Velvet, and SOAPdenovo had significantly more unique errors than the other assemblers, ranging from just over 2000 (SOAPdenovo) to 4000 (Bambus2). SGA had by far the fewest unique errors. Among the shared errors, ALLPATHS-LG and CABOG had the largest numbers, suggesting that these two assemblers might agree with one another and possibly that some of their errors might represent true haplotype differences. Finally, there were about 200 errors shared by all eight assemblers, indicating that these are likely true variations in the target genome rather than errors.
Figure 5.

Comparison of insertion and deletion errors among all eight assemblers for human chromosome 14. (Blue) The indel errors >5 bp in length that are unique to each assembler. (Red bars) Indel errors made by at least one other assembler. (Green bars) Indels shared by all assemblers, which might represent true differences between the target genome and the reference.
Conclusions
Figure 6 summarizes the results across the three genomes for which the true assembly is available. ALLPATHS-LG demonstrated consistently strong performance based on contig and scaffold size, with the best trade-off between size and error rate, as shown in the figure. MSR-CA also performed relatively well, although with more errors than ALLPATHS-LG. Bambus2 seems to be a very capable scaffolder, as shown in Figure 6, but its contigs contain numerous small errors. (An explanation for this result is that contig merging is a very recent addition to Bambus2, one that is still under development.) The latter two assemblers use parts of the CABOG assembler for many of their core functions, and in this respect their performance is not independent. SOAPdenovo produced results that initially seemed superior to most assemblers, but on closer inspection it generated many misassemblies that would be impossible to detect without access to a reference genome. Despite its poor performance on human, SOAPdenovo performed very well on the bacteria, creating contigs that were eight times larger than it built on the human data. Finally, Table 7 and Figure 6 show that Velvet had a particularly high error rate for its scaffolds, creating many more inversions and translocations than any other algorithm.
Figure 6.

Average contig (A) and scaffold (B) sizes, measured by N50 values, versus error rates, averaged over all three genomes for which the true assembly is known: S. aureus, R. sphaeroides, and human chromosome 14. Errors (vertical axis) are measured as the average distance between errors, in kilobases. N50 values represent the size N at which 50% of the genome is contained in contigs/scaffolds of length N or larger. In both plots, the best assemblers appear in the upper right.
As illustrated by the differences between the original and corrected N50 values in Tables 2–4, an assembler can produce a large N50 value by using an overly aggressive assembly strategy, which, in turn, will yield a higher number of errors. In contrast, more conservative assemblers might produce smaller contigs but fewer errors. For the genomes examined here, ALLPATHS-LG and CABOG stood out as assemblers capable of producing both high contiguity and high accuracy. SOAPdenovo often produced similar or larger N50 values, but it appears to achieve this by sacrificing correctness. For all three of the previously sequenced genomes, SOAPdenovo showed a higher rate of chaff, duplications, compressions, SNPs, indels, and misjoins than CABOG and ALLPATHS-LG. Considering all metrics, and with the caveat that it requires a precise recipe of input libraries, ALLPATHS-LG appears to be the most consistently performing assembler, both in terms of contiguity and correctness.
For all of the assemblers, contig sizes for the human chromosome assembly were smaller than contigs for either of the bacterial genomes. The problem would only be more difficult if we had used the entire genome rather than a single chromosome. We conclude that, despite very significant improvements in assembly technology, the problem of assembling a large genome from short reads remains very difficult. The remarkable gains in sequencing throughput of recent years will require further improvements, especially in read length and in paired-end protocols, before we are likely to see accurate, highly contiguous mammalian assemblies. Thanks to algorithmic improvements, the assemblers used in this study can handle very large data volumes, but they will need longer-range linking information if they are to match or exceed the quality of assemblies based on Sanger sequencing technology.
Finally, we should note that all of the assemblers considered here are under constant development, and many will be improved by the time this analysis appears. Evaluations of assemblers such as GAGE are useful snapshots of performance, but ongoing reevaluation will be necessary as algorithms and sequencing technology change. Assembly evaluations should also be reproducible, which requires that the complete recipes for running these complex programs should be provided, as we have done here for the first time.
Methods
Data for S. aureus were downloaded from the Sequence Read Archive (SRA) at NCBI, accession numbers SRX007714 and SRX016063. The R. sphaeroides data have SRA accessions SRX033397 and SRX016063. The SRA libraries downloaded had higher coverage than was needed for most experiments. Each library was therefore randomly sampled to create a data set with 45× genome coverage, giving a total of 90× coverage for each genome.
To create the human chromosome 14 data set, reads sequenced from cell line GM12878 were downloaded from the SRA under the following accession numbers: SRR067780, SRR067784, SRR067785, SRR067787, SRR067789, SRR067791–SRR067793, SRR067771, SRR067773, SRR067776–SRR067779, SRR067781, SRR067786, SRR068214, SRR068211, SRR068335. Reads came from one short fragment library (mean read length 101 bp, fragment size 155 bp), two short jump libraries (101-bp mean read length, 2536-bp mean insert size), and two fosmid libraries (76-bp mean read length, 35,295-bp mean insert length). The original set of >1 billion reads was mapped against the entire human genome (GRCh37/hg19) using Bowtie (Langmead et al. 2009); reads mapping to multiple locations were randomly distributed across those locations (parameters: -l 28 -n 3 -e 300 -3 20 -M 1-best). Only reads mapping to Hs14 were retained. Each read in a pair was mapped separately to allow for inclusion of real distribution of insert sizes (including chimeric reads) and to avoid excessively filtering the data so as to better reflect the distribution in the original data set. The overall coverage of Hs14 was 60×, as shown in Supplemental Figure 1, and the number of gaps in coverage was 108, with gap sizes ranging from 1 to 2412 bp.
The B. impatiens data were sequenced at the Keck Center for Comparative and Functional Genomics, University of Illinois and released for public use by Gene Robinson.
Reads were error-corrected using both Quake and the ALLPATHS-LG error corrector (for details, see the Supplemental Methods). All assemblers were run using multiple parameters and with corrected and uncorrected reads as input; the best assembly for each genome was chosen.
For the three previously finished genomes, N50 sizes were computed based on the known size of the genome. For the bumble bee, N50 sizes used the estimated genome size of 250 Mb. Contigs and scaffolds of 200 bp or longer were used for all computations.
Because N50 size might sometimes be a misleading statistic, we also computed another statistic, which we call E-size. The E-size is designed to answer the question: If you choose a location (a base) in the reference genome at random, what is the expected size of the contig or scaffold containing that location? This statistic is one way to answer the related question: How many genes will be completely contained within assembled contigs or scaffolds, rather than split into multiple pieces? E-size is computed as:
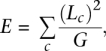 |
where LC is the length of contig C, and G is the genome length estimated by the sum of all contig lengths. E-size is computed similarly for scaffolds. To be consistent across all assemblies, we only considered contigs and scaffolds of 200 bp or longer in computing the E-size, and we used a constant value of G for all assemblies of a given genome. After computing E-sizes for all assemblies and all genomes, we found that they correlated very closely with N50 sizes in every case, validating our choice of N50 size as a representative assembly size metric. E-sizes for all assemblies can be found in Supplemental Table 1.
For evaluating correctness, alignment statistics and misassemblies were tallied using the program dnadiff (Phillippy et al. 2008) from MUMmer v3.23 (Kurtz et al. 2004). dnadiff operates by constructing local pairwise alignments between a reference and query genome using the Nucmer aligner. The aligned segments are then filtered to obtain a globally optimal mapping between the reference and query segments, while allowing for rearrangements, duplications, and inversions. This technique was later described in detail by Dubchak et al. (2009) as the SuperMap algorithm. Conveniently, this method identifies both a one-to-one mapping of segments as well as any duplicated sequences. When applied to assembly mapping, it can be used to measure the quantity and types of common misassemblies.
To create the alignments, contigs <200 bp were excluded, and the remainder were aligned using nucmer (Kurtz et al. 2004) with the options “-maxmatch -l 30 -banded -D 5.” Combined with its default options, this invocation requires a minimum exact-match anchor size of 30 bp and a minimum combined anchor length of 65 bp per cluster. Clusters are further required to have no more than 90 bp separation or more than five inserted bases between any two adjacent anchors. Acceptable clusters are then used to seed banded Smith-Waterman alignments (Smith and Waterman 1981). After running nucmer, alignments with <95% identity or >95% overlap with another alignment were discarded using delta-filter. dnadiff was then executed on the remaining alignments with default parameters, and correctness statistics were tabulated from its output (see the Supplemental Material).
For the scaffolds, we calculated three types of errors: indels, where there is an incorrect interleaving of multiple scaffolds; inversions, where a scaffold switches strands within a chromosome; and translocations, where a scaffold maps to multiple chromosomes in the reference. We also counted the number of gaps where the scaffold gap-size estimate is at least 1 kb off and the average absolute difference between the scaffold gap estimate and true gap size in each assembly. Details of how the scaffolds were aligned are in the Supplemental Material.
Any alignment-based metric is subject to the accuracy of the underlying alignments. Because complex repeat structures made the correct determination of alignment boundaries difficult in some cases, the figures presented here are to be taken only as estimates of the various features of each assembly. This is especially true of the misjoin features, which penalize small contig misassemblies just as severely as more major rearrangements. However, even allowing for some alignment-based error, the relative performance of each assembler would likely remain the same, and we should emphasize that all assemblies were analyzed with identical methods and against the same reference genomes.
Data access
All data sets, including error-corrected reads for each genome, are freely available from http://gage.cbcb.umd.edu/data.
Acknowledgments
This work was supported in part by NIH grants R01-LM006845 (S.L.S.), R01-HG006677 (S.L.S.), R01-HG04885 (M.P.), R01-HG002945 (J.A.Y. and A.Z.), USDA NRI grant 2009-35205-05209 (National Institute of Food and Agriculture) (S.L.S. and J.A.Y.), and was performed under Agreement No. HSHQDC-07-C-00020 (A.M.P.) awarded by the U.S. Department of Homeland Security for the management and operation of the National Biodefense Analysis and Countermeasures Center (NBACC), a Federally Funded Research and Development Center. The views and conclusions contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of the U.S. Department of Homeland Security.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.131383.111.
References
- Dalloul RA, Long JA, Zimin AV, Aslam L, Beal K, Blomberg Le A, Bouffard P, Burt DW, Crasta O, Crooijmans RP, et al. 2010. Multi-platform next-generation sequencing of the domestic turkey (Meleagris gallopavo): Genome assembly and analysis. PLoS Biol 8: e1000475 doi: 10.1371/journal.pbio.1000475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubchak I, Poliakov A, Kislyuk A, Brudno M 2009. Multiple whole-genome alignments without a reference organism. Genome Res 19: 682–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earl DA, Bradnam K, St John J, Darling A, Lin D, Faas J, Yu HO, Vince B, Zerbino DR, Diekhans M, et al. 2011. Assemblathon 1: A competitive assessment of de novo short read assembly methods. Genome Res 21: 2224–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnerre S, Maccallum I, Przybylski D, Ribeiro FJ, Burton JN, Walker BJ, Sharpe T, Hall G, Shea TP, Sykes S, et al. 2011. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci 108: 1513–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YS, Kim JI, Kim S, Hong D, Park H, Shin JY, Lee S, Lee WC, Yu SB, Park SS, et al. 2011. Extensive genomic and transcriptional diversity identified through massively parallel DNA and RNA sequencing of eighteen Korean individuals. Nat Genet 43: 745–752 [DOI] [PubMed] [Google Scholar]
- Kelley DR, Salzberg SL 2010. Detection and correction of false segmental duplications caused by genome mis-assembly. Genome Biol 11: R28 doi: 10.1186/gb-2010-11-3-r28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley DR, Schatz MC, Salzberg SL 2010. Quake: Quality-aware detection and correction of sequencing errors. Genome Biol 11: R116 doi: 10.1186/gb-2010-11-11-r116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Treangen TJ, Pop M 2011. Bambus 2: Scaffolding metagenomes. Bioinformatics 27: 2964–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL 2004. Versatile and open software for comparing large genomes. Genome Biol 5: R12 doi: 10.1186/gb-2004-5-2-r12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. International Human Genome Sequencing Consortium 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 doi: 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, et al. 2010a. The sequence and de novo assembly of the giant panda genome. Nature 463: 311–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. 2010b. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 20: 265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ III, Zody MC, et al. 2005. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438: 803–819 [DOI] [PubMed] [Google Scholar]
- Miller JR, Delcher AL, Koren S, Venter E, Walenz BP, Brownley A, Johnson J, Li K, Mobarry C, Sutton G 2008. Aggressive assembly of pyrosequencing reads with mates. Bioinformatics 24: 2818–2824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillippy AM, Schatz MC, Pop M 2008. Genome assembly forensics: Finding the elusive mis-assembly. Genome Biol 9: R55 doi: 10.1186/gb-2008-9-3-r55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz MC, Delcher AL, Salzberg SL 2010. Assembly of large genomes using second-generation sequencing. Genome Res 20: 1165–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster SC, Miller W, Ratan A, Tomsho LP, Giardine B, Kasson LR, Harris RS, Petersen DC, Zhao F, Qi J, et al. 2010. Complete Khoisan and Bantu genomes from southern Africa. Nature 463: 943–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Durbin R 2012. Efficient de novo assembly of large genomes using compressed data structures. Genome Res doi: 10.1101/gr.126953.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF, Waterman MS 1981. Identification of common molecular subsequences. J Mol Biol 147: 195–197 [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, et al. 2001. The sequence of the human genome. Science 291: 1304–1351 [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Zerbino DR, Birney E 2008. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]