

Abstract
Neuroregenerative therapies for central nervous system (CNS) injury, neurodegenerative disease, or stroke require axons of damaged neurons to grow and re-innervate their targets. However, mature mammalian CNS neurons do not regenerate their axons, limiting recovery in these diseases. Although neurons' intrinsic capacity for axon growth may depend in part on the panoply of expressed transcription factors, epigenetic factors such as the accessibility of DNA and organization of chromatin are required for downstream genes to be transcribed. Thus, a potential approach to overcoming regenerative failure focuses on the epigenetic mechanisms regulating regenerative gene expression in the CNS. Here we review molecular mechanisms regulating the epigenetic state of DNA through chromatin modifications, their implications for regulating axon and dendrite growth, and important new directions for this field of study.
Keywords: axon regeneration, axon growth, dendrite growth, transcription, epigenetics, chromatin, transcription factors, central nervous system
Introduction
Epigenetics is the regulation of gene expression beyond the heritable DNA sequence (Bird, 2007), by enzymes and their substrate modifications, which ultimately gate access to and transcription from DNA (Reik, 2007), and thus may play role in expression of regenerative genes in the nervous system. Central nervous system (CNS) regenerative failure may be attributable to the development of an inhibitory CNS environment by glial-associated inhibitory molecules (Yiu and He, 2006), and by various cell-autonomous factors (Sun and He, 2010). Intrinsic axon growth ability also declines developmentally (Li et al., 1995; Goldberg et al., 2002; Bouslama-Oueghlani et al., 2003; Blackmore and Letourneau, 2006) and is dependent on transcription (Moore et al., 2009). Several transcription factors have been identified as regulators of axon growth and regeneration, for example, Krüppel-like factors (KLFs), cAMP response element-binding protein (CREB), c-Jun, Smad1, c-EBP, SOX11, STAT3, ATF3, NFATs, and NFIL3 (Sun and He, 2010; Liu et al., 2011; Moore and Goldberg, 2011). Because access to DNA through chromatin remodeling is required for transcription factors to transcribe their target genes, epigenetic mechanisms may also play a significant role in regulating expression of pro-regenerative genes. Remodeling chromatin through protein and DNA modifications at specific loci not only passively gates accessibility of transcription factors to gene promoters, but can also actively recruit protein complexes which serve as scaffolds for specific transcription factors to promote or repress transcription (Strahl and Allis, 2000). Specific epigenetic factors are now emerging as regulators of chromatin remodeling that gate access to genes regulating axon and dendrite growth and regeneration. This review will provide critical analysis of the advances in the field of epigenetic regulation of axon growth and regeneration, focusing on chromatin remodeling enzymes which have been shown to play roles in neurite growth, and emphasizing the experimental challenges that need to be addressed.
Acetylation, HATs, and HDACs
In eukaryotes, histone proteins H2A, H2B, H3, and H4 wrap around DNA, and along with H1, form a nucleosome and gate transcription factors' access to DNA. Post-translational modifications of histone tails protruding from the nucleosome, and of histones in the nucleosome core, regulate DNA accessibility through modifying nucleosome architecture and chromatin structure (Figure 1) (Kouzarides, 2007). Combinations of various post-translational histone modifications are associated with differential regulation of chromatin and gene expression and comprise the “histone code,” (Turner, 2007) which could in principle encode axon regeneration programs regulating multiple pro-regenerative genes. The most prominent type of post-translational histone modification is the acetylation of lysine residues in histone tails. Thus, one of the key mechanisms regulating transcription factors access to target gene promoters is regulation of post-translational acetylation and deacetylation of histone tails by histone acetylases (HATs) and histone deacetylases (HDACs), respectively (Kouzarides, 2007). Epigenetic regulation of survival in neurodegenerative conditions by HATs and HDACs has been well described (Glozak et al., 2005; Saha and Pahan, 2006; Gaub et al., 2010), and their role in regulation of axon and dendrite growth is becoming more apparent (Gaub et al., 2010, 2011).
Figure 1.
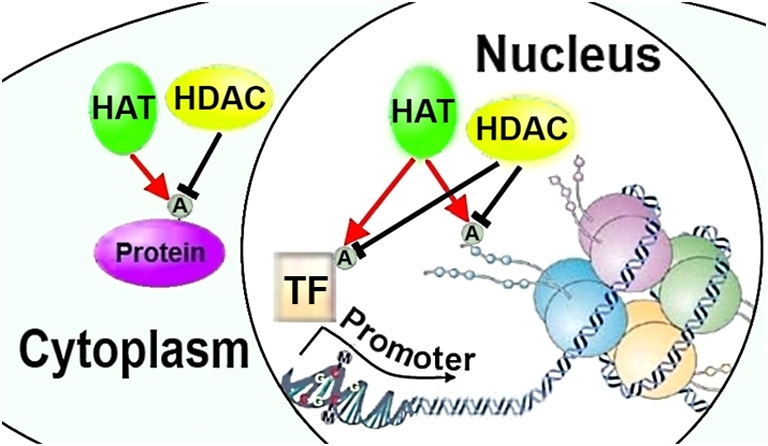
HATs and HDACs can regulate neurite growth through chromatin remodeling or post-translational modifications of transcription factors and cytoplasmic proteins. HAT, histone acetylase; HDAC, histone deacetylase; A, acetylation; TF, transcription factor (Figures 1–3 adapted in part from Levenson and Sweatt (2005)).
Does epigenetic regulation of transcriptional activity in mammalian CNS neurons, which lose their intrinsic neurite growth capacity during maturation (Goldberg et al., 2002; Moore et al., 2009), change developmentally? This question could be addressed in part by assessing whether histone acetylation levels, which signify transcriptional activity, change developmentally in the mammalian CNS neurons. Immunoblotting showed that in at least two types of CNS neurons, cortical neurons, and cerebellar granule neurons, histone 3 (H3) K9/14 acetylation decreases developmentally (Gaub et al., 2010). Because postnatal and adult cortical and cerebellar homogenates may include glial cells that are almost absent from the embryonic homogenates, it is possible that the decrease in the H3K9/14 immunoblotting band was a result of glial protein fraction increase in the input material; it is not known whether there is developmental regulation of histone acetylation in glial cells. Comparing histone acetylation in acutely purified homogeneous cultures—devoid of glial cells—of embryonic versus mature neurons using immunoblotting and in brain sections using immunohistochemistry would corroborate this finding. The developmental regulation of epigenetic regulators and whether they regulate intrinsic axon growth capacity remains to be further explored.
Are these epigenetic enzymes regulated after injury? The expression profile of epigenetic regulators points to attenuation of transcription following stroke in neurons exhibiting axonal sprouting (Li et al., 2010). Cortical neurons that grow short neurites after stroke and adjacent neurons that did not grow neurites were laser-dissected, and a comparison of their gene expression profiles revealed that HDAC4 expression was upregulated whereas p300, a HAT, was downregulated in the axon sprouting neurons (Li et al., 2010). It remains to be investigated whether this trend in expression change of epigenetic regulators functionally affects transcriptional activity and neurite growth, and whether this leads to differential epigenetic regulation of axon growth programs or affects axon growth through non-chromatin remodeling factors. Although an HDAC inhibitor trichostatin A (TSA) was neuroprotective after stroke (Endres et al., 2000b), this may have been mediated by HDAC inhibition in neurons, glia, or immune cells to promote neurite growth or survival. Future investigations will shed light on this question.
Can neurite growth be controlled epigenetically? In support of epigenetic regulation of neurite growth, the HDAC class I and II inhibitor TSA increased neurite growth in postnatal day 7 cerebellar granule neurons on permissive and inhibitory substrates—similar to a known enhancer of axon growth cAMP analog (dbcAMP) (Neumann et al., 2002)—and enhanced expression of axonal outgrowth marker GAP43 (Tedeschi et al., 2009; Gaub et al., 2010). Furthermore, immunoblotting and immunofluorescence revealed that TSA induced H3K9/14 hyperacetylation (Gaub et al., 2010). Measuring H3 total levels would help resolve whether TSA increased H3 acetylation, expression, or both. Did TSA-induced HDAC inhibition increase neurite growth through facilitating increased transcription at one or more target genes? TSA failed to rescue neurite growth suppressed by transcriptional inhibitor actinomycin D, suggesting that transcription is required for TSA-induced neurite growth (Gaub et al., 2010). This question was explored further by investigating whether HATs known to acetylate H3 at K9/14 modulate normal and TSA-induced neurite outgrowth, and if so, whether this effect is associated with regulation of transcription. RT-PCR showed that cerebellar granule neurons cultured for 24 h on an inhibitory myelin substrate decreased expression of HATs CBP/p300 and P/CAF, whereas TSA increased their expression on both permissive and inhibitory substrates. Furthermore, chromatin immunoprecipitation (ChIP) revealed that TSA increased H3K9/14 acetylation on CBP/p300 and P/CAF promoters, but not on a promoter of a control RPL13a gene (Gaub et al., 2010). These findings suggest that TSA induces promoter-specific H3 hyperacetylation, and that CBP/p300 and P/CAF are potential mediators of the TSA-induced increase and myelin-induced decrease in neurite growth, highlighting the importance of transcriptional regulation in neurite growth. It remains to be clarified whether TSA's inhibiting HDACs affects neurite growth by epigenetically augmenting transcription, or by directly acetylating and regulating specific transcription factors, α-tubulin (Hubbert et al., 2002; Rivieccio et al., 2009; Kim et al., 2010; Tapia et al., 2010), or other proteins that also depend on some basal transcriptional activity suppressed by actinomycin D.
To address direct role of HATs in epigenetic transcriptional regulation of neurite growth, it is important investigate whether HAT activity is required for normal neurite growth and whether it is sufficient to increase neurite growth. Knocking-down expression of CBP/p300 or P/CAF in postnatal day 7 cerebellar granule neurons cultured for 24 h decreased neurite growth on an inhibitory substrate, and blocked TSA-induced increase in neurite growth on both permissive and inhibitory substrates (Gaub et al., 2010). Although immunofluorescence showed decreased H3K9/14 acetylation also on a permissive substrate without TSA, neurite growth did not decrease significantly after 24 h in culture. In contrast, p300 overexpression increased neurite growth in postnatal day 7 cerebellar granule neurons cultured for 24 h on both permissive and inhibitory substrates, similar to TSA, and in a follow-up experiment p300 overexpression in postnatal day 7 retinal ganglion cells (RGCs), a type of CNS neuron, increased neurite growth at three days in culture after transduction (Gaub et al., 2010, 2011). Other data suggests that p300 knockdown decreases axon but not dendrite growth, and p300 overexpression had no effect on neurite growth of postnatal day 6 cerebellar granule neurons cultured four days on a permissive substrate (Ikeuchi et al., 2009). It is possible that time in culture or technical differences accounted for the opposite results in cerebellar granule neurons; however, further investigation is warranted.
These experiments suggest that HATs CBP/p300 and P/CAF are required for the hyperacetylation-induced increase in neurite growth. It remains to be clarified whether HAT activity is required for normal neurite growth on a permissive substrate over 24 h in culture, and if so, whether it is axon-, dendrite, or neuronal cell type-specific, and whether the p300 overexpression or TSA-induced hyperacetylation effect on neurite growth is only transient, at least on a permissive substrate. Furthermore, because actinomycin D suppressed normal neurite growth even within 24 h, it is possible that other HATs are also involved in normal neurite growth; it is also possible that the low-level of transcriptional activity remaining after RNAi-mediated HATs knockdown may suffice for maintenance of normal neurite growth in the short-term. Identifying other HDACs and HATs that play a role in neurite growth and whether they could compensate for each other is another question which needs to be addressed. Furthermore, these findings need to be tested in other types of neurons, at different time-points, and importantly, in vivo.
Is there evidence for such epigenetic regulation in vivo? Looking at expression and acetylation levels by immunofluorescence in retinal sections, recent experiments revealed that p300 expression and H3K18 acetylation in RGCs increases during maturation after P0 and then decreases in adulthood, although CBP expression appeared to remain elevated at all time points (Gaub et al., 2011). Again measuring H3 total levels would address whether this change occurs in H3 acetylation, expression, or both, and staining for H3K9/14 would provide additional support consistent with the in vitro data from cortical and cerebellar granule neurons referenced above. Immunofluorescence analysis also showed that although H3K18 acetylation did not change 72 h after optic nerve crush, p300, and CBP levels decreased (Gaub et al., 2011); perhaps other HATs could compensate for H3K18 acetylation.
Could TSA-induced HDAC inhibition increase axon regeneration in vivo? Although TSA increased RGC survival 14 days after optic nerve crush, it had no significant effect on axon regeneration. Immunofluorescence analysis, however, revealed that TSA does not affect p300 expression but increases H3K18 acetylation and CBP expression (Gaub et al., 2011). Testing TSA effects on RGC axon growth in vitro could resolve whether its failure to enhance axon regeneration in vivo was due to an inhibitory in vivo environment, or due to cell-type differences.
Could upregulating p300 increase RGCs axon regeneration in vivo? Indeed, p300 overexpression increased the number of regenerating axons after optic nerve crush, along with H3K18 acetylation, although survival was not affected (Gaub et al., 2011). Furthermore, this effect was additive to a lens injury-induced regeneration, suggesting that either p300 affects neurite growth through a different mechanism, or augments lens injury-induced pathways, or both (Gaub et al., 2011). That HDAC inhibition affected only survival and p300 overexpression affected only axon regeneration raises the interesting and important premise that these enzymatic regulations activate specific cellular phenotypes. Importantly, ChIP analysis of retinas with p300 overexpression after optic nerve crush revealed increased H3K18 acetylation and p300 occupancy on the promoters of pro-growth genes GAP43, Coronin1b, and Sprr1a, and RT-PCR confirmed their upregulation, supporting the biological plausibility of acetylation-dependent epigenetic regulation of neurite growth (Gaub et al., 2011). However, immunofluorescence analysis of retinal section showed that p300 overexpression after optic nerve crush also led to hyperacetylation-dependent activation of axon-growth promoting transcription factors p53 and C/EBP (although C/EBP basal level was not controlled for) (Gaub et al., 2011), leaving unresolved, in this case, the contribution of epigenetic regulation vs. other acetylation-dependent effects to promoting axon growth and regeneration (see below).
SnoN chromatin remodeling factor
Differential histone acetylation regulating axon growth may also be modulated by SnoN, which directly interacts with p300 and can act as a transcriptional repressor (Figure 2) (Ikeuchi et al., 2009). The ability of SnoN to increase axon growth is dependent on p300, as overexpression of the active form of SnoN in cerebellar granule neurons failed to promote axon growth if p300 expression was knocked-down, raising a possibility that SnoN may play a role in chromatin remodeling in cooperation with p300 to differentially activate axon growth-promoting gene programs (Ikeuchi et al., 2009). It is possible that although p300 is necessary for SnoN to promote axon growth, the actual effector of SnoN that promotes axon growth may be transcription factors, for example Smads (Wu et al., 2002), or another non-chromatin remodeling factor. Thus, it remains to be investigated whether SnoN affects neurite growth through epigenetic mechanisms, traditional gene regulation, other ways, or a combination.
Figure 2.
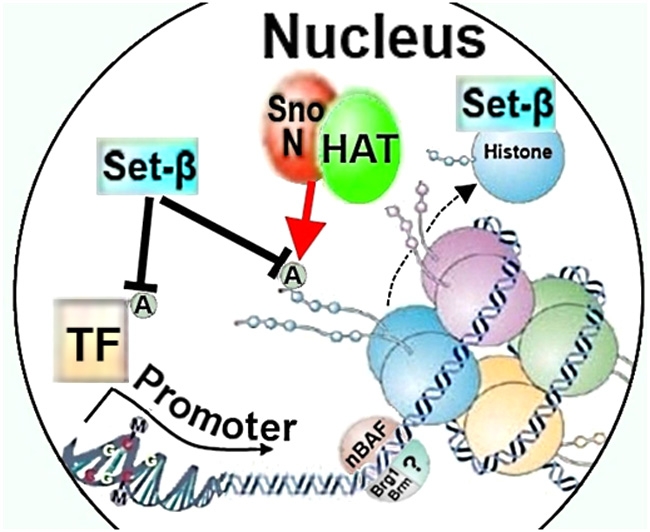
Chromatin remodeling factors SnoN, Set-β, and nBAF could affect neurite growth through regulating histone or transcription factor acetylation, histone chaperoning, or nucleosome structure. HAT, histone acetylase; A, acetylation; TF, transcription factor; nBAF, neuron-specific Brg/Brm-associated factor.
SET-β protein
The “patient SE translocation β ” (SET-β) oncogene (von Lindern et al., 1992) is another chromatin remodeling factor that may regulate axon growth (Trakhtenberg et al., 2011). Set-β could modify chromatin in multiple ways, by chaperoning histones, by governing access to chromatin (Gamble and Fisher, 2007) by preventing histone acetylation as part of the INHAT (inhibitor of acetyltransferases) complex (Seo et al., 2001; Loven et al., 2003), by recruiting HDACs directly (Li et al., 2005; Wagner et al., 2006) or by otherwise regulating transcription (Figure 2) (Okuwaki and Nagata, 1998). For example, glucocorticoid receptor transcription factor can displace Set-β from a histone, permitting histone acetylation and promoting transcription (Ichijo et al., 2008), whereas estrogen receptor (ER) transcription factor recruits Set-β to mask histone acetylation sites, thereby inhibiting transcription. Yet silencing Set-β also prevents ER from activating transcription because of consequent histone hypermethylation, suggesting that basal level of Set-β along with hypomethylation at the promoter are necessary for initiation of the ER-mediated transcription (Wagner et al., 2006).
Set-β can also regulate transcription factors directly, for example, Set-β interacts (Miyamoto et al., 2003) with the KLF family of transcription factors which regulate axon growth (Moore et al., 2009), and can modulate nuclear PP2A (Li et al., 1996; McCright et al., 1996) implicated in neuronal differentiation (Van Kanegan and Strack, 2009) and axon growth (Zhu et al.). Thus, it remains to be investigated whether Set-β regulates axon growth epigenetically—and if so through which mechanism of chromatin remodeling—or thorough regulating other nuclear factors. Interestingly, Set-β is developmentally upregulated in rat RGCs' nuclei, and overexpression of Set-β suppresses axon growth whereas its knockdown promotes axon growth (Trakhtenberg et al., 2011), supporting a hypothesis that Set-β is a physiologic regulator of axon growth during development. The molecular mechanisms for these effects are not yet known.
nBAF complex
SWI/SNF complex is a well-studied chromatin-remodeler (Wang et al., 1996). These enzymes all contain an ATPase catalytic center, and through altering nucleosome structure either move the nucleosome relative to DNA or even eject histones, thereby gating access to DNA (Varga-Weisz, 2001). One such SWI/SNF-like chromatin-remodeling enzymatic complex, neuron-specific Brg/Brm-associated factor (nBAF), regulates dendrite growth and axon myelination (Figure 2) (Wu et al., 2007). Knocking out neuronal subunit BAF53b of the nBAF complex in mice reduced dendrite growth and arborization in cortical neurons, cerebellar granule neurons, and hippocampal neurons in vivo (Wu et al., 2007). The effect was similar in hippocampal neurons in vitro. The knockout also reduced axonal myelination, with an observed reduction in the size of the myelinated axon bundles in the hippocampus. Electron microscopy revealed that substantially fewer axons were myelinated within these axon bundles (Wu et al., 2007). It is also possible that fewer axons grew in the knockout animals, requiring further examination of possible defects in axon morphogenesis and function. Because BAF53b is not expressed in the oligodendrocytes which make myelin (Olave et al., 2002; Wu et al., 2007), it is likely that the knockout in neurons caused axons to be undermyelinated, rather than a defect in oligodendrocytes. This could be due to a failure of neurons to secrete factors or maintain levels of electrical activity that support growth or differentiation of oligodendrocytes (Barres and Raff, 1993; Wu et al., 2007). It is also possible that BAF53b is necessary for axons to be receptive to myelination, or its absence in progenitors could result in differentiation of defective oligodendrocytes. Possible roles of nBAF in axon formation and oligodendrocyte differentiation require further examination and as above, it remains to be investigated whether discrete pro-regenerative programs are differentially affected by the nBAF complex or its subunits.
DNA and histone methylation
Along with histone and other protein acetylation, direct DNA methylation is a key mode of epigenetic regulation (Figure 3) (Kumar et al., 1994). Mammalian (cytosine-5) DNA methyltransferases 3a and 3b (Dnmt3a and -3b) catalyze de novo DNA methylation, whereas Dnmt1 is responsible for maintenance of DNA methylation (Kumar et al., 1994). Dnmt activity is high in normal adult brain neurons (Brooks et al., 1996) and DNA methylation levels are increased in the brain after ischemic insults, whereas reducing DNA methylation—particularly by decreasing expression of Dnmt1 (Endres et al., 2001)—is neuroprotective after stroke (Endres et al., 2000a; Qureshi and Mehler, 2010). The role of DNA methylation in axon and dendrite growth has been studied only scarcely. Dnmt3b positively regulates neurite growth in PC12 cells, induced by NGF to differentiate into a neuron-like phenotype, by recruiting HDAC—rather than by direct DNA methylation—to the promoter of the axon growth-suppressing gene T-cadherin (Fredette et al., 1996), thereby negatively regulating its expression, releasing the cells from T-cadherin-mediated inhibition, and thereby promoting neurite growth (Bai et al., 2006). Thus, a possibility of Dnmts affecting histones' post-translational modification needs to be addressed in investigation of their roles in axon growth. Double conditional knockout of Dnmt3a and Dnmt1 in postmitotic postnatal forebrain mice neurons, on the other hand, did not impair axonal development, although synaptic plasticity was defective (Feng et al., 2010).
Figure 3.
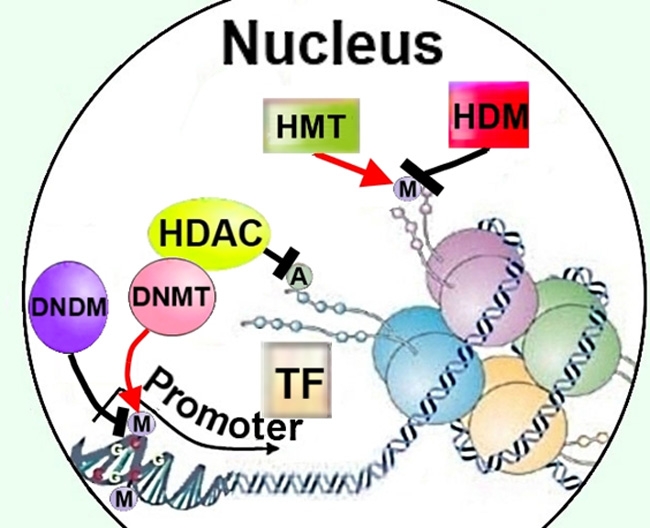
Regulation of cellular methylation could affect neurite growth though modifying DNA or histones, as well as by recruiting HDACs to promoters. HDAC, histone deacetylase; M, methylation; A, acetylation; TF, transcription factor; HMT, histone methyltransferase; HDM, histone demethylase; DNMT, DNA methyltransferase; DNDM, DNA demethylase.
Even if it is not critical for axon growth during development, it is still possible that diminished Dnmt3a expression or activity might augment CNS damage after injury and hinder regenerative therapies. For example, folic acid (FA) promotes CNS axon regeneration and a folate receptor isoform α (Folr1)—which can transport FA into neurons—is upregulated in dorsal root ganglia (DRG) neurons after spinal cord injury (Iskandar et al., 2010). Reduced Folr1 expression in Folr1+/− mice or inhibition of Dhfr—an enzyme catalyzing conversion of FA into tetrahydrofolate, which plays role in cellular metabolism—blocked FA-induced axon regeneration (Iskandar et al., 2010). Dnmt3a and Dnmt3b levels and global DNA methylation in the spinal cord decreased after spinal cord injury, but were restored by FA—a primary source for methyl donors utilized by Dnmts (Iskandar et al., 2010)—suggesting that a possible mechanism of action for FA-promoted regeneration is through DNA methylation. How FA increases Dnmt levels, and whether this depends on a pathway through which FA increased methyl donor substrate availability, remains to be investigated. Similarly, promoter methylation of the Gadd45a gene—which promoted neurite outgrowth in N1E-115 neuroblastoma cells (Yamauchi et al., 2007)—was increased after spinal cord injury and further hypermethylated by FA, suggesting specific epigenetic regulation of pro-regenerative gene expression by methylation (Figure 3). Because DNA hypermethylation is typically associated with silencing gene expression, it is important to examine how the expression of the Gadd45a gene product was affected. An inclusion of methylation assays for a non-axon growth-related gene promoters and several prominent axon regeneration promoting genes, as well as RT-PCR to measure effect on expression, could further support the implication that promoter methylation can differentially regulate pro-regenerative gene expression.
It is possible that in both cases, stroke and spinal cord injury, changes in DNA methylation were primarily due to changes in glial and immune cells' methylation, as the DNA from whole tissue was used in both cases. It is also possible that differences between effects of methylation on non-neuronal cells accounted for the opposite effects of methylation on neuroprotection and regeneration in these tissues. Because glia and immune cells play critical roles in the injured CNS (Trakhtenberg and Goldberg, 2011), it is possible that, for example, regulating DNA methylation in these cells modulated immunosuppression and ensuing inflammatory damage, which also inhibits axon regeneration. It is also possible that in both cases DNA methylation reflected primarily trends in neuronal DNA methylation, which could differ between brain stroke and spinal cord injury models or between the tissues. An interaction between the effects of neuronal and glial/immune cell DNA methylation patterns may also affect CNS differently after brain stroke and spinal cord injury. Thus, it is important to re-examine these questions with attention to changes in DNA methylation in specific cell types and include overexpression or knockdown of Dnmts, individually or in combinations.
Interestingly, histone methylation also modifies chromatin structure and gene regulation (Kouzarides, 2007; Verrier et al., 2011), and a methyltransferase which can methylate histones—protein arginine N-methyltransferase 1 (PRMT1)—was shown to be necessary for neurite outgrowth in the Neuro-2a cell line (Figure 3) (Miyata et al., 2008). Neural-specific histone lysine-specific demethylase 1 (nLSD1/KDM1) was also shown to regulate neurite growth of cortical neurons (Zibetti et al., 2010). nLSD1 was either knocked-down or overexpressed in cultured embryonic rat cortical neurons and neurite growth decreased or increased, respectively (Zibetti et al., 2010). It will be important to test nLSD1's axon regenerative potential in a CNS injury model in vivo. In Drosophila, deletion of another histone demethylase, DMEL/KDM4a, did not cause apparent abnormalities in axon or dendrite morphogenesis and growth during development. It remains to be investigated whether its mammalian homolog regulates neurite growth in the mammalian CNS (Lorbeck et al., 2010). In contrast to histone demethylases, DNA demethylases have not yet been studied extensively and investigation of their role in axon growth is clearly warranted. However, differentially regulating levels of histone methyltransferases vs. Dnmts could affect availability of methyl donors and, in turn, modulate methylation of targets which regulate axon growth (Mato and Lu, 2007).
Chromatin-dependent and -independent regulation of neurite growth
Most of the chromatin remodeling studies discussed in this review have converged on the importance of histone-associated chromatin modifications. Histone acetylation and methylation regulates chromatin remodeling, but acetylation is also a common mode of post-translational regulation of transcription factors and cytoplasmic proteins (Sadoul et al., 2011); regulation of transcription factors (e.g., STAT1) (Mowen et al., 2001) and other proteins by methylation is now also well-established (Paik et al., 2007). Manipulating cellular acetylation or methylation could thus work through chromatin remodeling as well as through regulation of various other proteins which could regulate neurite growth. For example, HDAC class I and HDAC6 also could directly regulate neurite growth independently of epigenetic effects on transcription through deacetylation of α-tubulin and other proteins (Hubbert et al., 2002; Rivieccio et al., 2009; Kim et al., 2010; Tapia et al., 2010). Furthermore, p300 acetylates transcription factor KLF4 (Evans et al., 2007) and might also promote neurite growth by increasing α-tubulin acetylation (Ferreira and Caceres, 1989; Veldman et al., 2010) through inhibiting cytoplasmic HDAC6, which normally deacetylates α-tubulin (Han et al., 2009). Set-β can also mask an acetylation site on KLF transcription factors and potentially regulate their effects on neurite growth-related transcription (Miyamoto et al., 2003). In yet another example, axon growth-promoting transcription factor p53 can be activated through accumulated acetylation after TSA inhibition of HDACs (Gaub et al., 2010), which may be in parallel to, or downstream of, observed concurrent histone-H3 acetylation (Gaub et al., 2010), again suggesting that acetylation of axon-growth promoting transcription factors may be an important component of the hyperacetylation effect on neurite growth. Such multiple roles of acetylation and methylation in other, non-chromatin-relevant cellular processes pose a challenge to experimental discernment of epigenetic and non-epigenetic regulation of neurite growth.
Discussion
Taken together, these findings raise the hypothesis that chromatin gatekeepers are as relevant for activation of pro-regenerative gene expression as the levels of pro-regenerative transcription factors themselves. It is interesting to hypothesize that epigenetic pathways may play wider range of roles in neurons than in other cell types, as mammalian neurons are not generally replaced through life and thus may be more reliant on such cellular regulatory mechanisms. This notion is supported by presence of neuron-specific epigenetic factors like nBAF and nLSD1 discussed above, which may contribute to unique neuronal adaptations such as transcription-dependent neuroplasticity and learning (Kandel, 2001; Wu et al., 2007) or axon and dendrite regeneration. Such a prospect is intriguing as current techniques for experimental manipulation of transcription factor expression in neurons allows the study of only one or a few transcription factors at a time. Unlocking the activity of multiple pro-regenerative transcription factors and allowing greater access—and maybe even active recruitment (Strahl and Allis, 2000)—to promoters of pro-regenerative genes, may prove a powerful approach to promoting regeneration; the same would be true of manipulating chromatin to block access to gene loci that inhibit regeneration. Exploring how manipulation of the upstream regulators of HATs/HDAC and Dnmts (Saha and Pahan, 2006), or other chromatin modifying factors, such as histone chaperones, ATP-dependent chromatin remodeling complexes, non-coding RNAs (Goldberg et al., 2007; Gupta et al., 2010), and regulators of histone methylation, phosphorylation, and ubiquitination (Kouzarides, 2007), should hold considerable promise for progress in the field of neuroregeneration.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge support from the National Institutes of Health (EY020913, NS061348 to Jeffrey L. Goldberg; P30-EY014081 to the University of Miami), the Walter G. Ross Distinguished Chair in Ophthalmic Research (Jeffrey L. Goldberg), an unrestricted grant from Research to Prevent Blindness (University of Miami), and the American Heart Association (11PRE7310069 to Ephraim F. Trakhtenberg). Ephraim F. Trakhtenberg was also funded by NIH (T32 NS007492).
References
- Bai S., Ghoshal K., Jacob S. T. (2006). Identification of T-cadherin as a novel target of DNA methyltransferase 3B and its role in the suppression of nerve growth factor-mediated neurite outgrowth in PC12 cells. J. Biol. Chem. 281, 13604–13611 10.1074/jbc.M513278200 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Barres B. A., Raff M. C. (1993). Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 361, 258–260 10.1038/361258a0 [DOI] [PubMed] [Google Scholar]
- Bird A. (2007). Perceptions of epigenetics. Nature 447, 396–398 10.1038/nature05913 [DOI] [PubMed] [Google Scholar]
- Blackmore M., Letourneau P. C. (2006). Changes within maturing neurons limit axonal regeneration in the developing spinal cord. J. Neurobiol. 66, 348–360 10.1002/neu.20224 [DOI] [PubMed] [Google Scholar]
- Bouslama-Oueghlani L., Wehrle R., Sotelo C., Dusart I. (2003). The developmental loss of the ability of Purkinje cells to regenerate their axons occurs in the absence of myelin: an in vitro model to prevent myelination. J. Neurosci. 23, 8318–8329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks P. J., Marietta C., Goldman D. (1996). DNA mismatch repair and DNA methylation in adult brain neurons. J. Neurosci. 16, 939–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M., Fan G., Hirt L., Fujii M., Matsushita K., Liu X., Jaenisch R., Moskowitz M. A. (2000a). Ischemic brain damage in mice after selectively modifying BDNF or NT4 gene expression. J. Cereb. Blood Flow Metab. 20, 139–144 10.1097/00004647-200001000-00018 [DOI] [PubMed] [Google Scholar]
- Endres M., Fan G., Meisel A., Dirnagl U., Jaenisch R. (2001). Effects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neurons. Neuroreport 12, 3763–3766 [DOI] [PubMed] [Google Scholar]
- Endres M., Meisel A., Biniszkiewicz D., Namura S., Prass K., Ruscher K., Lipski A., Jaenisch R., Moskowitz M. A., Dirnagl U. (2000b). DNA methyltransferase contributes to delayed ischemic brain injury. J. Neurosci. 20, 3175–3181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. M., Zhang W., Chen X., Yang J., Bhakat K. K., Liu C. (2007). Kruppel-like factor 4 is acetylated by p300 and regulates gene transcription via modulation of histone acetylation. J. Biol. Chem. 282, 33994–34002 10.1074/jbc.M701847200 [DOI] [PubMed] [Google Scholar]
- Feng J., Zhou Y., Campbell S. L., Le T., Li E., Sweatt J. D., Silva A. J., Fan G. (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13, 423–430 10.1038/nn.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A., Caceres A. (1989). The expression of acetylated microtubules during axonal and dendritic growth in cerebellar macroneurons which develop in vitro. Brain Res. Dev. Brain Res. 49, 205–213 [DOI] [PubMed] [Google Scholar]
- Fredette B. J., Miller J., Ranscht B. (1996). Inhibition of motor axon growth by T-cadherin substrata. Development 122, 3163–3171 [DOI] [PubMed] [Google Scholar]
- Gamble M. J., Fisher R. P. (2007). SET and PARP1 remove DEK from chromatin to permit access by the transcription machinery. Nat. Struct. Mol. Biol. 14, 548–555 10.1038/nsmb1248 [DOI] [PubMed] [Google Scholar]
- Gaub P., Joshi Y., Wuttke A., Naumann U., Schnichels S., Heiduschka P., Di Giovanni S. (2011). The histone acetyltransferase p300 promotes intrinsic axonal regeneration. Brain 134, 2134–2148 10.1093/brain/awr142 [DOI] [PubMed] [Google Scholar]
- Gaub P., Tedeschi A., Puttagunta R., Nguyen T., Schmandke A., Di Giovanni S. (2010). HDAC inhibition promotes neuronal outgrowth and counteracts growth cone collapse through CBP/p300 and P/CAF-dependent p53 acetylation. Cell Death Differ. 17, 1392–1408 10.1038/cdd.2009.216 [DOI] [PubMed] [Google Scholar]
- Glozak M. A., Sengupta N., Zhang X., Seto E. (2005). Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23 10.1016/j.gene.2005.09.010 [DOI] [PubMed] [Google Scholar]
- Goldberg A. D., Allis C. D., Bernstein E. (2007). Epigenetics: a landscape takes shape. Cell 128, 635–638 10.1016/j.cell.2007.02.006 [DOI] [PubMed] [Google Scholar]
- Goldberg J. L., Klassen M. P., Hua Y., Barres B. A. (2002). Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science 296, 1860–1864 10.1126/science.1068428 [DOI] [PubMed] [Google Scholar]
- Gupta R. A., Shah N., Wang K. C., Kim J., Horlings H. M., Wong D. J., Tsai M. C., Hung T., Argani P., Rinn J. L., Wang Y., Brzoska P., Kong B., Li R., West R. B., van de Vijver M. J., Sukumar S., Chang H. Y. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076 10.1038/nature08975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Jeong H. M., Jin Y. H., Kim Y. J., Jeong H. G., Yeo C. Y., Lee K. Y. (2009). Acetylation of histone deacetylase 6 by p300 attenuates its deacetylase activity. Biochem. Biophys. Res. Commun. 383, 88–92 10.1016/j.bbrc.2009.03.147 [DOI] [PubMed] [Google Scholar]
- Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X. F., Yao T. P. (2002). HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 10.1038/417455a [DOI] [PubMed] [Google Scholar]
- Ichijo T., Chrousos G. P., Kino T. (2008). Activated glucocorticoid receptor interacts with the INHAT component Set/TAF-Ibeta and releases it from a glucocorticoid-responsive gene promoter, relieving repression: implications for the pathogenesis of glucocorticoid resistance in acute undifferentiated leukemia with Set-Can translocation. Mol. Cell. Endocrinol. 283, 19–31 10.1016/j.mce.2007.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi Y., Stegmuller J., Netherton S., Huynh M. A., Masu M., Frank D., Bonni S., Bonni A. (2009). A SnoN-Ccd1 pathway promotes axonal morphogenesis in the mammalian brain. J. Neurosci. 29, 4312–4321 10.1523/JNEUROSCI.0126-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iskandar B. J., Rizk E., Meier B., Hariharan N., Bottiglieri T., Finnell R. H., Jarrard D. F., Banerjee R. V., Skene J. H., Nelson A., Patel N., Gherasim C., Simon K., Cook T. D., Hogan K. J. (2010). Folate regulation of axonal regeneration in the rodent central nervous system through DNA methylation. J. Clin. Invest. 120, 1603–1616 10.1172/JCI40000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel E. R. (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038 10.1126/science.1067020 [DOI] [PubMed] [Google Scholar]
- Kim J. Y., Shen S., Dietz K., He Y., Howell O., Reynolds R., Casaccia P. (2010). HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat. Neurosci. 13, 180–189 10.1038/nn.2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. (2007). Chromatin modifications and their function. Cell 128, 693–705 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- Kumar S., Cheng X., Klimasauskas S., Mi S., Posfai J., Roberts R. J., Wilson G. G. (1994). The DNA (cytosine-5) methyltransferases. Nucleic Acids Res. 22, 1–10 10.1093/nar/22.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson J. M., Sweatt J. D. (2005). Epigenetic mechanisms in memory formation. Nat. Rev. Neurosci. 6, 108–118 10.1038/nrn1604 [DOI] [PubMed] [Google Scholar]
- Li D., Field P. M., Raisman G. (1995). Failure of axon regeneration in postnatal rat entorhinohippocampal slice coculture is due to maturation of the axon, not that of the pathway or target. Eur. J. Neurosci. 7, 1164–1171 [DOI] [PubMed] [Google Scholar]
- Li D., Yea S., Li S., Chen Z., Narla G., Banck M., Laborda J., Tan S., Friedman J. M., Friedman S. L., Walsh M. J. (2005). Kruppel-like factor-6 promotes preadipocyte differentiation through histone deacetylase 3-dependent repression of DLK1. J. Biol. Chem. 280, 26941–26952 10.1074/jbc.M500463200 [DOI] [PubMed] [Google Scholar]
- Li M., Makkinje A., Damuni Z. (1996). The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J. Biol. Chem. 271, 11059–11062 [DOI] [PubMed] [Google Scholar]
- Li S., Overman J. J., Katsman D., Kozlov S. V., Donnelly C. J., Twiss J. L., Giger R. J., Coppola G., Geschwind D. H., Carmichael S. T. (2010). An age-related sprouting transcriptome provides molecular control of axonal sprouting after stroke. Nat. Neurosci. 13, 1496–1504 10.1038/nn.2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K., Tedeschi A., Park K. K., He Z. (2011). Neuronal intrinsic mechanisms of axon regeneration. Annu. Rev. Neurosci. 34, 131–152 10.1146/annurev-neuro-061010-113723 [DOI] [PubMed] [Google Scholar]
- Lorbeck M. T., Singh N., Zervos A., Dhatta M., Lapchenko M., Yang C., Elefant F. (2010). The histone demethylase Dmel\textbackslash Kdm4A controls genes required for life span and male-specific sex determination in Drosophila. Gene 450, 8–17 10.1016/j.gene.2009.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven M. A., Muster N., Yates J. R., Nardulli A. M. (2003). A novel estrogen receptor alpha-associated protein, template-activating factor Ibeta, inhibits acetylation and transactivation. Mol. Endocrinol. 17, 67–78 [DOI] [PubMed] [Google Scholar]
- Mato J. M., Lu S. C. (2007). Role of S-adenosyl-L-methionine in liver health and injury. Hepatology 45, 1306–1312 10.1002/hep.21650 [DOI] [PubMed] [Google Scholar]
- McCright B., Rivers A. M., Audlin S., Virshup D. M. (1996). The B56 family of protein phosphatase 2A (PP2A) regulatory subunits encodes differentiation-induced phosphoproteins that target PP2A to both nucleus and cytoplasm. J. Biol. Chem. 271, 22081–22089 [DOI] [PubMed] [Google Scholar]
- Miyamoto S., Suzuki T., Muto S., Aizawa K., Kimura A., Mizuno Y., Nagino T., Imai Y., Adachi N., Horikoshi M., Nagai R. (2003). Positive and negative regulation of the cardiovascular transcription factor KLF5 by p300 and the oncogenic regulator SET through interaction and acetylation on the DNA-binding domain. Mol. Cell. Biol. 23, 8528–8541 10.1128/MCB.23.23.8528-8541.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata S., Mori Y., Tohyama M. (2008). PRMT1 and Btg2 regulates neurite outgrowth of Neuro2a cells. Neurosci. Lett. 445, 162–165 10.1016/j.neulet.2008.08.065 [DOI] [PubMed] [Google Scholar]
- Moore D. L., Blackmore M. G., Hu Y., Kaestner K. H., Bixby J. L., Lemmon V. P., Goldberg J. L. (2009). KLF family members regulate intrinsic axon regeneration ability. Science 326, 298–301 10.1126/science.1175737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D. L., Goldberg J. L. (2011). Multiple transcription factor families regulate axon growth and regeneration. Dev. Neurobiol. 71, 1186–1211 10.1002/dneu.20934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowen K. A., Tang J., Zhu W., Schurter B. T., Shuai K., Herschman H. R., David M. (2001). Arginine methylation of STAT1 modulates IFNalpha/beta-induced transcription. Cell 104, 731–741 10.1016/S0092-8674(01)00269-0 [DOI] [PubMed] [Google Scholar]
- Neumann S., Bradke F., Tessier-Lavigne M., Basbaum A. I. (2002). Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34, 885–893 10.1016/S0896-6273(02)00702-X [DOI] [PubMed] [Google Scholar]
- Okuwaki M., Nagata K. (1998). Template activating factor-I remodels the chromatin structure and stimulates transcription from the chromatin template. J. Biol. Chem. 273, 34511–34518 10.1074/jbc.273.51.34511 [DOI] [PubMed] [Google Scholar]
- Olave I., Wang W., Xue Y., Kuo A., Crabtree G. R. (2002). Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes Dev. 16, 2509–2517 10.1101/gad.992102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik W. K., Paik D. C., Kim S. (2007). Historical review: the field of protein methylation. Trends Biochem. Sci. 32, 146–152 10.1016/j.tibs.2007.01.006 [DOI] [PubMed] [Google Scholar]
- Qureshi I. A., Mehler M. F. (2010). Emerging role of epigenetics in stroke: part 1: DNA methylation and chromatin modifications. Arch. Neurol. 67, 1316–1322 10.1001/archneurol.2010.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reik W. (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447, 425–432 10.1038/nature05918 [DOI] [PubMed] [Google Scholar]
- Rivieccio M. A., Brochier C., Willis D. E., Walker B. A., D'Annibale M. A., Mclaughlin K., Siddiq A., Kozikowski A. P., Jaffrey S. R., Twiss J. L., Ratan R. R., Langley B. (2009). HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. U.S.A. 106, 19599–19604 10.1073/pnas.0907935106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoul K., Wang J., Diagouraga B., Khochbin S. (2011). The tale of protein lysine acetylation in the cytoplasm. J. Biomed. Biotechnol. 2011, 970382 10.1155/2011/970382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha R. N., Pahan K. (2006). HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 13, 539–550 10.1038/sj.cdd.4401769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S. B., Mcnamara P., Heo S., Turner A., Lane W. S., Chakravarti D. (2001). Regulation of histone acetylation and transcription by INHAT, a human cellular complex containing the set oncoprotein. Cell 104, 119–130 10.1016/S0092-8674(01)00196-9 [DOI] [PubMed] [Google Scholar]
- Strahl B. D., Allis C. D. (2000). The language of covalent histone modifications. Nature 403, 41–45 10.1038/47412 [DOI] [PubMed] [Google Scholar]
- Sun F., He Z. (2010). Neuronal intrinsic barriers for axon regeneration in the adult CNS. Curr. Opin. Neurobiol. 20, 510–518 10.1016/j.conb.2010.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapia M., Wandosell F., Garrido J. J. (2010). Impaired function of HDAC6 slows down axonal growth and interferes with axon initial segment development. PLoS One 5:e12908 10.1371/journal.pone.0012908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A., Nguyen T., Puttagunta R., Gaub P., Di Giovanni S. (2009). A p53-CBP/p300 transcription module is required for GAP-43 expression, axon outgrowth, and regeneration. Cell Death Differ. 16, 543–554 10.1038/cdd.2008.175 [DOI] [PubMed] [Google Scholar]
- Trakhtenberg E. F., Goldberg J. L. (2011). Immunology. Neuroimmune communication. Science 334, 47–48 10.1126/science.1213099 [DOI] [PubMed] [Google Scholar]
- Trakhtenberg E. F., Wang Y., Lapins A., Yang S., Goldberg J. L. (2011). Set-β regulation of axon growth in central nervous system. Soc. Neurosci. (Wash.) 438, B27 [Google Scholar]
- Turner B. M. (2007). Defining an epigenetic code. Nat. Cell Biol. 9, 2–6 10.1038/ncb0107-2 [DOI] [PubMed] [Google Scholar]
- Van Kanegan M. J., Strack S. (2009). The protein phosphatase 2A regulatory subunits B'beta and B'delta mediate sustained TrkA neurotrophin receptor autophosphorylation and neuronal differentiation. Mol. Cell. Biol. 29, 662–674 10.1128/MCB.01242-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga-Weisz P. (2001). ATP-dependent chromatin remodeling factors: nucleosome shufflers with many missions. Oncogene 20, 3076–3085 10.1038/sj.onc.1204332 [DOI] [PubMed] [Google Scholar]
- Veldman M. B., Bemben M. A., Goldman D. (2010). Tuba1a gene expression is regulated by KLF6/7 and is necessary for CNS development and regeneration in zebrafish. Mol. Cell. Neurosci. 43, 370–383 10.1016/j.mcn.2010.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrier L., Vandromme M., Trouche D. (2011). Histone demethylases in chromatin crosstalks. Biol. Cell 103, 381–401 10.1042/BC20110028 [DOI] [PubMed] [Google Scholar]
- von Lindern M., Van Baal S., Wiegant J., Raap A., Hagemeijer A., Grosveld G. (1992). Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes: characterization of the set gene. Mol. Cell. Biol. 12, 3346–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner S., Weber S., Kleinschmidt M. A., Nagata K., Bauer U. M. (2006). SET-mediated promoter hypoacetylation is a prerequisite for coactivation of the estrogen-responsive pS2 gene by PRMT1. J. Biol. Chem. 281, 27242–27250 10.1074/jbc.M605172200 [DOI] [PubMed] [Google Scholar]
- Wang W., Cote J., Xue Y., Zhou S., Khavari P. A., Biggar S. R., Muchardt C., Kalpana G. V., Goff S. P., Yaniv M., Workman J. L., Crabtree G. R. (1996). Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 15, 5370–5382 [PMC free article] [PubMed] [Google Scholar]
- Wu J. I., Lessard J., Olave I. A., Qiu Z., Ghosh A., Graef I. A., Crabtree G. R. (2007). Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56, 94–108 10.1016/j.neuron.2007.08.021 [DOI] [PubMed] [Google Scholar]
- Wu J. W., Krawitz A. R., Chai J., Li W., Zhang F., Luo K., Shi Y. (2002). Structural mechanism of Smad4 recognition by the nuclear oncoprotein Ski: insights on Ski-mediated repression of TGF-beta signaling. Cell 111, 357–367 10.1016/S0092-8674(02)01006-1 [DOI] [PubMed] [Google Scholar]
- Yamauchi J., Miyamoto Y., Murabe M., Fujiwara Y., Sanbe A., Fujita Y., Murase S., Tanoue A. (2007). Gadd45a, the gene induced by the mood stabilizer valproic acid, regulates neurite outgrowth through JNK and the substrate paxillin in N1E-115 neuroblastoma cells. Exp. Cell Res. 313, 1886–1896 10.1016/j.yexcr.2007.02.019 [DOI] [PubMed] [Google Scholar]
- Yiu G., He Z. (2006). Glial inhibition of CNS axon regeneration. Nat. Rev. Neurosci. 7, 617–627 10.1038/nrn1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L. Q., Zheng H. Y., Peng C. X., Liu D., Li H. L., Wang Q., Wang J. Z. (2010). Protein phosphatase 2A facilitates axonogenesis by dephosphorylating CRMP2. J. Neurosci. 30, 3839–3848 10.1523/JNEUROSCI.5174-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zibetti C., Adamo A., Binda C., Forneris F., Toffolo E., Verpelli C., Ginelli E., Mattevi A., Sala C., Battaglioli E. (2010). Alternative splicing of the histone demethylase LSD1/KDM1 contributes to the modulation of neurite morphogenesis in the mammalian nervous system. J. Neurosci. 30, 2521–2532 10.1523/JNEUROSCI.5500-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]