

Abstract
Unlike type 2 diabetes which is caused by the loss of insulin sensitivity, type 1 diabetes (T1D) is manifested by the absolute deficiency of insulin secretion due to the loss of β mass by autoimmune response against β-cell self-antigens. Although significant advancement has been made in understanding the pathoetiology for type 1 diabetes, the exact mechanisms underlying autoimmune-mediated β-cell destruction, however, are yet to be fully addressed. Accumulated evidence demonstrates that endoplasmic reticulum (ER) stress plays an essential role in autoimmune-mediated β-cell destruction. There is also evidence supporting that ER stress regulates the functionality of immune cells relevant to autoimmune progression during T1D development. In this paper, we intend to address the role of ER stress in autoimmune-mediated β-cell destruction during the course of type 1 diabetes. The potential implication of ER stress in modulating autoimmune response will be also discussed. We will further dissect the possible pathways implicated in the induction of ER stress and summarize the potential mechanisms underlying ER stress for mediation of β-cell destruction. A better understanding of the role for ER stress in T1D pathoetiology would have great potential aimed at developing effective therapeutic approaches for the prevention/intervention of this devastating disorder.
1. Introduction
Recent epidemiologic studies revealed that the incidence of type 1 diabetes (T1D) in most regions worldwide has been increasing by 2% to 5% [1]. Particularly, in some developing countries such as China, the rapid economic development along with changes in lifestyle and presumably the living environment has rendered this country with an annual increase of 7.4% for T1D prevalence [2]. Given that T1D is typically developed in children and juveniles, its impact on the quality of life is far more significant than that of type 2 diabetes, in which it usually occurs in adults. Although exogenous insulin therapy partly compensates the function of β cells, it cannot regulate blood glucose as accurately as the action of endogenous insulin. As a result, long-term improperly control of blood glucose homeostasis predisposes T1D patients to the development of diverse complications such as diabetic retinopathy [3–5], nephropathy [6, 7], neuropathy [8–10], foot ulcers [11–13], and cardiovascular diseases [14–16]. Although the underlying mechanisms leading to T1D have yet to be fully addressed, extensive studies have consistently demonstrated that endoplasmic reticulum (ER) stress plays a critical role in autoimmune-mediated β-cell destruction during the course of T1D development.
The pancreatic β-cells are equipped with highly developed endoplasmic reticulum (ER) to fulfill the requirement of secreting a large amount of insulin. This physiological feature renders β cells particularly vulnerable to ER stress [17]. Exhaustion of β cells is essential for the onset of T1D, which requires the residual β cells for compensated insulin secretion. While this compensated action is beneficial for control of blood glucose homeostasis, it also increases ER burden associated with the induction of unfolded protein response (UPR) and ER stress, which further exacerbates β-cell death. Although the implication of ER stress in β-cell death has been extensively emphasized, the underlying mechanisms, however, are yet to be fully elucidated. As such, understanding the role of ER stress in the loss of β mass and dissecting the mechanisms underlying ER stress would be important for developing therapeutic approaches aimed at prevention and intervention of type 1 diabetes. In the present paper, we will first intend to address the overall role of ER stress in autoimmune-mediated β-cell destruction based on published genetic and experimental data. The impact of ER stress on modulation of autoimmune response during the course of T1D development will be next discussed. We will finally focus on the possible pathways implicated in the induction of ER stress and summarize the potential mechanisms underlying ER stress for mediation of β-cell destruction.
2. The Endoplasmic Reticulum (ER)
ER is a membranous network of tubules, vesicles, and cisternae that are interconnected by the cytoskeleton in the cytoplasm of eukaryotic cells. ER is responsible for many general cellular functions, including the facilitation of protein folding and assembly [18–20], manufacture of the membranes [21], biosynthesis of lipid and sterol, storage of intracellular Ca2+, and transport of synthesized proteins in cisternae.
ER can be categorized into rough endoplasmic reticulum (RER) and smooth endoplasmic reticulum (SER). RER is responsible for protein synthesis, while SER is in charge of the synthesis of lipids and steroids, regulation of calcium concentration, attachment of receptors on cell membrane proteins, and detoxification of drugs. As featured by its name, RER bears ribosomes on the outer surfaces of the cisternae and looks bumpy and rough under a microscope. The newly synthesized proteins by RER are sequestered in cisternae and sent to Golgi complex or membrane via small vesicles. In contrast, SER does not have ribosomes on its cisternae and appears to have a smooth surface under the microscope. SER is found commonly in places such as in the liver and muscle. It is important for the liver to detoxify poisonous substances. Sarcoplasmic reticulum (SR) is a special type of SER, which is found in smooth and striated muscle. SR is responsible for the regulation of calcium levels. It sequesters a large store of calcium and releases them when the muscle cell is stimulated.
3. ER Stress
ER stress is the cellular responses to the disturbances of normal function of ER. The most focused and well-studied ER stress is that caused by protein misfolding. The accumulation of unfolded proteins leads to a protective pathway to restore ER function, termed as unfolded protein response (UPR). ER employs a type of special proteins called chaperones as a quality control mechanism. Chaperones attach to the newly synthesized proteins and assist them to fold into their native conformations. In addition, chaperones also help to break down unfolded or incorrectly folded proteins in the ER via a process called ER-associated degradation (ERAD). Protein folding requires a serial of ER-resident protein folding machinery. Exhaustion of those protein folding machineries or insufficient energy supply increases the accumulation of unfolded or misfolded proteins in ER, leading to the activation of UPR. Various physiological and pathological insults such as increased general protein synthesis, failure of posttranslational modifications, hypoxia, nutrient/glucose starvation, and alterations in calcium homeostasis can result in the accumulation of unfolded or misfolded proteins in ER which then causes ER stress [22]. For example, altered expression of antithrombin III [23, 24] or blood coagulation factor VIII [25, 26] results in the exhaustion of protein-folding machinery and thus induces UPR. Some physiological processes such as the differentiation of B lymphocytes into plasma cells along with the development of highly specialized secretory capacity can also cause accumulation of unfolded proteins and induce UPR [27–29]. In response to certain physiological and pathological insults, cells undergo UPR to get rid of the unfolded or misfolded proteins. Therefore, UPR is a protective mechanism by which it monitors and maintains the homeostasis of ER. For instance, UPR increases the folding capacity by upregulating ER chaperones and foldases, and attenuates the biosynthetic burden of secretory pathway through downregulating the expression of secreted proteins [30–32]. In addition, UPR also activates ERAD to eliminate unfolded proteins [33–35] (Figure 1). However, once the stress is beyond the compensatory capacity of UPR, the cells would undergo apoptosis. As such, UPR and ER stress are reported to be implicated in a variety of pathological processes, including diabetes, neurodegenerative diseases, pathogenic infections, atherosclerosis, and ischemia [22, 36].
Figure 1.
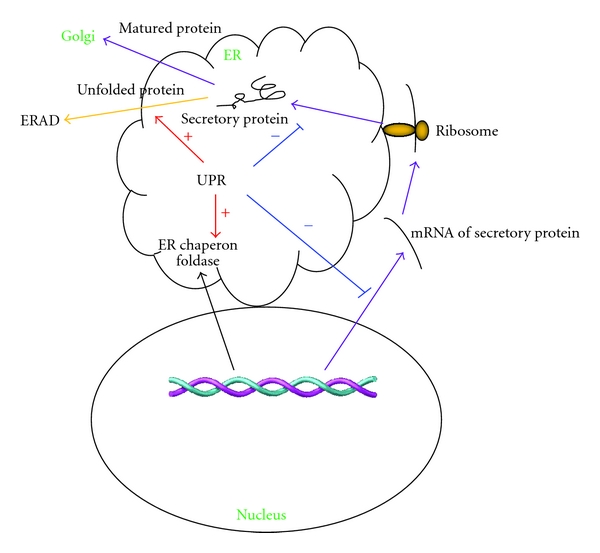
The regulatory role of unfolded protein response (UPR). Various physiological and pathological insults can result in the accumulation of unfolded proteins which then induces UPR and ER stress. In response to stressful insults, UPR regulates secretory pathway via following mechanisms: (1) enhancing (red arrow) the expression of ER chaperones and foldases to increase the folding capacity of ER; (2) attenuating (blue) the biosynthetic burden of secretory pathway through downregulating the expression of secreted proteins (purple arrow); (3) promoting the clearance of unfolded proteins by activating ERAD (orange arrow).
As aforementioned, there is a monitoring mechanism to ensure the correct protein folding in ER. The unfolded proteins usually have a higher number of hydrophobic surface patches than that of proteins with native conformation [37]. Thus, unfolded proteins are prone to aggregate with each other in a crowed environment and directed to degradative pathway [38]. Molecular chaperones in ER are the major mechanisms to promote protein folding. They preferentially interact with hydrophobic surface patches on unfolded proteins and create a private folding environment by preventing unfolded proteins from interaction and aggregation with other unfolded proteins. In addition, the concentration of Ca2+ in ER also impairs protein folding by inhibiting the activity of ER-resident chaperones and foldases [39–42]. ER is the major site for Ca2+ storage in mammalian cells. The concentration of Ca2+ in ER is thousands times higher than that in the cytosol [43]. Most chaperones and foldases in ER are vigorous Ca2+ binding proteins. Their activity, therefore, is affected by the concentration of Ca2+ in ER. A variety of posttranslational modifications including N-linked glycosylation, disulfide bond formation, lipidation, hydroxylation, and oligomerization occur in ER. Disruption of those posttranslational modifications can also result in the accumulation of incorrectly folded proteins and thereby induce UPR or ER stress. For example, glucose deprivation impairs the process for N-linked protein glycosylation and thus leads to ER stress [44].
UPR is mediated by three major pathways, which are initiated by the three transmembrane signaling proteins located on the ER membrane. Those transmembrane proteins function as a bridge to link cytosol and ER with their C-terminal in the cytosol and N-terminal in the ER lumen. The N-terminal is usually engaged by an ER-resident chaperone BiP (Grp78) to avoid aggregation. When unfolded proteins accumulate in ER, chaperons are occupied by unfolded proteins and release the transmembrane signaling proteins: which include the following three axes of signals: the pancreatic endoplasmic reticulum kinase (PERK), the inositol-requiring enzyme 1 (IRE1), and the activating transcription factor 6 (ATF6). The release of these proteins triggers UPR and ER stress (Figure 2). PERK is a Ser/Thr protein kinase uniquely present in ER. Once released from BiP, PERK becomes oligomerized and autophosphorylated. PERK inactivates eukaryotic initiation factor 2α (eIF2α) by phosphorylation of Ser51 to reduce mRNA translation and protein load on ER. Deficiency of PERK results in an abnormally elevated protein synthesis in response to the accumulation of unfolded proteins in ER. IRE1 is another axis of signal involved in UPR. IRE1 increases the production of X box protein-1 (XBP-1), a bZIP-family transcriptional factor, by promoting its mRNA splicing [45]. XBP-1 heterodimerizes with NF-Y and enhances gene transcription by binding to the ER stress enhancer (ERSE) and unfolded protein response element (UPRE) in the promoters of targeted genes. Unlike PERK and IRE1 which oligomerize upon UPR, when released from BiP, ATF6, the third axis of signal, translocates into the Golgi apparatus where its transmembrane domain is cleaved [46]. The cleaved ATF6 is then relocated into the nucleus to regulate the expression of targeted genes. For example, once released from the ER membrane, ATF6 enhances the transcription of XBP-1 mRNA which is further regulated by IRE1 [45].
Figure 2.
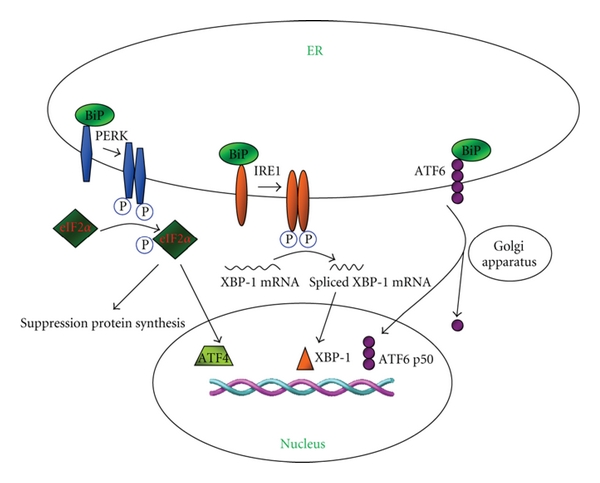
Signaling pathways relevant to UPR. PERK, IRE1, and ATF6 act as ER stress sensors by binding to the ER chaperone BiP, and by which they remain inactive under normal condition. Upon the accumulation of unfolded proteins, BiP preferentially binds to the unfolded proteins, which results in the release of PERK, IRE1, and ATF6. Once released from BiP, PERK becomes activated and dimerized. Activated PERK phosphorylates eIF2α to suppress the overall transcription of mRNAs while selectively enhance the transcription of genes implicated in UPR such as the ATF4 mRNA, and through which ATF4 initiates the transcription of UPR target genes. Similar to PERK, IRE1 is dimerized and activated after detached from BiP. IRE1 induces XBP-1 by promoting the splicing of its mRNA. XBP-1 activates the transcription of its target genes to enhance UPR. The release of ATF6 from BiP results in the translocation of ATF6 to the Golgi apparatus, where ATF6 is cleaved and then translocates into the nucleus, and by which ATF6 initiates the transcription of target genes.
4. ER Stress in Autoimmune-Mediated β-Cell Destruction
Accumulative evidence supports that ER stress is implicated in autoimmune-mediated β-cell destruction in type 1 diabetes [47, 48]. It was noted that loss of β cells is the direct causing factor for insufficient insulin secretion in T1D patients. As described earlier, pancreatic β cells have a very well-developed ER to fulfill their biological function for secreting insulin and other glycoproteins, and therefore, β cells are highly sensitive to ER stress and the subsequent unfolded protein response (UPR). Severe or long-term ER stress would direct β cells undergoing apoptosis [47]. For example, mice deficient in PERK, a molecule responsible for regulating UPR, are extremely susceptible to diabetes. The null mice display a progressive loss of β mass and hyperglycemia with aging [49]. Consistent with the observations in these mice, some infant-onset diabetes in humans have also been confirmed to be associated with the mutations in PERK. For example, loss of EIF2AK3 (the gene encodes PERK) develops Wolcott-Rallison syndrome, an autosomal recessive disorder characterized by early infancy insulin-dependent diabetes and multisystemic manifestations including growth retardation, hepatic/renal dysfunction, mental retardation, and cardiovascular abnormalities [50, 51]. Similarly, disruption of UPR by mutating eIF2α, a protein that controls mRNA translation upon ER stress, enhances the sensitivity to ER stress-induced apoptosis and results in defective gluconeogenesis. Mice carrying a homozygous Ser51Ala mutation for eIF2α show defective in pancreatic β cells manifested by the smaller core of insulin-secreting β cells and attenuated insulin secretion [52]. Altogether, defects in PERK/eIF2α signaling render β cells highly vulnerable to ER stress in both humans and mice [53, 54].
In type 1 diabetes, ER stress in the pancreatic β cells is primarily induced by proinflammatory cytokines produced by infiltrated immune cells, which then contributes to β-cell destruction. During the course of autoimmunity, pro-inflammatory cytokines are secreted by the infiltrated autoreactive immune cells in the milieu of pancreatic islets. For example, nitric oxygen (NO) is an inflammatory mediator resulted from autoimmune response during the course of type 1 diabetes. Studies have shown that excessive NO production induces β-cell apoptosis in a CHOP-dependent manner [55]. Other than ER stress caused by autoimmunity, misfolding of insulin in β cells can also directly induce chronic ER stress as evidenced by the observations in Akita mice. The Akita mouse carries a mutation for the Ins2 gene which disrupts a disulfide bond between the α and β chain of proinsulin, leading to the mis-folding of the mutated insulin, and by which the mutated insulin induces ER stress in β cells to cause diabetes [56].
It is likely that inflammatory cytokines produced by islet-infiltrated autoreactive immune cells are the major factors causing β-cell death in type 1 diabetes [57]. In the early stage of type 1 diabetes, the autoreactive immune cells such as macrophages and T lymphocytes infiltrate into the pancreatic islets along with the secretion of inflammatory cytokines such as IL-1β, IFN-γ, and TNF-α, which then induce ER stress to mediate β-cell destruction. The damaged or dying β cells also release danger signals such as high-mobility group box 1 and heat shock proteins (HSPs), to alert the immune system for the presence of β-cell injury, which in turn further promotes autoimmune progression [57–60]. Studies have shown that stimulation of β cells with IL-1β and IFN-γ induces the expression of death protein 5 (DP5), and through which these cytokines mediate β-cell apoptosis via ER stress [61]. Knockdown of DP5 provides protection for β cells against inflammatory cytokine-induced ER stress [61]. Insult of β cells with IL-1β and IFN-γ has also been found to decrease the expression of sarcoendoplasmic reticulum pump Ca2+ ATPase (SERCA) 2b, which controls the storage of ER Ca2+ [62]. It has been well demonstrated that altered ER Ca2+ concentration induces the accumulation of unfolded proteins in ER associated with the induction of UPR and ER stress in β cells [63].
Given that hyperglycemia only occurs when β cells fail to compensate the increased demand for insulin, β cells are usually “exhausted” in T1D patients [54]. Therefore, other than the ER stress induced by autoimmune response, β cells in T1D patients are also under ER stress caused by altered insulin synthesis. In later case, the increased insulin demand requires the remaining functional β cells to increase insulin synthesis to compensate the decrease of β mass. While this process in short term is beneficial for control of blood glucose homeostasis, it also induces ER stress, which in turn exacerbates β-cell dysfunction to promote disease progression and diabetes onset. Collectively, there is convincing evidence that ER stress plays an essential role in β-cell destruction during the course of T1D development.
5. The Impact of ER Stress on Modulation of Autoimmune Response
Unlike its well-defined effect on autoimmune-mediated β-cell destruction in type 1 diabetes, the impact of ER stress in modulating autoimmune response during the course of type 1 diabetes, however, remains poorly elucidated. There is evidence supporting that other than its critical roles played in β-cell destruction, ER stress also modulates the functionality of immune cells with implications in autoimmune response in type 1 diabetes.
It has been well accepted that the presence of β-cell-specific autoantibodies serves as a marker for the initiation and progression of autoimmunity in type 1 diabetes [64]. Studies have shown that IRE1, a key molecule in UPR, modulates the differentiation of antibody-producing B lymphocytes. Deficiency of IRE1 hampers pro-B cells differentiating into pre-B cells [65], and XBP-1, an IRE1 downstream molecule, is required for antibody production by mature B cells [66]. It was found that the engagement of B-cell receptor (BCR) induces ubiquitin-mediated degradation of BCL-6, a repressor for B-lymphocyte-induced maturation protein 1 (BLIMP1) [67], while BLIMP1 negatively regulates the expression of B-cell-lineage-specific activator protein (BSAP) [68], and BSAP is suggested to function as a repressor for XBP-1 [69]. In line with these results, B lymphocytes deficient in BLIMP1 failed to express XBP-1 in response to LPS stimulation [66].
Recent studies highlighted the importance of innate immunity in the pathogenesis of type 1 diabetes [59, 60], while elements of the UPR pathway are found to regulate innate immune response [70]. The expression of CREBH, an ER stress-associated transcription factor, can be induced by inflammatory cytokines such as IL-1β and IL-6, which in turn regulates the transcription of serum amyloid P-component and C-reactive protein, the two critical factors implicated in innate immune responses [71]. Furthermore, the differentiation of dendritic cells (DCs), the most critical innate immune cells, is regulated by UPR signaling element, XBP-1 [72]. High levels of mRNA splicing for XBP-1 are found in DCs, and mice deficient in XBP-1 show altered development of both conventional and plasmacytoid DCs. Loss of XBP-1 renders DCs vulnerable to ER stress-induced apoptosis [72]. Moreover, the capacity for DCs secretion of inflammatory cytokine IL-23 is regulated by CHOP, a UPR mediator. CHOP can directly bind to the IL-23 gene and regulate its transcription. ER stress combined with Toll-like receptor (TLR) agonists was found to markedly increase the mRNA of IL-23 p19 subunit and the secretion of IL-23, while knockdown of CHOP suppressed the induction of IL-23 by ER stress and TLR signaling [73].
Richardson and coworkers reported that innate immune response induced by P. aeruginosa infection causes ER stress in C. elegans, and mutations with loss of function for XBP-1 lead to larval lethality [74]. In consistent with this result, the polymorphisms within the XBP-1 gene were found to be associated with Crohn's disease and ulcerative colitis in humans [75], and the two autoimmune diseases share similar properties as type 1 diabetes. Loss of XBP-1 in intestinal epithelial cells induces Paneth cell dysfunction and overactive epithelium, leading to impaired mucosal defense to Listeria monocytogenes and increased sensitivity to colitis [75].
Other than the IRE1/XBP-1 axis, the PERK/eIF2α/ATF4 axis of UPR is also found to be associated with innate response. TLR signaling, the most important innate signaling pathway, is reported to induce selective suppression of the ATF-4/CHOP axis of UPR pathway [76]. TLR signaling decreases eIF2α-induced ATF4 translation. For example, pretreatment of LPS, an agonist for TLR4, suppressed ATF4/CHOP signaling and prevented systemic ER stress-induced apoptosis in macrophages, renal tubule cells, and hepatocytes [76]. In contrast, loss of Toll-IL-1R-containing adaptor inducing IFN-β (TRIF), an important adapter for TLR signaling, abrogated the protective effect of LPS on systemic ER stress-induced renal dysfunction and hepatosteatosis, suggesting that TLR signaling suppresses ATF4/CHOP via a TRIF-dependent pathway [76].
6. Pathways for Cytokines Induction of ER Stress
Upon the insults of pathogens, mutated self-antigens, or tissue damage, the immune system initiates inflammatory response by releasing copious amount of cytokines. UPR and ER stress are interconnected with inflammatory cytokines through multiple mechanisms including reactive oxygen species (ROS), NFκB, and JNK (Figure 3). ROS are highly reactive small molecules with unpaired electrons. They are important mediators of inflammatory response. The accumulation of ROS, referred to as oxidative stress, was confirmed to be associated with ER stress [77]. Oxidizing condition is required for the disulphide bond formation during the process of protein folding [78]. Increased protein folding load may lead to oxidative stress. The PERK axis of UPR signaling is reported to be able to activate antioxidant pathway by promoting ATF4 and nuclear factor-erythroid-derived 2-related factor 2 (NRF2) [79, 80]. Therefore, loss of PERK markedly increases ROS accumulation induced by toxic chemicals [79, 81]. The IRE1/TRAF2 axis of UPR can recruit IκB kinase (IKK), leading to the activation of NFκB, a key regulator in inflammation [82]. As a result, NFκB activation and TNF-α production are reduced in cells lacking IRE1 [82]. Furthermore, the IRE1/TRAF2 axis can activate JNK, and by which it induces the expression of inflammatory genes by activating activator protein 1 (AP1) [83]. ATF6, the third axis of UPR signaling, can also activate NFκB pathway, in which suppression of ATF6 reduces NFκB activation caused by BiP degradation [84].
Figure 3.
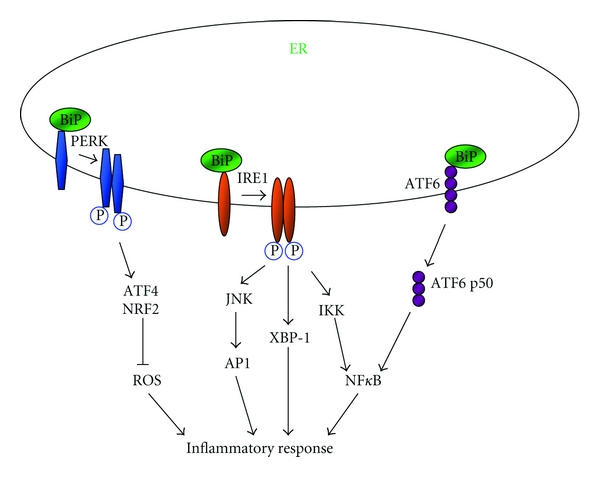
The possible implication of UPR in inflammatory response. UPR is associated with inflammation via a variety of mechanisms involving ROS, JNK, and NFκB. PERK promotes ATF4 and NRF2, which then suppress ROS production by activating antioxidant pathway. Upon activation, IRE1/TRAF2 recruits IKK, leading to the phosphorylation of IκBα and subsequent activation of NFκB. IRE1/TRAF2 can also activate AP1, resulting in the activation of JNK. XBP-1 induced by IRE1 can further induce the expression of various genes implicated inflammation. Furthermore, ATF6, the other axis of UPR signaling, can promote inflammation via activating NFκB.
Other than the above described pathways, cytokines may also induce ER stress via inducible nitric oxide (NO) synthase (iNOS) and JNK pathway. JNK pathway is activated by IL-1β. Suppression of JNK by its inhibitor SP600125 protected β cells from IL-1β-induced apoptosis [85]. Cytokines have been evidenced to induce the expression of iNOS, leading to excessive NO production. Stimulation with IL-1β and IFN-γ activates ER stress pathway and induces β-cell apoptosis via NO synthesis [62]. NO has been suggested to be an important mediator of β-cell death in type 1 diabetes. Inflammatory cytokines including IL-1β, IFN-γ, and TNF-α can induce iNOS expression in β cells which then produces copious amount of NO [50]. Excessive NO induces DNA damage and thus results in β-cell apoptosis through p53 pathway or necrosis through poly(ADP-ribose) polymerase (PARP) pathway [54]. Moreover, NO depletes ER Ca2+ stores via activating Ca2+ channels or inhibiting Ca2+ pumps [86–88]. Depletion of Ca2+ then leads to ER stress and apoptosis in β cells via the induction of CHOP signaling [55, 89].
7. Mechanisms Underlying ER-Stress-Induced β-Cell Death
ER stress is a key mediator for β-cell death in type 1 diabetes. The primary purpose of ER stress or UPR is to compensate the damage caused by the disturbances of normal ER function. However, continuous ER dysfunction would eventually render cells undergoing apoptosis. The mechanisms by which ER stress induces cell death are not fully elucidated, due to the fact that multiple potential participants involved but little clarity on the dominant death effectors in a particular cellular context. In general, ER stress induction of cell death can be illustrated in three phases: adaptation, alarm, and apoptosis [44].
The phase for adaptation response is initiated to restore the homeostasis of ER and to protect cells from damage induced by the disturbances of ER function. As described earlier, the signaling for UPR involves three axes of responses: IRE1, PERK, and ATF6. These axes interact between each other and form a feedback regulatory mechanism to control the activity of UPR. The accumulation of unfolded proteins in ER results in the engagement of ER-resident chaperon BiP, and as a consequence, IRE1, PERK, and ATF6 are released from BiP. Therefore, overexpression of BiP can prevent cell death induced by oxidative stress, Ca2+ disturbances, and hypoxia [90]. PERK is oligomerized and phosphorylated when released from BiP. Activated PERK inactivates eIF2α to reduce mRNA translation and protein load on ER. Therefore, PERK deficiency results in an abnormally elevated protein synthesis in response to the accumulation of unfolded proteins in ER, which renders cells highly sensitive to ER stress and ER stress-induced apoptosis [91]. Similarly, as a downstream molecule of PERK, eIF2α is required for cell survival upon the insult of ER stress, and a mutation at the phosphorylation site of eIF2α (Ser51Ala) abolishes the translational suppression in response to ER stress [52]. Similar as PERK, IRE1 becomes dimerized and activated once released from BiP. IRE1 induces XBP-1 by promoting the splicing of its mRNA [45]. XBP-1 is a transcriptional factor belonging to the bZIP-family and is responsible for the transcription of many adaptation genes implicated in UPR. Unlike PERK and IRE1, ATF6 translocates into the Golgi apparatus upon the release from BiP. The transmembrane domain of ATF6 is cleaved in the Golgi apparatus and is then relocated into the nucleus, by which it regulates gene expression [46].
During the alarm phase, many signal pathways are activated, and the expression of responsive genes has been induced to alert the system. For example, the cytoplasmic part of IRE1 binds to TNF receptor-associated factor 2 (TRAF2), a key adaptor for TNF-mediated innate immune signaling. TRAF2 would then activate NFκB pathway via activating IKK and activate the signaling for c-Jun N-terminal kinases (JNK) by apoptosis signal-regulating kinase 1 (Ask1). Studies have shown that dominant negative TRAF2 suppresses the activation of JNK by IRE1 in response to ER stress [92]. Importantly, TRAF2 is also a critical component for E3 ubiquitin-protein ligase complex [93], which binds to Ubc13 and promotes the noncanonical ubiquitination of substrates. The Ubc13-dependent ubiquitination of TRAF2 is suggested to be required for the activation of JNK [94]. In addition, IRE1 can further activate JNK signaling through interacting with c-Jun N-terminal inhibitory kinase (JIK) [95].
Although the purpose for the initiation of adaptation response is to restore the homeostasis of ER, apoptosis however could occur, once the accumulation of unfolded proteins exceeds the cellular regulatory capacity. The action for apoptosis is initiated by the activation of several proteases such as caspase-12, caspase-4, caspase-2, and caspase-9. Studies in rodents provided evidence supporting that caspase-12 is involved in ER stress-induced apoptosis. Caspase-12 is activated by IRE1 upon the insult of ER stress. Mice deficient in caspase-12 are resistant to ER stress-induced apoptosis, but remain susceptible to apoptosis induced by other stimuli [96]. There is evidence that caspase-12 can also be activated by interacting with TRAF2, a signaling molecule downstream of IRE1 [95]. In response to ER stress, caspase-7 is translocated from the cytosol to the ER surface, which then activates procaspase-12 as well [97]. The human caspase-4 is the closest paralog of rodent caspase-12, which is normally located on the ER membrane. However, caspase-4 can only be activated by ER stress-inducing reagents not by the other apoptotic reagents, and knockdown of caspase-4 by siRNA reduces ER stress-induced apoptosis in neuroblastoma cells [98]. Similarly, caspase-2 and caspase-9 are found to be activated in the early phase of ER stress and inhibition of their activation either by inhibitors or siRNA reduces ER stress-induced apoptosis [99]. Studies also suggest that some members of inhibitor of apoptosis protein family prevent ER stress-induced cell death via interacting with caspase-2 and caspase-9 [99].
Other than the implication of caspases, Ask1 kinase and CCAAT/enhancer binding protein (C/EBP) homologous protein (CHOP) are also critical mediators for ER stress-induced cell death. IRE1/TRAF2 complex recruits Ask1 and activates subsequent JNK signaling. Studies have shown that the activation of JNK inhibits antiapoptotic protein BCL-2 [100] and induces proapoptotic protein Bim [101, 102]. Loss of Ask1 suppresses ER stress-induced JNK activation and provides protection for cells against ER stress-induced death [103]. CHOP is a transcription factor belonging to basic leucine zipper transcription factor (bZIP) family. Many inducers of UPR including ATF4, ATF6, and XBP-1 up-regulate CHOP expression, and phosphorylation of CHOP at ser78 and ser81 by p38 MAPK enhances its transcriptional activity [44, 104]. Upon its activation, CHOP suppresses anti-apoptotic protein BCL-2 which in turn induces cells undergoing apoptosis [105–107].
8. Conclusion and Future Directions
There is convincing evidence that ER stress plays an essential role in autoimmune-mediated β-cell destruction. Feasible evidence also supports that ER stress modulates autoimmune response during T1D development (Table 1). ER stress in β cells can be either triggered by autoimmune responses against β-cell self-antigens and/or by the increase of compensated insulin synthesis. During the course of type 1 diabetes, autoreactive immune cells secrete copious amount of inflammatory cytokines such as IL-1β, TNF-α, and IFN-γ into the islet milieu, which stimulate excessive production of NO in β cells and mediate β-cell destruction by inducing ER stress. Recent studies further suggest that ER stress also modulates the functionality of immune cells with implications in autoimmune progression. The absolute insulin deficiency in T1D patients renders the residual β cells for compensated insulin secretion to meet the demands of insulin for maintaining blood glucose homeostasis. This increase in insulin biosynthesis could overwhelm the folding capacity of ER, leading to UPR and ER stress in β cells, which in turn exacerbates β-cell dysfunction and T1D onset.
Table 1.
Publications relevant to ER stress in the regulation of immune response and β-cell destruction.
| Author | Defective/mutant gene | Species | Major finding | Reference |
|---|---|---|---|---|
| Harding et al. | PERK−/− | Mouse | PERK-deficient mice are extremely susceptible to diabetes. They display a progressive β-cell loss and hyperglycemia with aging. | [49] |
| Delépine et al. | PERK−/− | Human | Deficiency of PERK in human results in Wolcott-Rallison syndrome, which is characterized by early infancy insulin-dependent diabetes and multisystemic dysfunction. | [50] |
| Scheuner et al. | eIF2α mutant (Ser51Ala) | Mouse | Ser51Ala mutation of eIF2α shows a deficiency in pancreatic β cells manifested by the smaller core of insulin-secreting β cells and attenuated insulin secretion, and the mice die from hypoglycemia at their early infancy. | [52] |
| Ron et al. | Ins2 mutation | Mouse | Ins2 mutation in Akita mice disrupts disulfide bond between the α and β chain of proinsulin, which leads to the mis-folding of the mutated insulin and further induces ER stress in β cells and diabetes. | [56] |
| Zhang et al. | IRE1−/− | Mouse | Pro-B cells failed to differentiate into pre-B cells when deficient for IRE1. | [65] |
| Iwakoshi et al. | XBP-1−/− | Mouse | Deficiency of XBP-1 results in the impacted development of both conventional and plasmacytoid DCs. Loss of XBP-1 renders DCs vulnerable to ER stress-induced apoptosis. | [72] |
| Goodall et al. | CHOP knockdown | Knockdown of CHOP suppressed the production of IL-23 induced by ER stress and TLR signaling. | [73] | |
| Richardson et al. | XBP-1 mutation | C. elegans | Innate immune response induced by P. aeruginosa infection causes ER stress in C. elegans, and mutations with loss of function for XBP-1 lead to larval lethality. | [74] |
| Kaser et al. | XBP-1 polymorphisms | Human | Loss of XBP-1 in intestinal epithelial cells induces Paneth cell dysfunction and overactive epithelium, leading to impaired mucosal defense to Listeria monocytogenes and increased sensitivity to colitis, an inflammatory disease sharing similar properties with T1D. The polymorphisms within the XBP-1 gene are associated with Crohn's disease and ulcerative colitis in humans. | [75] |
| Nakagawa et al. | Caspase-12−/− | Mouse | Caspase-12 is involved in ER stress-induced apoptosis. Mice deficient in caspase-12 are resistant to ER stress-induced apoptosis, but remain susceptible to apoptosis induced by other stimuli. | [96] |
| Hitomi et al. | Caspase-4 knockdown | Human | Human caspase-4, the closest paralog of rodent caspase-12, is involved in ER stress-induced apoptosis. Knockdown of caspase-4 by siRNA reduces ER stress-induced apoptosis. | [98] |
| Nishitoh et al. | Ask1−/− | Mouse | Loss of Ask1 suppresses ER stress-induced JNK activation and protects cells from ER stress-induced death. | [103] |
It should be kept in mind that the mechanisms underlying autoimmune-mediated β-cell destruction in type 1 diabetes are complex, and ER stress is unlikely the exclusive mechanism implicated in disease process. Despite recent significant advancement in this field, there are still many questions yet to be addressed. Can ER stress be served as a biomarker for β-cell destruction and autoimmune progression in the clinic setting? Are there additional factors for induction of ER stress in β cells during T1D development? Does modulation of ER stress in immune cells attenuate autoimmune progression? Does blockade of ER stress protect β cells from autoimmune-mediated destruction? Future studies aimed at dissecting these questions would provide a broadened insight for T1D pathogenesis and would have great potential for developing novel therapeutic strategies against this devastating disorder.
Acknowledgments
Our research is supported by grants from the Juvenile Diabetes Research Foundation International (JDRFI), the EFSD/CDC/Lilly Program for Collaborative Diabetes Research between China and Europe, the Synergy Award from the Diabetes and Obesity Discovery Institute (DODI), and the National Natural Science Foundation of China (81130014/H0704 and 81101553/H1604). The authors declare that they have no financial conflict of interests.
Abbreviations
- AP1:
Activator protein 1
- Ask1:
Apoptosis signal-regulating kinase 1
- ATF6:
Activating transcription factor 6
- BCR:
B-cell receptor
- BLIMP1:
B-lymphocyte-induced maturation protein 1
- BSAP:
B-cell-lineage-specific activator protein
- bZIP:
Basic leucine zipper transcription factor
- C/EBP:
CCAAT/enhancer binding protein
- CHOP:
C/EBP homologous protein
- CPA:
Cyclopiazonic acid
- CREBH:
Cyclic-AMP-responsive-element-binding protein H
- DC:
Dendritic cell
- DP5:
Death protein 5
- eIF2α:
Eukaryotic initiation factor 2α
- ER:
Endoplasmic reticulum
- ER stress:
Endoplasmic reticulum stress
- ERAD:
ER-associated degradation
- ERSE:
ER stress enhancer
- HMGB1:
High-mobility group box 1
- HSPs:
Heat shock proteins
- IKK:
IκB kinase
- iNOS:
Inducible nitric oxide synthase
- IRE1:
Inositol-requiring enzyme 1
- IRS-1:
Insulin receptor substrate-1
- JIK:
c-Jun N-terminal inhibitory kinase
- JNK:
c-Jun N-terminal kinases
- NO:
Nitric oxygen
- NRF2:
Nuclear factor-erythroid-derived 2-related factor 2
- PARP:
Poly(ADP-ribose) polymerase
- PERK:
Pancreatic endoplasmic reticulum kinase
- RER:
Rough endoplasmic reticulum
- ROS:
Reactive oxygen species
- SER:
Smooth endoplasmic reticulum
- SERCA:
Sarcoendoplasmic reticulum pump Ca2+ ATPase
- SR:
Sarcoplasmic reticulum
- T1D:
Type 1 diabetes
- TLR:
Toll-like receptor
- TRAF2:
TNF receptor-associated factor 2
- TRIF:
Toll-IL-1R-containing adaptor inducing IFN-β
- UPR:
Unfolded protein response
- UPRE:
Unfolded protein response element
- XBP-1:
X box protein-1.
References
- 1.Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. Epidemiology of type 1 diabetes. Endocrinology and Metabolism Clinics of North America. 2010;39(3):481–497. doi: 10.1016/j.ecl.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang P, Li H-L, Wang C-Y. FUT2 nonfunctional variant: a "missing link" between genes and environment in type 1 diabetes? Diabetes. 2011;60(11):2685–2687. doi: 10.2337/db11-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bandurska-Stankiewicz E, Wiatr D. Programme preventing vision loss due to diabetes. Klinika Oczna. 2007;109(7–9):359–362. [PubMed] [Google Scholar]
- 4.Cheung N, Wong TY. Diabetic retinopathy and systemic vascular complications. Progress in Retinal and Eye Research. 2008;27(2):161–176. doi: 10.1016/j.preteyeres.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Studholme S. Diabetic retinopathy. Journal of Perioperative Practice. 2008;18(5):205–210. doi: 10.1177/175045890801800504. [DOI] [PubMed] [Google Scholar]
- 6.Monhart V. Diabetes mellitus, hypertension and kidney. Vnitrni Lekarstvi. 2008;54(5):499–507. [PubMed] [Google Scholar]
- 7.Navarro-González JF, Mora-Fernández C. The role of inflammatory cytokines in diabetic nephropathy. Journal of the American Society of Nephrology. 2008;19(3):433–442. doi: 10.1681/ASN.2007091048. [DOI] [PubMed] [Google Scholar]
- 8.Boulton AJ. Diabetic neuropathy: classification, measurement and treatment. Current Opinion in Endocrinology, Diabetes and Obesity. 2007;14(2):141–145. doi: 10.1097/MED.0b013e328014979e. [DOI] [PubMed] [Google Scholar]
- 9.Cornell RS, Ducic I. Painful diabetic neuropathy. Clinics in Podiatric Medicine and Surgery. 2008;25(3):347–360. doi: 10.1016/j.cpm.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Otto-Buczkowska E, Kazibutowska Z, Sołyk J, Machnica Ł. Neuropathy and type 1 diabetes mellitus. Endokrynologia, Diabetologia i Choroby Przemiany Materii Wieku Rozwojowego. 2008;14(2):109–116. [Google Scholar]
- 11.Gardner SE, Frantz RA. Wound bioburden and infection-related complications in diabetic foot ulcers. Biological Research for Nursing. 2008;10(1):44–53. doi: 10.1177/1099800408319056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malgrange D. Physiopathology of the diabetic foot. Revue de Medecine Interne. 2008;29(supplement 2):S231–S237. doi: 10.1016/S0248-8663(08)73950-X. [DOI] [PubMed] [Google Scholar]
- 13.Ochoa O, Torres FM, Shireman PK. Chemokines and diabetic wound healing. Vascular. 2007;15(6):350–355. doi: 10.2310/6670.2007.00056. [DOI] [PubMed] [Google Scholar]
- 14.Anselmino M, Gohlke H, Mellbin L, Rydén L. Cardiovascular prevention in patients with diabetes and prediabetes. Herz. 2008;33(3):170–177. doi: 10.1007/s00059-008-3105-5. [DOI] [PubMed] [Google Scholar]
- 15.Inoguchi T, Takayanagi R. Role of oxidative stress in diabetic vascular complications. Fukuoka Igaku Zasshi. 2008;99(3):47–55. [PubMed] [Google Scholar]
- 16.Marwick TH. Diabetic heart disease. Postgraduate Medical Journal. 2008;84(990):188–192. doi: 10.1136/hrt.2005.067231. [DOI] [PubMed] [Google Scholar]
- 17.D'Hertog W, Maris M, Ferreira GB, et al. Novel insights into the global proteome responses of insulin-producing INS-1E cells to different degrees of endoplasmic reticulum stress. Journal of Proteome Research. 2010;9(10):5142–5152. doi: 10.1021/pr1004086. [DOI] [PubMed] [Google Scholar]
- 18.Hubbard SC, Ivatt RJ. Synthesis and processing of asparagine-linked oligosaccharides. Annual Review of Biochemistry. 1981;50:555–583. doi: 10.1146/annurev.bi.50.070181.003011. [DOI] [PubMed] [Google Scholar]
- 19.Kornfeld R, Kornfeld S. Assembly of asparagine-linked oligosaccharides. Annual Review of Biochemistry. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]
- 20.Fewell SW, Travers KJ, Weissman JS, Brodsky JL. The action of molecular chaperones in the early secretory pathway. Annual Review of Genetics. 2001;35:149–191. doi: 10.1146/annurev.genet.35.102401.090313. [DOI] [PubMed] [Google Scholar]
- 21.Paltauf F, Kohlwein SD, Henry SA. Regulation and compartmentalization of lipid synthesis in yeast. In: Jones EW, Broach JR, editors. The Molecular and Cellular Biology of the Yeast Saccharomyces. New York, NY, USA: Cold Spring Harbor Laboratory Press; 1992. pp. 415–500. [Google Scholar]
- 22.Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends in Biochemical Sciences. 2001;26(8):504–510. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 23.Schröder M, Friedl P. Overexpression of recombinant human antithrombin III in Chinese hamster ovary cells results in malformation and decreased secretion of recombinant protein. Biotechnology and Bioengineering. 1997;53(6):547–559. doi: 10.1002/(SICI)1097-0290(19970320)53:6<547::AID-BIT2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 24.Schröder M, Schäfer R, Friedl P. Induction of protein aggregation in an early secretory compartment by elevation of expression level. Biotechnology and Bioengineering. 2002;78(2):131–140. doi: 10.1002/bit.10206. [DOI] [PubMed] [Google Scholar]
- 25.Dorner AJ, Wasley LC, Kaufman RJ. Increased synthesis of secreted proteins induces expression of glucose-regulated proteins in butyrate-treated Chinese hamster ovary cells. Journal of Biological Chemistry. 1989;264(34):20602–20607. [PubMed] [Google Scholar]
- 26.Kaufman RJ, Wasley LC, Dorner AJ. Synthesis, processing, and secretion of recombinant human factor VIII expressed in mammalian cells. Journal of Biological Chemistry. 1988;263(13):6352–6362. [PubMed] [Google Scholar]
- 27.Schröder M, Kaufman RJ. ER stress and the unfolded protein response. Mutation Research. 2005;569(1-2):29–63. doi: 10.1016/j.mrfmmm.2004.06.056. [DOI] [PubMed] [Google Scholar]
- 28.Reimold AM, Iwakoshi NN, Manis J, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412(6844):300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 29.Iwakoshi NN, Lee AH, Vallabhajosyula P, Otipoby KL, Rajewsky K, Glimcher LH. Plasma cell differentiation and the unfolded protein response intersect at the transcription factor XBP-I. Nature Immunology. 2003;4(4):321–329. doi: 10.1038/ni907. [DOI] [PubMed] [Google Scholar]
- 30.Martínez IM, Chrispeels MJ. Genomic analysis of the unfolded protein response in Arabidopsis shows its connection to important cellular processes. Plant Cell. 2003;15(2):561–576. doi: 10.1105/tpc.007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pakula TM, Laxell M, Huuskonen A, Uusitalo J, Saloheimo M, Penttilä M. The effects of drugs inhibiting protein secretion in the filamentous fungus Trichoderma reesei. Evidence for down-regulation of genes that encode secreted proteins in the stressed cells. Journal of Biological Chemistry. 2003;278(45):45011–45020. doi: 10.1074/jbc.M302372200. [DOI] [PubMed] [Google Scholar]
- 32.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic- reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 33.Casagrande R, Stern P, Diehn M, et al. Degradation of proteins from the ER of S. cerevisiae requires an intact unfolded protein response pathway. Molecular Cell. 2000;5(4):729–735. doi: 10.1016/s1097-2765(00)80251-8. [DOI] [PubMed] [Google Scholar]
- 34.Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nature Cell Biology. 2000;2(7):379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- 35.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101(3):249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 36.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. Journal of Clinical Investigation. 2002;110(10):1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens FJ, Argon Y. Protein folding in the ER. Seminars in Cell and Developmental Biology. 1999;10(5):443–454. doi: 10.1006/scdb.1999.0315. [DOI] [PubMed] [Google Scholar]
- 38.Schröder M. Endoplasmic reticulum stress responses. Cellular and Molecular Life Sciences. 2008;65(6):862–894. doi: 10.1007/s00018-007-7383-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suzuki CK, Bonifacino JS, Lin AY, Davis MM, Klausner RD. Regulating the retention of T-cell receptor α chain variants within the endoplasmic reticulum: Ca2+-dependent association with BiP. Journal of Cell Biology. 1991;114(2):189–205. doi: 10.1083/jcb.114.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li LJ, Li X, Ferrario A, et al. Establishment of a Chinese hamster ovary cell line that expresses grp78 antisense transcripts and suppresses A23187 induction of both GRP78 and GRP94. Journal of Cellular Physiology. 1992;153(3):575–582. doi: 10.1002/jcp.1041530319. [DOI] [PubMed] [Google Scholar]
- 41.Corbett EF, Oikawa K, Francois P, et al. Ca2+ regulation of interactions between endoplasmic reticulum chaperones. Journal of Biological Chemistry. 1999;274(10):6203–6211. doi: 10.1074/jbc.274.10.6203. [DOI] [PubMed] [Google Scholar]
- 42.Zhang JX, Braakman I, Matlack KES, Helenius A. Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins with C-terminal truncations. Molecular Biology of the Cell. 1997;8(10):1943–1954. doi: 10.1091/mbc.8.10.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nature Reviews Molecular Cell Biology. 2003;4(7):552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- 44.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. Journal of Clinical Investigation. 2005;115(10):2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee K, Tirasophon W, Shen X, et al. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes and Development. 2002;16(4):452–466. doi: 10.1101/gad.964702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Molecular Cell. 2000;6(6):1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 47.Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocrine Reviews. 2008;29(1):42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 48.Laybutt DR, Preston AM, Åkerfeldt MC, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50(4):752–763. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- 49.Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in Perk-/- mice reveals a role for translational control in secretory cell survival. Molecular Cell. 2001;7(6):1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 50.Delépine M, Nicolino M, Barrett T, Golamaully M, Mark Lathrop G, Julier C. EIF2AK3, encoding translation initiation factor 2-α kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nature Genetics. 2000;25(4):406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- 51.Araki E, Oyadomari S, Mori M. Endoplasmic reticulum stress and diabetes mellitus. Internal Medicine. 2003;42(1):7–14. doi: 10.2169/internalmedicine.42.7. [DOI] [PubMed] [Google Scholar]
- 52.Scheuner D, Song B, McEwen E, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Molecular Cell. 2001;7(6):1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- 53.Mathis D, Vence L, Benoist C. β-cell death during progression to diabetes. Nature. 2001;414(6865):792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 54.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death and Differentiation. 2004;11(4):381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 55.Oyadomari S, Takeda K, Takiguchi M, et al. Nitric oxide-induced apoptosis in pancreatic β cells is mediated by the endoplasmic reticulum stress pathway. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(19):10845–10850. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ron D. Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. Journal of Clinical Investigation. 2002;109(4):443–445. doi: 10.1172/JCI15020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and β-cell loss in type 1 diabetes. Nature Reviews Endocrinology. 2009;5(4):219–226. doi: 10.1038/nrendo.2009.21. [DOI] [PubMed] [Google Scholar]
- 58.Zhong J, Yang P, Wang C. Type 1 Diabetes Mellitus: Etiology, Diagnosis and Treatment. Nova Science; 2011. Environmental triggers and endogenous alarmins linking innate immunity to the pathogenesis of type 1 diabetes; pp. 177–206. [Google Scholar]
- 59.Han J, Zhong J, Wei W, et al. Extracellular high-mobility group box 1 acts as an innate immune mediator to enhance autoimmune progression and diabetes onset in NOD mice. Diabetes. 2008;57(8):2118–2127. doi: 10.2337/db07-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang S, Zhong J, Yang P, Gong F, Wang CY. HMGB1, an innate alarmin, in the pathogenesis of type 1 diabetes. International Journal of Clinical and Experimental Pathology. 2010;3(1):24–38. [PMC free article] [PubMed] [Google Scholar]
- 61.Gurzov EN, Ortis F, Cunha DA, et al. Signaling by IL-1β+IFN-γ and ER stress converge on DP5/Hrk activation: a novel mechanism for pancreatic β-cell apoptosis. Cell Death and Differentiation. 2009;16(11):1539–1550. doi: 10.1038/cdd.2009.99. [DOI] [PubMed] [Google Scholar]
- 62.Cardozo AK, Ortis F, Storling J, et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic β-cells. Diabetes. 2005;54(2):452–461. doi: 10.2337/diabetes.54.2.452. [DOI] [PubMed] [Google Scholar]
- 63.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends in Cell Biology. 2004;14(1):20–28. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 64.Baekkeskov S, Nielsen JN, Marner B. Autoantibodies in newly diagnosed diabetic children immunoprecipitate human pancreatic islet cell proteins. Nature. 1982;298(5870):167–169. doi: 10.1038/298167a0. [DOI] [PubMed] [Google Scholar]
- 65.Zhang K, Wong HN, Song B, Miller CN, Scheuner D, Kaufman RJ. The unfolded protein response sensor IRE1α is required at 2 distinct steps in B cell lymphopoiesis. Journal of Clinical Investigation. 2005;115(2):268–281. doi: 10.1172/JCI21848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shaffer AL, Shapiro-Shelef M, Iwakoshi NN, et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity. 2004;21(1):81–93. doi: 10.1016/j.immuni.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 67.Niu H, Ye BH, Dalla-Favera R. Antigen receptor signaling induces MAP kinase-mediated phosphorylation and degradation of the BCL-6 transcription factor. Genes and Development. 1998;12(13):1953–1961. doi: 10.1101/gad.12.13.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lin KI, Angelin-Duclos C, Kuo TC, Calame K. Blimp-1-dependent repression of Pax-5 is required for differentiation of B cells to immunoglobulin M-secreting plasma cells. Molecular and Cellular Biology. 2002;22(13):4771–4780. doi: 10.1128/MCB.22.13.4771-4780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reimold AM, Ponath PD, Li YS, et al. Transcription factor B cell lineage-specific activator protein regulates the gene for human X-box binding protein 1. Journal of Experimental Medicine. 1996;183(2):393–401. doi: 10.1084/jem.183.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao L, Ackerman SL. Endoplasmic reticulum stress in health and disease. Current Opinion in Cell Biology. 2006;18(4):444–452. doi: 10.1016/j.ceb.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 71.Zhang K, Shen X, Wu J, et al. Endoplasmic reticulum stress activates cleavage of CREBH to induce a systemic inflammatory response. Cell. 2006;124(3):587–599. doi: 10.1016/j.cell.2005.11.040. [DOI] [PubMed] [Google Scholar]
- 72.Iwakoshi NN, Pypaert M, Glimcher LH. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. Journal of Experimental Medicine. 2007;204(10):2267–2275. doi: 10.1084/jem.20070525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goodall JC, Wu C, Zhang Y, et al. Endoplasmic reticulum stress-induced transcription factor, CHOP, is crucial for dendritic cell IL-23 expression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(41):17698–17703. doi: 10.1073/pnas.1011736107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Richardson CE, Kooistra T, Kim DH. An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature. 2010;463(7284):1092–1095. doi: 10.1038/nature08762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134(5):743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Woo CW, Cui D, Arellano J, et al. Adaptive suppression of the ATF4-CHOP branch of the unfolded protein response by toll-like receptor signalling. Nature cell biology. 2009;11(12):1473–1480. doi: 10.1038/ncb1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Malhotra JD, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants and Redox Signaling. 2007;9(12):2277–2293. doi: 10.1089/ars.2007.1782. [DOI] [PubMed] [Google Scholar]
- 78.Tu BP, Weissman JS. Oxidative protein folding in eukaryotes: mechanisms and consequences. Journal of Cell Biology. 2004;164(3):341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harding HP, Zhang Y, Zeng H, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Molecular Cell. 2003;11(3):619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 80.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Molecular and Cellular Biology. 2003;23(20):7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cullinan SB, Diehl JA. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. Journal of Biological Chemistry. 2004;279(19):20108–20117. doi: 10.1074/jbc.M314219200. [DOI] [PubMed] [Google Scholar]
- 82.Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Molecular and Cellular Biology. 2006;26(8):3071–3084. doi: 10.1128/MCB.26.8.3071-3084.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 84.Yamazaki H, Hiramatsu N, Hayakawa K, et al. Activation of the Akt-NF-κB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. Journal of Immunology. 2009;183(2):1480–1487. doi: 10.4049/jimmunol.0900017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang Q, Zhang H, Zhao B, Fei H. IL-1β caused pancreatic β-cells apoptosis is mediated in part by endoplasmic reticulum stress via the induction of endoplasmic reticulum Ca2+ release through the c-Jun N-terminal kinase pathway. Molecular and Cellular Biochemistry. 2009;324(1-2):183–190. doi: 10.1007/s11010-008-9997-9. [DOI] [PubMed] [Google Scholar]
- 86.Messmer UK, Brüne B. Nitric oxide-induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochemical Journal. 1996;319(1):299–305. doi: 10.1042/bj3190299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schöneich C. Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochemical Journal. 1999;340(3):657–669. [PMC free article] [PubMed] [Google Scholar]
- 88.Xu KY, Huso DL, Dawson TM, Bredt DS, Becker LC. Nitric oxide synthase in cardiac sarcoplasmic reticulum. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(2):657–662. doi: 10.1073/pnas.96.2.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (Ryanodoine receptor) by poly-S-nitrosylation. Science. 1998;279(5348):234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 90.Liu H, Bowes RC, III, Van De Water B, Sillence C, Nagelkerke JF, Stevens JL. Endoplasmic reticulum chaperones GRP78 and calreticulin prevent oxidative stress, Ca2+ disturbances, and cell death in renal epithelial cells. Journal of Biological Chemistry. 1997;272(35):21751–21759. doi: 10.1074/jbc.272.35.21751. [DOI] [PubMed] [Google Scholar]
- 91.Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Molecular Cell. 2000;5(5):897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- 92.Urano F, Wang X, Bertolotti A, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287(5453):664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 93.Zheng C, Kabaleeswaran V, Wang Y, Cheng G, Wu H. Crystal structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 complexes: affinity, specificity, and regulation. Molecular Cell. 2010;38(1):101–113. doi: 10.1016/j.molcel.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Habelhah H, Takahashi S, Cho SG, Kadoya T, Watanabe T, Ronai Z. Ubiquitination and translocation of TRAF2 is required for activation of JNK but not of p38 or NF-κB. EMBO Journal. 2004;23(2):322–332. doi: 10.1038/sj.emboj.7600044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoneda T, Imaizumi K, Oono K, et al. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2-dependent mechanism in response to the ER stress. Journal of Biological Chemistry. 2001;276(17):13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- 96.Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β . Nature. 2000;403(6765):98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 97.Rao RV, Hermel E, Castro-Obregon S, et al. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. Journal of Biological Chemistry. 2001;276(36):33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- 98.Hitomi J, Katayama T, Eguchi Y, et al. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Aβ-induced cell death. Journal of Cell Biology. 2004;165(3):347–356. doi: 10.1083/jcb.200310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cheung HH, Lynn Kelly N, Liston P, Korneluk RG. Involvement of caspase-2 and caspase-9 in endoplasmic reticulum stress-induced apoptosis: a role for the IAPs. Experimental Cell Research. 2006;312(12):2347–2357. doi: 10.1016/j.yexcr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 100.Yamamoto K, Ichijo H, Korsmeyer SJ. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G2/M. Molecular and Cellular Biology. 1999;19(12):8469–8478. doi: 10.1128/mcb.19.12.8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Putcha GV, Le S, Frank S, et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38(6):899–914. doi: 10.1016/s0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 102.Lei K, Davis RJ. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2432–2437. doi: 10.1073/pnas.0438011100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes and Development. 2002;16(11):1345–1355. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang X, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP kinase. Science. 1996;272(5266):1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 105.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bc12 and perturbing the cellular redox state. Molecular and Cellular Biology. 2001;21(4):1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matsumoto M, Minami M, Takeda K, Sakao Y, Akira S. Ectopic expression of CHOP (GADD153) induces apoptosis in M1 myeloblastic leukemia cells. FEBS Letters. 1996;395(2-3):143–147. doi: 10.1016/0014-5793(96)01016-2. [DOI] [PubMed] [Google Scholar]
- 107.Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes and Development. 1998;12(7):982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]