

Abstract
Down syndrome (DS) is the most genetic cause of mental retardation and is caused by the triplication of chromosome 21. In addition to the disabilities caused early in life, DS is also noted as causing Alzheimer's-disease-like pathological changes in the brain, leading to 50–70% of DS patients showing dementia by 60–70 years of age. Inflammation is a complex process that has a key role to play in the pathogenesis of Alzheimer's disease. There is relatively little understood about inflammation in the DS brain and how the genetics of DS may alter this inflammatory response and change the course of disease in the DS brain. The goal of this review is to highlight our current understanding of inflammation in Alzheimer's disease and predict how inflammation may affect the pathology of the DS brain based on this information and the known genetic changes that occur due to triplication of chromosome 21.
1. Introduction
Down syndrome (DS) is the most common chromosomal anomaly among live-born infants and is the most frequent genetic cause of mental retardation [1, 2], with an incidence of one per 733 live births in the United States [3]. DS is caused by a triplication of chromosome 21 (a full list of genes located on chromosome 21 can be found in [4]). Due to the extensive number of genes triplicated, there is an extremely high incidence of congenital cardiac and gastrointestinal abnormalities [5]. DS is usually detected during pregnancy through first-trimester screening tests followed up by confirmation through amniocentesis, chorionic villus sampling, or percutaneous umbilical blood sampling [6].
Alzheimer's disease (AD) is the leading cause of dementia and is characterized clinically by a progressive loss of memory and cognition. An absolute diagnosis of AD can only occur after pathological analysis is performed on the brain tissue. There are two signature pathological lesions required for diagnosis; neuritic plaques composed of aggregated amyloid-β (Aβ) peptides, and neurofibrillary tangles composed of hyperphosphorylated, aggregated tau protein [7]. AD is usually considered a disease of aging, where currently 1 in 8 Americans over the age of 65 have AD yet half of those over 85 years have AD (data obtained from the Alzheimer's Association; www.alz.org).
In DS, Aβ deposits begin to accumulate in childhood and increase progressively with age [8]. There is an acceleration of this pathology between the ages of 35–45 years when other AD pathologies begin to occur, most importantly neurofibrillary tangles and inflammation [9]. Despite the certainty of developing AD-like pathologies in DS by mid-life, the onset of dementia is less certain. The consensus from a number of studies is that 50–70% of DS individuals will develop dementia by ages 60–70 years [10–13]. The reason individuals with DS develop Aβ deposits early in life is primarily due to the presence of some AD-related genes on chromosome 21, and hence these genes are triplicated in most cases of DS. Of the AD-associated genes triplicated in DS, the critical ones are amyloid precursor protein (APP) and β-amyloid cleavage enzyme 2 (BACE2). Aβ peptide is a cleavage product of APP. APP is a transmembrane protein and is differentially cleaved by enzymes called secretases of which there exist α-secretase, β-secretase (BACE), and γ-secretase. When β-secretase and γ-secretase cleave APP Aβ is a product, when α-secretase cleaves, this occurs in the middle of the Aβ portion and other peptides are produced.
Inflammation is known to occur in the brains of both AD and DS patients in response to the presence of neuritic plaques and neurofibrillary tangles. This inflammation is primarily mediated by microglial cells, although other glial cells and even neurons participate in this inflammatory response. It is becoming increasingly clear in the AD field that inflammation can directly influence plaques and tangles in the same way that plaques and tangles can directly influence inflammation. The purpose of this review is to discuss the evolving understanding of neuroinflammation in AD and determine how this may relate to the pathophysiology of DS.
2. Neuroinflammation in Alzheimer's Disease
Neuroinflammation is a complex process with many phenotypically varied states. The primary inflammatory cell in the brain is the microglial cell, which was first identified as a unique cell subtype by Del Rio Hortega in the 1920s. The microglial cell has been described as an ameboid-like cell that can be labeled immunocytochemically using macrophage cell surface markers [14, 15]. Other cells in the brain can contribute to the inflammatory response as well as microglia, although this contribution is considered to be significantly less than that of the microglia. Astrocytes and neurons can participate in the neuroinflammatory process as well as oligodendrocytes and vascular pericytes [16].
The view of neuroinflammation in the brain, and in disorders of the brain, has evolved over time, and continues to evolve as our understanding of the capabilities of the system grows. While once considered “immunologically privileged,” the brain is now known to exhibit an almost complete spectrum of inflammatory responses given the correct stimuli and environment. While once considered a cytotoxic loop [17], there are now examples of harnessing the inflammatory system of the brain to ameliorate AD pathologies and improve outcomes (see further discussion later in this section).
In AD, microglia expressing some classic activation markers such as MHC-II (associated with antigen presentation), CD68 (a lysosomal protein), and CD36 (a class B scavenger receptor) are highly localized to the area immediately surrounding an amyloid plaque or neurofibrillary tangle [18]. While this led some to hypothesize that this reaction was contributing to the toxicity of these pathologies, others suggested that the microglia may be performing a beneficial function in removing the abnormal protein deposits from the brain. As yet, there is no consensus, and it is likely that both phenomena are occurring to differing degrees. To better understand these processes, researchers turned to the assessment of cytokines to determine the function(s) of these microglial cells.
In AD, many cytokines have been found to be altered. Among those, the most common are IL-1β, IL-6, TNFα, and TGFβ. IL-1β was first shown by Griffin et al. in 1995 to be associated with the development of neuritic amyloid plaques from diffused deposits using human postmortem tissue [19]. Later, Griffin et al. expanded their findings to develop a “cytokine cycle” hypothesis that suggested the IL-1β production in response to amyloid deposits initiated a series of events including increased APP production and processing by neurons, recruitment of astrocytes, and activation of these astrocytes leading to signaling in the microglia inducing yet further IL-1β [20]. IL-1β induces S100β production in astrocytes [21], which is a cytokine that promotes neurite growth [22]. Most recently, serum IL-1β has been found to be elevated in cases of mild cognitive impairment (MCI) that has a higher risk for conversion to dementia, possibly indicating that serum IL-1β may be useful for identifying those MCI patients at risk for converting to AD [23]. Also, there are genome-wide association studies (GWASs) that have identified IL-1β polymorphisms associated with AD (reviewed in [24]). It will require further studies and analyses to determine whether these polymorphisms, are, in fact, associated with AD risk. However, in contrast to the negative data presented with respect to IL-1β, there is more recent data showing that IL-1β overexpression in the hippocampus of transgenic mice results in amelioration of amyloid pathology. IL-1β was increased specifically in a single hippocampus of an APP/PS1 transgenic mouse by genetic means and this hippocampus showed a 50% reduction in plaque load [25].
IL-6 is another cytokine that mediates immune responses and inflammatory reactions [26]. While microglia are the main source of IL-6 in the CNS, astrocytes, neurons, and endothelial cells are all capable of producing the cytokine [27–29]. In AD, brain tissue IL-6 has been shown to be elevated in pathologically relevant regions [30]. While much of the focus on IL-6 has been on its destructive effects such as induction of acute-phase proteins, increasing vascular permeability, activation of lymphocytes, and antibody synthesis (reviewed in [31]), there are some positive effects of IL-6 that may play a role in AD. This includes enhancing neuronal survival [32–34] and suppressing demyelination in a model of multiple sclerosis [35]. Moreover, in a mouse model of amyloid deposition, Chakrabarty et al. showed that overexpression of IL-6 enhanced microglial phagocytosis of amyloid deposits and, therefore, ameliorated amyloid burden [36].
TNFα is another cytokine that has been shown to have both beneficial and detrimental effects in the CNS. It acts as a highly potent proinflammatory and cytotoxic molecule in conditions of the CNS [37–40]. In contrast, TNFα has been shown to have trophic effects on hippocampal neurons [41] and provide protection from free-radical damage in primary neurons [42]. It is thought that the source of such dichotomous effects is the receptor subtype through which the TNFα is acting. There are two primary receptors for TNFα in the CNS; TNFα receptor 1 (TNFR1) and TNFα receptor 2 (TNFR2) [43]. TNFR1 mediates neuronal death via the TNF-receptor-associated death domain protein and caspase-8-activated apoptosis [44, 45]. TNFR2 is thought to mediate the beneficial, prosurvival action of TNFα through the nuclear factor-κB- (NFκB-) mediated antiapoptotic pathway [46]. This is likely an oversimplified view of the actions of TNFα through its receptors and there have been many subtleties of these systems described in the literature. In AD, it has been shown that expression of TNFR1 is elevated in the brain while levels of TNFR2 are decreased [44]. In addition, clinical trials are ongoing for the treatment of AD with etanercept, a fusion protein combining TNFR2 and the Fc portion of IgG used to treat Crohn's disease and arthritis as well as other autoimmune disorders [47]. Etanercept acts as a decoy receptor for TNF, reducing the effects of TNF at the biologically active receptors. Preliminary studies showed that perispinal delivery of etanercept in a small number of AD patients improved cognition [48]. In addition, thalidomide is also currently in clinical trials for AD based on its anti-TNFα effects. In transgenic mice, thalidomide has been shown to improve learning and memory [49].
Finally, TGFβ is a growth factor that has been shown to play a prominent role in tissue development, homeostasis, and repair [50]. Unlike the cytokines discussed to this point, TGFβ is associated mostly with repair mechanisms and is not known for its damaging or cytotoxic actions in the CNS. Instead, it is mostly associated with the formation of a glial scar [51] and upregulation of extracellular matrix proteins [52–54]. In AD, TGFβ levels are increased in the brain [55] but decreased in serum [56]. In APP transgenic mice, overproduction of TGFβ by astrocytes results in lower parenchymal amyloid deposits but increased deposition of amyloid in the cerebrovasculature [57]. Most recently, Tesseur et al. have shown that deficiencies exist in TGFβ signaling in the human AD brain, and these deficiencies can lead to enhanced AD pathology and associated neurodegeneration [58].
There is a rapidly growing interest in better characterizing the inflammatory state in the brain, and especially in AD. A paper by Colton et al. in 2006 described “classical activation” and “alternative activation” of microglia in the brain [59]. Classical activation was used to describe the Th1 cytokines such as IFNγ, IL-1β, TNFα, and IL-6. Alternative activation was used to describe a state associated with anti-inflammatory, repair, and wound healing effects mediated by IL-10, TGFβ, IL-4, IL-13, arginase 1 (AG1), and tissue remodeling factors Found in Inflammatory Zone 1 (FIZZ1) and chitinase 3-like 3 (YM1). This paper showed that cultured microglial cells, transgenic mouse models of AD, and postmortem tissue from human AD brains all showed expression of both classical and alternative activation markers. Most interesting was that alternative activation markers were expressed to the same degree, sometimes more than the classical activation markers commonly associated with an inflammatory response.
We have now expanded on the concept of multiple activation states to include a full spectrum of macrophage responses. Shown in Figure 1 are the four distinct inflammatory states we are currently studying in the brain. These states are well characterized in the peripheral macrophage literature (reviewed in [60, 61]). The M1 response is stimulated by IFNγ and/or TNFα and is characterized by traditional inflammatory cytokines IL-1β, IL-6, and IL-12. Broadly, the M2 response represents the alternative activation state described by Colton et al. We can further categorize this state into M2a, M2b, and M2c. Each subtype of M2 response has distinct stimuli and responses. IL-4 and/or IL-13 initiate an M2a response that is characterized by tissue remodeling factors FIZZ and YM1 as well as AG1 and mannose receptor C1 (MRC1). Immune complexes stimulate an M2b response, which is a specific response that has components of both M1 and M2a states. Finally, IL-10 stimulates an M2c response, which is sometimes called an acquired deactivation state. The M2c response is characterized by a series of markers that actively antagonize M1 signaling pathways. By categorizing the inflammatory response into these distinct types where each stimuli and marker is established, we can better understand what role(s) each state plays in AD progression and therapy.
Figure 1.
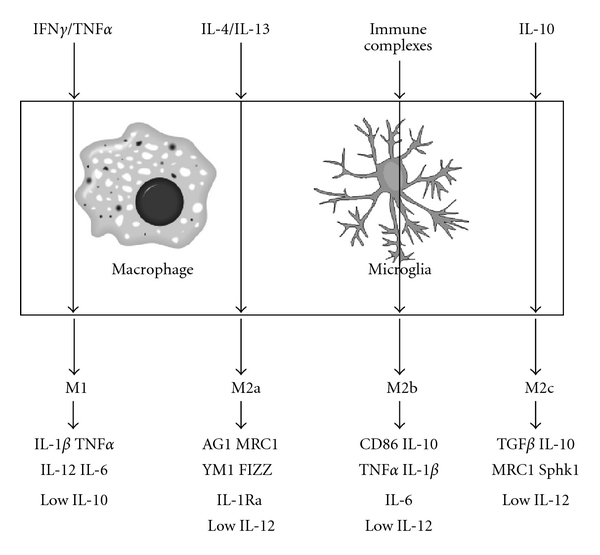
Schematic showing the four distinct states of inflammation possible in response to a stimuli in microglial/macrophage cells.
Drug development for the treatment of AD has recently been harnessing the inflammatory component of the disease for treatment. The most interesting approach is immunotherapy for AD. First demonstrated in 1999 [62], immunotherapy uses either an active vaccination approach or passive immunization to introduce anti-Aβ antibodies in patients (reviewed in [63]). These anti-Aβ antibodies then result in reductions in Aβ in the brain and ultimately, at least in transgenic mouse models, improvements in learning and memory [64, 65]. Injection of anti-Aβ antibodies directly into the brains of transgenic mouse models showed a dependence of amyloid removal on microglial activation [66, 67]. Later studies systemically administering anti-Aβ antibodies also showed a transient activation of microglia [68] and a reduced efficacy when the antibody was deglycosylated; a process that renders the IgG molecule incapable of interacting with effector cells such as microglia [69].
Another approach that targets the inflammatory response is the administration of nonsteroidal anti-inflammatory drugs (NSAIDs). NSAIDs showed great promise in retrospective epidemiological studies finding significant protection from AD with long-term NSAID use [70]. However, a prospective clinical trial performed by NIA/NIH, called the ADAPT trial, failed to show any significant benefit [71]. The NSAID story was furthermore clouded because some NSAIDs also possessed γ-secretase modifying properties that shifted APP cleavage to promote Aβ38 production, as opposed to Aβ40 or Aβ42 [72]. The NSAIDs found to have this activity were not included in the NIA/NIH trial. However, it was recently found that a subset of patients in the ADAPT trial did, in fact, benefit from NSAID use. Naproxen attenuated cognitive decline in a subgroup of AD patients termed “slow decliners,” whereas cognitive decline was accelerated in those termed “fast decliners” [73]. It is unclear why this would be the case, however, it is possible that different inflammatory states may exist in these different AD cases; some benefit from NSAIDs while some do not. Future studies will examine whether this is, indeed, the case.
3. Neuroinflammation in Down's Syndrome
While many of the pathways of inflammation described for AD will be directly relevant to DS, there are some critical inflammatory genes on chromosome 21 that will be triplicated in DS and may, therefore, influence the inflammatory state of the DS brain. We will discuss those factors in this review and take our current knowledge of inflammatory states in other neurological disorders to predict how these may be playing a role in DS. Table 1 shows the inflammatory-associated genes that are found on chromosome 21 and are triplicated in most DS patients. We will discuss each of these factors and their impact on the inflammatory balance of the brain.
Table 1.
A summary of the inflammation-related genes located on chromosome 21.
| Gene | Protein | Function | Ref |
|---|---|---|---|
| CXADR | Coxsackie virus and adenovirus receptor | Activation of JNK and p38-MAPK pathways leading to production of M1 cytokines. | [76] |
| ADAMTS1 | ADAM metalloproteinase with thrombospondin type 1 motif, 1 | Secreted protease known to be induced by IL-1β | [81] |
| ADAMTS5 | ADAM metalloproteinase with thrombospondin type 1 motif, 5 | Secreted protease known to be induced by IL-1β and TGFβ. | [82] |
| TIAM1 | T-cell lymphoma invasion and metastasis 1 | Necessary for cytokine-mediated generation of oxidative species through NADPH oxidase. | [87] |
| SOD1 | Superoxide dismuatose 1 | Scavenges superoxide radicals producing H2O2 and O2. | [109] |
| IFNAR2 | Interferon (alpha, beta, and omega) receptor 2 | Activates JAK/STAT-mediated pathway in response to IFNα/β. | [110] |
| IFNAR1 | Interferon (alpha, beta, and omega) receptor 1 | Activates JAK/STAT-mediated pathway in response to IFNα/β. | [110] |
| IFNGR2 | Interferon gamma receptor 2 | Activates JAK/STAT-mediated pathway in response to IFNγ. | [111] |
| RIPK4 | Receptor-interacting serine-threonine kinase 4 | Necessary for signaling through TNFR1 | [96] |
| CBS | Cystathione-beta-synthase | Production of hydrogen sulfide (H2S); a regulator of inflammation | [112] |
| S100B | S100 calcium binding protein B | Constitutive expression by astrocytes, released in response to TNFα | [113] |
| PRMT2 | Protein arginine methyltransferase 2 | Blocks the actions of NFκB in the nucleus | [114] |
CXADR is a gene encoding for a protein called coxsackie virus and adenovirus receptor (herein abbreviated CXADR). CXADR has a dual function as a viral receptor and an adhesion molecule associated with tight junctions. It is highly expressed in brain as well as systemic secretory organs such as the pancreas, testis, and small intestine [74]. In the heart, CXADR is increased in models of myocardial inflammation and cardiac injury in the absence of viral infection suggesting that there is an innate role of this protein in the inflammatory response [75]. Recently, it was shown that CXADR can induce stress-activated mitogen-activated protein kinase (MAPK) pathways in the heart leading to increased production of IFNγ, IL-12, IL-1β, TNFα, and IL-6 [76]. One can predict then that increased expression of CXADR in Down's syndrome may contribute to an overactivated M1 inflammatory response, since all of these inflammatory cytokines induced by CXADR are associated with an M1 response. In addition, CXADR has a significant role in tight junction function where, in endothelial cells, it facilitates transendothelial migration of neutrophils [77]. If CXADR expression is altered on the endothelial cells of the cerebrovasculature in DS patients, then there may be altered infiltration of peripheral inflammatory cells into the brain influencing the inflammatory response.
Two members of the ADAMTS (a disintegrin and metalloproteinase with thrombospondin motif) family are located on chromosome 21 and, therefore, subject to triplication in DS, ADAMTS1, and ADAMTS5. ADAMTS1 contains a signal peptide in the N-terminal region indicating it is secreted [78]. It acts as a proteinase degrading extracellular matrix proteoglycans such as aggrecan and versican [79]. ADAMTS5 is also a proteinase and shares the same substrates as ADAMTS1 [80]. Both ADAMTS1 and ADAMTS5 can be induced by IL-1β, indicating a dependence on an inflammatory response [81, 82]. It has been shown in DS that ADAMTS1 is five-fold overexpressed at the protein level, while ADAMTS5 was not significantly increased by Western blot measurements [83]. Given the induction by inflammatory cytokine IL-1β, one could hypothesize that the triplication of these proteinases would lead to exacerbated degradation of extracellular matrix proteins in response to an inflammatory insult. In addition, Griffin et al. showed that DS brain has greater IL-1β immunoreactivity indicating that there is more IL-1β present in the DS brain to stimulate the ADAMTSs [84].
T-cell lymphoma invasion and metastasis 1 (TIAM1) is a guanine nucleotide exchange factor for Rac1 [85] and, therefore, contributes to the activation of Rac1, which is necessary for the activation of NADPH oxidase [86]. Most recently, Tiam1 was found to be a critical regulatory factor in cytokine-induced induction of NADPH oxidase, more specifically, induction by IL-1β [87]. While these data used pancreatic β-cells, one could predict that overexpression of Tiam1 in the DS brain could lead to increased oxidative stress in response to an inflammatory insult that involves IL-1β. Indeed, it has been shown that Tiam1 protein expression is increased in fetal DS brain compared to control fetal brain [88].
Superoxide dismutase 1 (SOD1) binds copper and zinc and is a potent endogenous antioxidant. The enzyme is a soluble cytoplasmic and mitochonidral interspace protein that converts superoxide radicals to molecular oxygen and hydrogen peroxide [89]. Mutations in the SOD1 gene are commonly associated with genetic susceptibility to anterolateral sclerosis (ALS) [90]. While the hypothesis for the role of these mutations centered on the potential loss of function, and, therefore, increased oxidative stress, there has been increasing evidence to discount this hypothesis including the lack of ALS symptoms or pathology in SOD1 knockout mice [91]. It is unclear what the consequence is of overexpression of nonmutant SOD1 as would occur in DS. In a model of retinitis pigmentosa, it was found that loss of SOD1 worsened the outcomes. However, when SOD1 was overexpressed in this model, the levels of oxidative damage were actually worse. The authors found that in the absence of a peroxide-detoxifying enzyme in the same cellular compartment, overexpression of SOD1 actually causes more oxidative stress [92]. It could be suggested that the same may be the case in DS if the triplication of SOD1 results in overexpression of the protein in the absence of an increased level of peroxide-detoxifying enzyme.
Interferon receptors IFNAR1, IFNAR2, and IFNGR2 are all located on chromosome 21 and are, therefore, all subject to triplication in most cases of DS. IFNAR1 and IFNAR2 both respond to IFNα, IFNβ, or IFNο and, upon ligand binding, activate the JAK/STAT signaling pathway leading to induction of proinflammatory gene expression such as IL-1β, TNFα, and IL-6. IFNGR2 uses the same signaling pathway but responds to IFNγ specifically. A mouse model for the study of DS, the trisomy 16 mouse, includes triplication of IFNGR2 and IFNAR2. These mice develop significant pathology in utero and rarely survive to birth. Studies in these mice have shown that anti-IFN IgG treatment of fetuses improves the mouse phenotype suggesting the triplication of the IFN receptors significantly contributes to the severe pathology present in these mice [93]. The same group later showed that introducing a partial knockout of the IFNAR2 and IFNGR2 can improve growth and viability of cultured neurons derived from the trisomy 16 mouse fetuses [94]. Since these genes are triplicated in DS, it is likely that there is a hyperresponsiveness to IFN in the DS patient that may lead to an increased inflammatory response, both in the brain and systemically.
Receptor-interacting serine-threonine kinase 4 (RIPK4) is a protein kinase involved in multiple cell signaling pathways. One of these pathways is the signaling pathway for the activation of NFkB [95]. In addition, RIPK4 is involved in the signaling cascade of the TNFα receptor TNFR1 [96]. It is important to note that the TNFR1 is most heavily implicated with the toxic effects of TNFα and it could be predicted that overexpression of RIPK4 may increase responsiveness of TNFR1 to TNFα exacerbating the effects of TNFR1. At this time, however, this is purely speculative.
Cytathione beta synthase (CBS) is a cytosolic enzyme that catalyzes the desulfhydration of cysteine-producing hydrogen sulfide (H2S). H2S is now recognized as an atypical cellular messenger that has many normal physiological functions [97]. CBS binds NO or CO in its heme pocket and this binding modulates the activity of the enzyme [98]. H2S is a complicated signaling molecule with an apparent bimodal action on inflammation, where low levels appear to be anti-inflammatory, yet high levels may exacerbate inflammation in some instances. There are several extensive reviews on H2S signaling that discuss this phenomenon in great detail (see [99, 100]). It remains unclear how the overexpression of CBS in DS influences the DS pathology and whether the amount of H2S produced in DS patients is of the anti-inflammatory or proinflammatory concentrations.
S100β is a protein localized primarily to the brain where it is expressed by astrocytes. It is secreted by astrocytes in response to IL-1β and cyclic-AMP [101]. S100β is another inflammatory mediator with dichotomous actions. At low concentrations, it appears to enhance survival of neurons [102] and stimulate neurite outgrowth [22]. In contrast, high concentrations of S100β increases cell death [103] and causes apoptosis [104]. It has been shown in DS brains that S100 is greatly increased compared to control brain. The concentrations would place the levels of S100β in the toxic category, suggesting that the overexpression of S100β in DS brain plays a negative role in the aging pathology [84].
Protein arginine methytranferase 2 (PRMT2) is an enzyme that catalyzes the methylation of arginine. It has been shown that arginine methylation is a means of regulation of the JAK-STAT signaling pathway, which is key for many inflammatory processes including IFNγ, IFNα, and IL-6 [105]. In addition, natural degradation of proteins containing methylated arginine results in the production of asymmetric dimethylarginine (ADMA) [106]. ADMA is an endogenous inhibitor of nitric oxide synthase (NOS), a key player in normal cell signaling and inflammation [107]. It is unclear whether the triplication of PRMT2 results in significant changes in ADMA concentrations in the brain, however, DS patients with pulmonary hypertension do show increased ADMA concentrations compared to non-DS patients with pulmonary hypertension [108]. If this were also true for the brain, one could predict that there would be decreased production of NO and increased activation of the JAK-STAT pathway, both factors could influence the inflammatory state of the brain.
4. Inflammation Hypothesis and Future Directions
We hypothesize that the triplication of chromosome 21 as occurs in DS will result in a greatly exacerbated M1 inflammatory response. The basis for this hypothesis is the range of genes that are found on chromosome 21 and, therefore, triplicated. We have discussed each of the genes that are relevant to inflammation above and have summarized what these may mean to inflammation in Figure 2. Since most of the genes are primarily associated with the M1 inflammatory response, we predict that this is the main state that will be enhanced in the DS brain. Triplication of the major interferon receptors IFNAR1, IFNAR2, and IFNGR2 means that there will be enhanced interferon signaling. In turn, this enhanced signaling will increase production of M1 markers IL-1β, TNFα, and IL-6. While these components are known to result in oxidative stress, the triplication of TIAM1, SOD1, and PRMT2 will greatly exacerbate this oxidative stress. TIAM1 enhances oxidation by inducing NADPH oxidase, SOD1 at high concentrations has been shown to enhance oxidation, and PRMT2 inhibits nitric oxide production, which acts as an antioxidant in the brain at physiologic concentrations. All of these factors will combine to enhance neurodegeneration in the DS brain in response to primary pathologies such as amyloid plaques and neurofibrillary tangles.
Figure 2.
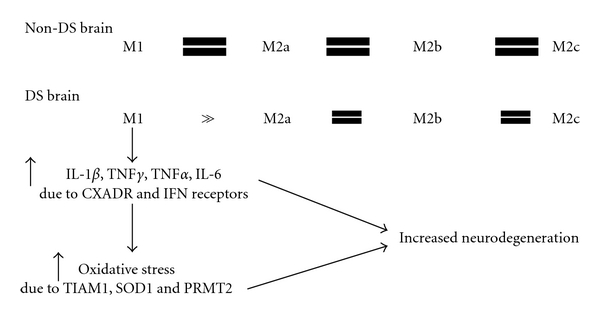
Schematic illustrating our hypothesis for the role of inflammation in Down syndrome.
In considering inflammation in DS, there is a relative lack of data relative to other disorders. While AD provides us with significant background information on the role of inflammation in the disease, it is clear that the condition of DS, and the triplication of so many inflammatory-associated genes, creates a unique inflammatory environment worthy for further study. The data obtained through the study of inflammation in DS will be essential to further not only the study of DS but also, in turn, the normal inflammatory pathways in neurodegenerative disorders.
References
- 1.Yang Q, Rasmussen SA, Friedman JM. Mortality associated with Down’s syndrome in the USA from 1983 to 1997: a population-based study. Lancet. 2002;359(9311):1019–1025. doi: 10.1016/s0140-6736(02)08092-3. [DOI] [PubMed] [Google Scholar]
- 2.Canfield MA, Honein MA, Yuskiv N, et al. National estimates and race/ethnic-specific variation of selected birth defects in the United States, 1999–2001. Birth Defects Research Part A. 2006;76(11):747–756. doi: 10.1002/bdra.20294. [DOI] [PubMed] [Google Scholar]
- 3.Improved national prevalence estimates for 18 selected major birth defects—United States, 1999–2001. Morbidity and Mortality Weekly Report. 2006;54(51):1301–1305. [PubMed] [Google Scholar]
- 4.Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 5.Van Trotsenburg ASP, Heymans HSA, Tijssen JGP, De Vijlder JJM, Vulsma T. Comorbidity, hospitalization, and medication use and their influence on mental and motor development of young infants with Down syndrome. Pediatrics. 2006;118(4):1633–1639. doi: 10.1542/peds.2006-1136. [DOI] [PubMed] [Google Scholar]
- 6.Kahn KR, Bett GCL. Triple-marker prenatal screening program for chromosomal defects. Obstetrics and Gynecology. 2009;114(5):p. 1147. doi: 10.1097/AOG.0b013e3181bf5744. [DOI] [PubMed] [Google Scholar]
- 7.Newell KL, Hyman BT, Growdon JH, Hedley-Whyte ET. Application of the National Institute on Aging (NIA)-Reagan Institute criteria for the neuropathological diagnosis of Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 1999;58(11):1147–1155. doi: 10.1097/00005072-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Leverenz JB, Raskind MA. Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: a regional quantitative analysis. Experimental Neurology. 1998;150(2):296–304. doi: 10.1006/exnr.1997.6777. [DOI] [PubMed] [Google Scholar]
- 9.Wisniewski KE, Wisniewski HM, Wen GY. Occurrence of neuropathological changes and dementia of Alzheimer’s disease in Down’s syndrome. Annals of Neurology. 1985;17(3):278–282. doi: 10.1002/ana.410170310. [DOI] [PubMed] [Google Scholar]
- 10.Oliver C, Crayton L, Holland A, Hall S, Bradbury J. A four year prospective study of age-related cognitive change in adults with Down’s syndrome. Psychological Medicine. 1998;28(6):1365–1377. doi: 10.1017/s0033291798007417. [DOI] [PubMed] [Google Scholar]
- 11.Lai F, Williams RS. A prospective study of Alzheimer disease in Down syndrome. Archives of Neurology. 1989;46(8):849–853. doi: 10.1001/archneur.1989.00520440031017. [DOI] [PubMed] [Google Scholar]
- 12.Johannsen P, Christensen JEJ, Mai J. The prevalence of dementia in Down syndrome. Dementia. 1996;7(4):221–225. doi: 10.1159/000106883. [DOI] [PubMed] [Google Scholar]
- 13.Janicki MP, Dalton AJ. Prevalence of dementia and impact on intellectual disability services. Mental Retardation. 2000;38(3):276–288. doi: 10.1352/0047-6765(2000)038<0276:PODAIO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 14.Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends in Neurosciences. 1988;11(6):273–277. doi: 10.1016/0166-2236(88)90110-5. [DOI] [PubMed] [Google Scholar]
- 15.Ling EA. Some aspects of amoeboid microglia in the corpus callosum and neighbouring regions of neonatal rats. Journal of Anatomy. 1976;121(1):29–45. [PMC free article] [PubMed] [Google Scholar]
- 16.Colton CA, Wilcock DM. Assessing activation states in microglia. CNS and Neurological Disorders - Drug Targets. 2010;9(2):174–191. doi: 10.2174/187152710791012053. [DOI] [PubMed] [Google Scholar]
- 17.McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neuroscience Letters. 1987;79(1-2):195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- 18.Xiang Z, Haroutunian V, Ho L, Purohit D, Pasinetti GM. Microglia activation in the brain as inflammatory biomarker of Alzheimer’s disease neuropathology and clinical dementia. Disease Markers. 2006;22(1-2):95–102. doi: 10.1155/2006/276239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Griffin WST, Sheng JG, Roberts GW, Mrak RE. Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. Journal of Neuropathology and Experimental Neurology. 1995;54(2):276–281. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- 20.Griffin WST, Sheng JG, Royston MC, et al. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ’cytokine cycle’ in disease progression. Brain Pathology. 1998;8(1):65–72. doi: 10.1111/j.1750-3639.1998.tb00136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sheng JG, Mrak RE, Rovnaghi CR, Kozlowska E, Van Eldik LJ, Griffin WST. Human brain S100β and S100β mRNA expression increases with age: pathogenic implications for Alzheimer’s disease. Neurobiology of Aging. 1996;17(3):359–363. doi: 10.1016/0197-4580(96)00037-1. [DOI] [PubMed] [Google Scholar]
- 22.Kligman D, Marshak DR. Purification and characterization of a neurite extension factor from bovine brain. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(20):7136–7139. doi: 10.1073/pnas.82.20.7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Forlenza OV, Diniz BS, Talib LL, et al. Increased serum IL-1β level in Alzheimer’s disease and mild cognitive impairment. Dementia and Geriatric Cognitive Disorders. 2009;28(6):507–512. doi: 10.1159/000255051. [DOI] [PubMed] [Google Scholar]
- 24.Di Bona D, Plaia A, Vasto S, et al. Association between the interleukin-1β polymorphisms and Alzheimer’s disease: a systematic review and meta-analysis. Brain Research Reviews. 2008;59(1):155–163. doi: 10.1016/j.brainresrev.2008.07.003. [DOI] [PubMed] [Google Scholar]
- 25.Shaftel SS, Kyrkanides S, Olschowka JA, Miller JNH, Johnson RE, O’Banion MK. Sustained hippocampal IL-1β overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. Journal of Clinical Investigation. 2007;117(6):1595–1604. doi: 10.1172/JCI31450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vallières L, Rivest S. Regulation of the genes encoding interleukin-6, its receptor, and gp130 in the rat brain in response to the immune activator lipopolysaccharide and the proinflammatory cytokine interleukin-1β . Journal of Neurochemistry. 1997;69(4):1668–1683. doi: 10.1046/j.1471-4159.1997.69041668.x. [DOI] [PubMed] [Google Scholar]
- 27.März P, Cheng JG, Gadient RA, et al. Sympathetic neurons can produce and respond to interleukin 6. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(6):3251–3256. doi: 10.1073/pnas.95.6.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hampel H, Haslinger A, Scheloske M, et al. Pattern of interleukin-6 receptor complex immunoreactivity between cortical regions of rapid autopsy normal and Alzheimer’s disease brain. European Archives of Psychiatry and Clinical Neuroscience. 2005;255(4):269–278. doi: 10.1007/s00406-004-0558-2. [DOI] [PubMed] [Google Scholar]
- 29.Frei K, Malipiero UV, Leist TP, Zinkernagel RM, Schwab ME, Fontana A. On the cellular source and function of interleukin 6 produced in the central nervous system in viral diseases. European Journal of Immunology. 1989;19(4):689–694. doi: 10.1002/eji.1830190418. [DOI] [PubMed] [Google Scholar]
- 30.Strauss S, Bauer J, Ganter U, Jonas U, Berger M, Volk B. Detection of interleukin-6 and α2-macroglobulin immunoreactivity in cortex and hippocampus of Alzheimer's disease patients. Laboratory Investigation. 1992;66(2):223–230. [PubMed] [Google Scholar]
- 31.Gadient RA, Otten UH. Interleukin-6 (IL-6)—a molecule with both beneficial and destructive potentials. Progress in Neurobiology. 1997;52(5):379–390. doi: 10.1016/s0301-0082(97)00021-x. [DOI] [PubMed] [Google Scholar]
- 32.Kushima Y, Hama T, Hatanaka H. Interleukin-6 as a neurotrophic factor for promoting the survival of cultured catecholaminergic neurons in a chemically defined medium from fetal and postnatal rat midbrains. Neuroscience Research. 1992;13(4):267–280. doi: 10.1016/0168-0102(92)90039-f. [DOI] [PubMed] [Google Scholar]
- 33.Hama T, Kushima Y, Miyamoto M, Kubota M, Takei N, Hatanaka G. Interleukin-6 improves the survival of mesencephalic catecholaminergic and septal cholinergic neurons from postnatal, two-week-old rats in cultures. Neuroscience. 1991;40(2):445–452. doi: 10.1016/0306-4522(91)90132-8. [DOI] [PubMed] [Google Scholar]
- 34.Kushima Y, Hatanaka H. Interleukin-6 and leukemia inhibitory factor promote the survival of acetylcholinesterase-positive neurons in culture from embryonic rat spinal cord. Neuroscience Letters. 1992;143(1-2):110–114. doi: 10.1016/0304-3940(92)90244-2. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez M, Pavelko KD, McKinney CW, Leibowitz JL. Recombinant human IL-6 suppresses demyelination in a viral model of multiple sclerosis. Journal of Immunology. 1994;153(8):3811–3821. [PubMed] [Google Scholar]
- 36.Chakrabarty P, Jansen-West K, Beccard A, et al. Massive gliosis induced by interleukin-6 suppresses Aβ deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB Journal. 2010;24(2):548–559. doi: 10.1096/fj.09-141754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mullberg J, Durie FH, Otten-Evans C, et al. A metalloprotease inhibitor blocks shedding of the IL-6 receptor and the p60 TNF receptor. Journal of Immunology. 1995;155(11):5198–5205. [PubMed] [Google Scholar]
- 38.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84(2):299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 39.Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4(4):387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 40.Antel JP, Becher B, Owens T. Immunotherapy for multiple sclerosis: from theory to practice. Nature Medicine. 1996;2(10):1074–1075. doi: 10.1038/nm1096-1074. [DOI] [PubMed] [Google Scholar]
- 41.Barger SW, Harmon AD. Microglial activation by alzhelmer amyloid precursor protein and modulation by apolipoprotein E. Nature. 1997;388(6645):878–881. doi: 10.1038/42257. [DOI] [PubMed] [Google Scholar]
- 42.Barger SW, Horster D, Furukawa K, Goodman Y, Krieglstein J, Mattson MP. Tumor necrosis factors α and β protect neurons against amyloid β- peptide toxicity: evidence for involvement of a κB-binding factor and attenuation of peroxide and Ca2+ accumulation. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(20):9328–9332. doi: 10.1073/pnas.92.20.9328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quintana A, Molinero A, Florit S, et al. Diverging mechanisms for TNF-α receptors in normal mouse brains and in functional recovery after injury: from gene to behavior. Journal of Neuroscience Research. 2007;85(12):2668–2685. doi: 10.1002/jnr.21126. [DOI] [PubMed] [Google Scholar]
- 44.Zhao M, Cribbs DH, Anderson AJ, et al. The induction of the TNFα death domain signaling pathway in Alzheimer’s disease brain. Neurochemical Research. 2003;28(2):307–318. doi: 10.1023/a:1022337519035. [DOI] [PubMed] [Google Scholar]
- 45.Depuydt B, Van Loo G, Vandenabeele P, Declercq W. Induction of apoptosis by TNF receptor 2 in a T-cell hybridoma is FADD dependent and blocked by caspase-8 inhibitors. Journal of Cell Science. 2005;118(3):497–504. doi: 10.1242/jcs.01640. [DOI] [PubMed] [Google Scholar]
- 46.Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115(1):1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silva LCR, Ortigosa LCM, Benard G. Anti-TNF-α agents in the treatment of immune-mediated inflammatory diseases: mechanisms of action and pitfalls. Immunotherapy. 2010;2(6):817–833. doi: 10.2217/imt.10.67. [DOI] [PubMed] [Google Scholar]
- 48.Tobinick EL, Gross H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. Journal of Neuroinflammation. 2008;5, article no. 2 doi: 10.1186/1742-2094-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alkam T, Nitta A, Mizoguchi H, et al. Restraining tumor necrosis factor-alpha by thalidomide prevents the Amyloid beta-induced impairment of recognition memory in mice. Behavioural Brain Research. 2008;189(1):100–106. doi: 10.1016/j.bbr.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 50.Border WA, Noble NA. TGF-β in kidney fibrosis: a target for gene therapy. Kidney International. 1997;51(5):1388–1396. doi: 10.1038/ki.1997.190. [DOI] [PubMed] [Google Scholar]
- 51.Logan A, Berry M, Gonzalez AM, Frautschy SA, Sporn MB, Baird A. Effects of transforming growth factor β1 on scar production in the injured central nervous system of the rat. European Journal of Neuroscience. 1994;6(3):355–363. doi: 10.1111/j.1460-9568.1994.tb00278.x. [DOI] [PubMed] [Google Scholar]
- 52.Ignotz RA, Massague J. Transforming growth factor-β stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. Journal of Biological Chemistry. 1986;261(9):4337–4345. [PubMed] [Google Scholar]
- 53.Roberts AB, Sporn MB, Assoian RK. Transforming growth factor type β: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(12):4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wyss-Coray T, Feng L, Masliah E, et al. Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-β1. American Journal of Pathology. 1995;147(1):53–67. [PMC free article] [PubMed] [Google Scholar]
- 55.Flanders KC, Lippa CF, Smith TW, Pollen DA, Sporn MB. Altered expression of transforming growth factor-β in Alzheimer’s disease. Neurology. 1995;45(8):1561–1569. doi: 10.1212/wnl.45.8.1561. [DOI] [PubMed] [Google Scholar]
- 56.De Servi B, La Porta CAM, Bontempelli M, Comolli R. Decrease of TGF-β1 plasma levels and increase of nitric oxide synthase activity in leukocytes as potential biomarkers of Alzheimer’s disease. Experimental Gerontology. 2002;37(6):813–821. doi: 10.1016/s0531-5565(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 57.Wyss-Coray T, Lin C, Yan F, et al. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nature Medicine. 2001;7(5):612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- 58.Tesseur I, Zou K, Esposito L, et al. Deficiency in neuronal TGF-β signaling promotes neurodegeneration and Alzheimer’s pathology. Journal of Clinical Investigation. 2006;116(11):3060–3069. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. Journal of Neuroinflammation. 2006;3, article no. 27 doi: 10.1186/1742-2094-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology. 2004;25(12):677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 62.Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-β attenuates Alzheimer disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 63.Wilcock DM, Colton CA. Anti-amyloid-β immunotherapy in Alzheimer’s disease: relevance of transgenic mouse studies to clinical trials. Journal of Alzheimer’s Disease. 2008;15(4):555–569. doi: 10.3233/jad-2008-15404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilcock DM, Gordon MN, Ugen KE, et al. Number of Aβ inoculations in APP+PS1 transgenic mice influences antibody titers, microglial activation, and congophilic plaque levels. DNA and Cell Biology. 2001;20(11):731–736. doi: 10.1089/10445490152717596. [DOI] [PubMed] [Google Scholar]
- 65.Morgan D, Diamond DM, Gottschall PE, et al. A β peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408(6815):982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 66.Wilcock DM, Munireddy SK, Rosenthal A, Ugen KE, Gordon MN, Morgan D. Microglial activation facilitates Aβ plaque removal following intracranial anti-Aβ antibody administration. Neurobiology of Disease. 2004;15(1):11–20. doi: 10.1016/j.nbd.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 67.Wilcock DM, DiCarlo G, Henderson D, et al. Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. Journal of Neuroscience. 2003;23(9):3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wilcock DM, Rojiani A, Rosenthal A, et al. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. Journal of Neuroscience. 2004;24(27):6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilcock DM, Alamed J, Gottschall PE, et al. Deglycosylated anti-amyloid-β antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. Journal of Neuroscience. 2006;26(20):5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Szekely CA, Thorne JE, Zandi PP, et al. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer’s disease: a systematic review. Neuroepidemiology. 2004;23(4):159–169. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- 71.Meinert CL, McCaffrey LD, Breitner JC. Alzheimer’s Disease Anti-inflammatory Prevention Trial: design, methods, and baseline results. Alzheimer’s & Dementia. 2009;5(2):93–104. doi: 10.1016/j.jalz.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weggen S, Eriksen JL, Das P, et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature. 2001;414(6860):212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 73.Leoutsakos J-MS, Muthen BO, Breitner JC, Lyketsos CG. Effects of non-steroidal anti-inflammatory drug treatments on cognitive decline vary by phase of pre-clinical Alzheimer disease: findings from the randomized controlled Alzheimer's Disease Anti-inflammatory Prevention Trial. doi: 10.1002/gps.2723. International Journal of Geriatric Psychiatry. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tomko RP, Xu R, Philipson L. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(7):3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ito M, Kodama M, Masuko M, et al. Expression of coxsackievirus and adenovirus receptor in hearts of rats with experimental autoimmune myocarditis. Circulation Research. 2000;86(3):275–280. doi: 10.1161/01.res.86.3.275. [DOI] [PubMed] [Google Scholar]
- 76.Yuen S, Smith J, Caruso L, Balan M, Opavsky MA. The coxsackie-adenovirus receptor induces an inflammatory cardiomyopathy independent of viral infection. Journal of Molecular and Cellular Cardiology. 2011;50(5):826–840. doi: 10.1016/j.yjmcc.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 77.Zen K, Liu Y, McCall IC, et al. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Molecular Biology of the Cell. 2005;16(6):2694–2703. doi: 10.1091/mbc.E05-01-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei P, Zhao YG, Zhuang L, Hurst DR, Ruben S, Sang QXA. Protein engineering and properties of human metalloproteinase and thrombospondin 1. Biochemical and Biophysical Research Communications. 2002;293(1):478–488. doi: 10.1016/S0006-291X(02)00255-3. [DOI] [PubMed] [Google Scholar]
- 79.Kuno K, Okada Y, Kawashima H, et al. ADAMTS-1 cleaves a cartilage proteoglycan, aggrecan. FEBS Letters. 2000;478(3):241–245. doi: 10.1016/s0014-5793(00)01854-8. [DOI] [PubMed] [Google Scholar]
- 80.Tortorella MD, Liu RQ, Burn T, Newton RC, Arner E. Characterization of human aggrecanase 2 (ADAM-TS5): substrate specificity studies and comparison with aggrecanase 1 (ADAM-TS4) Matrix Biology. 2002;21(6):499–511. doi: 10.1016/s0945-053x(02)00069-0. [DOI] [PubMed] [Google Scholar]
- 81.Kuno K, Kanada N, Nakashirma E, Fujiki F, Ichimura F, Matsushima K. Molecular cloning of a gene encoding a new type of metalloproteinase- disintegrin family protein with thrombospondin motifs as an inflammation associated gene. Journal of Biological Chemistry. 1997;272(1):556–562. doi: 10.1074/jbc.272.1.556. [DOI] [PubMed] [Google Scholar]
- 82.Yamanishi Y, Boyle DL, Clark M, et al. Expression and regulation of aggrecanase in arthritis: the role of TGF-β . Journal of Immunology. 2002;168(3):1405–1412. doi: 10.4049/jimmunol.168.3.1405. [DOI] [PubMed] [Google Scholar]
- 83.Miguel RF, Pollak A, Lubec G. Metalloproteinase ADAMTS-1 but not ADAMTS-5 is manifold overexpressed in neurodegenerative disorders as Down syndrome, Alzheimer’s and Pick’s disease. Molecular Brain Research. 2005;133(1):1–5. doi: 10.1016/j.molbrainres.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 84.Griffin WST, Stanley LC, Ling C, et al. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(19):7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Veluthakal R, Palanivel R, Zhao Y, McDonald P, Gruber S, Kowluru A. Ceramide induces mitochondrial abnormalities in insulin-secreting INS-1 cells: potential mechanisms underlying ceramide-mediated metabolic dysfunction of the β cell. Apoptosis. 2005;10(4):841–850. doi: 10.1007/s10495-005-0431-4. [DOI] [PubMed] [Google Scholar]
- 86.Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circulation Research. 2006;98(4):453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- 87.Subasinghe W, Syed I, Kowluru A. Phagocyte-like NADPH oxidase promotes cytokine-induced mitochondrial dysfunction in pancreatic β-cells: evidence for regulation by Rac1. American Journal of Physiology. 2011;300(1):R12–R20. doi: 10.1152/ajpregu.00421.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cheon MS, Kim SH, Yaspo ML, et al. Protein levels of genes encoded on chromosome 21 in fetal Down syndrome brain: challenging the gene dosage effect hypothesis (Part I) Amino Acids. 2003;24(1-2):111–117. doi: 10.1007/s00726-002-0336-2. [DOI] [PubMed] [Google Scholar]
- 89.Rakhit R, Chakrabartty A. Structure, folding, and misfolding of Cu,Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochimica et Biophysica Acta. 2006;1762(11-12):1025–1037. doi: 10.1016/j.bbadis.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 90.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362(6415):59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 91.Reaume AG, Elliott JL, Hoffman EK, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nature Genetics. 1996;13(1):43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 92.Usui S, Oveson BC, Iwase T, et al. Overexpression of SOD in retina: Need for increase in H2O2-detoxifying enzyme in same cellular compartment. Free Radical Biology and Medicine. 2011;51(7):1347–1354. doi: 10.1016/j.freeradbiomed.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maroun LE. Anti-interferon immunoglobulins can improve the trisomy 16 mouse phenotype. Teratology. 1995;51(5):329–335. doi: 10.1002/tera.1420510509. [DOI] [PubMed] [Google Scholar]
- 94.Maroun LE, Heffernan TN, Hallam DM. Partial IFN-α/β and IFN-γ receptor knockout trisomy 16 mouse fetuses show improved growth and cultured neuron viability. Journal of Interferon and Cytokine Research. 2000;20(2):197–203. doi: 10.1089/107999000312612. [DOI] [PubMed] [Google Scholar]
- 95.Meylan E, Martinon F, Thome M, Gschwendt M, Tschopp J. RIP4 (DIK/PKK), a novel member of the RIP kinase family, activates NF-κB and is processed during apoptosis. EMBO Reports. 2002;3(12):1201–1208. doi: 10.1093/embo-reports/kvf236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rountree RB, Willis CR, Dinh H, et al. RIP4 regulates epidermal differentiation and cutaneous inflammation. Journal of Investigative Dermatology. 2010;130(1):102–112. doi: 10.1038/jid.2009.223. [DOI] [PubMed] [Google Scholar]
- 97.Barañano DE, Ferris CD, Snyder SH. Atypical neural messengers. Trends in Neurosciences. 2001;24(2):99–106. doi: 10.1016/s0166-2236(00)01716-1. [DOI] [PubMed] [Google Scholar]
- 98.Taoka S, Banerjee R. Characterization of NO binding to human cystathionine β-synthase: possible implications of the effects of CO and NO binding to the human enzyme. Journal of Inorganic Biochemistry. 2001;87(4):245–251. doi: 10.1016/s0162-0134(01)00335-x. [DOI] [PubMed] [Google Scholar]
- 99.Li L, Bhatia M, Moore PK. Hydrogen sulphide—a novel mediator of inflammation? Current Opinion in Pharmacology. 2006;6(2):125–129. doi: 10.1016/j.coph.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 100.Whiteman M, Le Trionnaire SL, Chopra M, Fox B, Whatmore J. Emerging role of hydrogen sulfide in health and disease: critical appraisal of biomarkers and pharmacological tools. Clinical Science. 2011;121(11):459–488. doi: 10.1042/CS20110267. [DOI] [PubMed] [Google Scholar]
- 101.Zimmer DB, Van Eldik LJ. Analysis of the calcium-modulated proteins, S100 and calmodulin, and their target proteins during C6 glioma cell differentiation. Journal of Cell Biology. 1989;108(1):141–151. doi: 10.1083/jcb.108.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Van Eldik LJ, Christie-Pope B, Bolin LM, Shooter EM, Whetsell WO. Neurotrophic activity of S-100β in cultures of dorsal root ganglia from embryonic chick and fetal rat. Brain Research. 1991;542(2):280–285. doi: 10.1016/0006-8993(91)91579-p. [DOI] [PubMed] [Google Scholar]
- 103.Selinfreund RH, Barger SW, Pledger WJ, Van Eldik LJ. Neurotrophic protein S100β stimulates glial cell proliferation. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(9):3554–3558. doi: 10.1073/pnas.88.9.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sorci G, Riuzzi F, Agneletti AL, Marchetti C, Donato R. S100B causes apoptosis in a myoblast cell line in a RAGE-independent manner. Journal of Cellular Physiology. 2004;199(2):274–283. doi: 10.1002/jcp.10462. [DOI] [PubMed] [Google Scholar]
- 105.Mowen KA, Tang J, Zhu W, et al. Arginine methylation of STAT1 modulates IFNα/β-induced transcription. Cell. 2001;104(5):731–741. doi: 10.1016/s0092-8674(01)00269-0. [DOI] [PubMed] [Google Scholar]
- 106.McDermott JR. Studies on the catabolism of Ng-methylarginine, Ng, Ng-dimethylarginine and Ng, Ng-dimethylarginine in the rabbit. The Biochemical Journal. 1976;154(1):179–184. doi: 10.1042/bj1540179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vallance P, Leone A, Calver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339(8793):572–575. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- 108.Cua CL, Rogers LK, Chicoine LG, et al. Down syndrome patients with pulmonary hypertension have elevated plasma levels of asymmetric dimethylarginine. European Journal of Pediatrics. 2011;170(7):859–863. doi: 10.1007/s00431-010-1361-x. [DOI] [PubMed] [Google Scholar]
- 109.Hong J-H, Kim M-J, Park M-R, et al. Effects of vitamin E on oxidative stress and membrane fluidity in brain of streptozotocin-induced diabetic rats. Clinica Chimica Acta. 2004;340(1-2):107–115. doi: 10.1016/j.cccn.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 110.Kim SH, Cohen B, Novick D, Rubinstein M. Mammalian type I interferon receptors consists of two subunits: IFNaR1 and IFNaR2. Gene. 1997;196(1-2):279–286. doi: 10.1016/s0378-1119(97)00240-0. [DOI] [PubMed] [Google Scholar]
- 111.Boselli D, Ragimbeau J, Orlando L, et al. Expression of IFNγR2 mutated in a dileucine internalization motif reinstates IFNγ signaling and apoptosis in human T lymphocytes. Immunology Letters. 2010;134(1):17–25. doi: 10.1016/j.imlet.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 112.Sen U, Givvimani S, Abe OA, Lederer ED, Tyagi SC. Cystathionine β-synthase and cystathionine γ-lyase double gene transfer ameliorate homocysteine-mediated mesangial inflammation through hydrogen sulfide generation. American Journal of Physiology. 2011;300(1):C155–C163. doi: 10.1152/ajpcell.00143.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Donato R, Sorci G, Riuzzi F, et al. S100B’s double life: intracellular regulator and extracellular signal. Biochimica et Biophysica Acta. 2009;1793(6):1008–1022. doi: 10.1016/j.bbamcr.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 114.Ganesh L, Yoshimoto T, Moorthy NC, et al. Protein methyltransferase 2 inhibits NF-κB function and promotes apoptosis. Molecular and Cellular Biology. 2006;26(10):3864–3874. doi: 10.1128/MCB.26.10.3864-3874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]