

Abstract
Nitric oxide and its metabolites undergo nitration reactions with unsaturated fatty acids during oxidative inflammatory conditions, forming electrophilic nitro-fatty acid derivatives. These endogenous electrophilic mediators activate anti-inflammatory signaling reactions, serving as high-affinity ligands for peroxisome proliferator-activated receptor γ (PPARγ). Here we examined the therapeutic effects of 9- or 10-nitro-octadecenoic oleic acid (OA-NO2) and native oleic acid (OA) in a mouse model of colitis. OA-NO2 reduced the disease activity index and completely prevented dextran sulfate sodium-induced colon shortening and the increase in colonic p65 expression. Increased PPARγ expression was observed in colon samples as well as in cells after OA-NO2 administration, whereas no effect was seen with OA. This induction of PPARγ expression was completely abolished by the PPARγ antagonist GW9662. 5-Aminosalicylic acid, an anti-inflammatory drug routinely used in the management of inflammatory bowel disease, also increased PPARγ expression but to a lesser extent. Altogether, these findings demonstrate that administration of OA-NO2 attenuates colonic inflammation and improves clinical symptoms in experimental inflammatory bowel disease. This protection involves activation of colonic PPARγ.
Keywords: Nitrated fatty acids, PPARγ, Experimental colitis, Nitric oxide, Free radicals
Ulcerative colitis and Crohn disease are chronic relapsing inflammatory intestinal disorders of unknown etiology. Current treatments for inflammatory bowel disease (IBD)2 include anti-inflammatory corticosteroids, aminosalicylates, and various immune modulators as well as the more recently developed anti-tumor necrosis factor-α [1]. Although therapies for IBD are improving they are still far from optimal for long-term disease management, and adverse side effects are common. In the search for novel therapeutic interventions attempts are being made to explore and selectively target the specific proinflammatory pathways that are central in IBD pathogenesis. Recent studies have indicated a role for peroxisome proliferator-activated receptor γ (PPARγ) in IBD [2].
PPARγ is a member of the nuclear receptor superfamily of transcription factors involved in the control of inflammation, cell proliferation, apoptosis, and metabolic functions. Ligands for PPARγ include natural compounds with relatively low affinity such as polyunsaturated fatty acids, oxidized low-density lipoprotein, certain eicosanoids, α, β-unsaturated keto derivatives of fatty acids, 15-deoxy-Δ12,14-PGJ2, and drugs including the thiazolidinedione derivatives troglitazone, rosiglitazone, and pioglitazone used for the treatment of type 2 diabetes [3]. PPARγ+/− heterozygous mice exhibit an increased susceptibility to experimentally induced colitis [4], indicating a key role for PPARγ in maintaining large intestine homeostasis. The fact that PPARγ is highly expressed in colonic epithelium [5] makes it an attractive drug target for IBD therapy. Indeed, activation of PPARγ was recently shown to mediate the effects of aminosalicylates [6], anti-inflammatory agents routinely used in the treatment of IBD. In addition, thiazolidinedione depresses inflammation in murine models of IBD [7–9] and has also shown some benefit in recent clinical trials [9,10].
The free radical gas nitric oxide (NO) is generated by the inducible NO synthase during inflammation [11], and colonic NO formation is greatly enhanced in patients with active IBD [12]. It has been reported that nitration products of unsaturated fatty acids are formed via NO-dependent oxidative reactions [13]. These derivatives were initially viewed to be, like nitrotyrosine, a “footprint” of NO-dependent redox reactions [13,14]. More recently, it has been observed that electrophilic nitroalkene derivatives of unsaturated fatty acids mediate pluripotent cell signaling actions. In vitro cell studies have revealed potent anti-inflammatory actions of nitroalkenes via interaction with numerous pathways, including nuclear factor-κB (NF-κB), heme oxygenase-1, xanthine oxidase, and signal transducer and activator of transcription (STAT) signaling [15]. In addition, these electrophilic mediators were recently shown to be potent activators of PPARγ [16–18]. Although in vitro studies support potential anti-inflammatory effects of nitrated fatty acids, little is known about their effects in vivo.
Here we studied the therapeutic effects of nitrated oleic acid (OA-NO2) in a murine model of IBD. We also explored whether any of the anti-inflammatory actions of OA-NO2 involve activation of PPARγ-dependent signaling.
Material and methods
Materials
Dextran sulfate sodium (DSS; 45 kDa) was from TdB Consultancy (Uppsala, Sweden). OA-NO2 was synthesized as described previously [16]. 5-Aminosalicylic acid (5-ASA) and GW9662 were from Sigma (St. Louis, MO, USA). Fugene transfection reagent was from Roche (Mannheim, Germany). The Dual-Luciferase Reporter Assay System was from Promega (Madison, WI, USA). Trizol reagent and M-MLV reverse transcriptase were from Invitrogen (Carlsbad, CA, USA). SYBR green master mix was from Applied Biosystems (Foster City, CA, USA).
Animals
All experimental work on animals was approved by the Ethics Committee for Animal Experiments at the Karolinska Institutet. Female BALB/c mice, 7–8 weeks of age and weighing 19–22 g, were housed under temperature- and humidity-controlled conditions with a 12:12 h light:dark cycle and fed a standard pellet diet and tap water ad libitum. The mice were acclimatized for 2 weeks before the study was started.
Animal treatments and assessment of colitis
Mice were divided in four groups (eight animals per group: control, DSS, DSS + OA (native oleic acid), and DSS + OA-NO2). DSS, DSS + OA, and DSS + OA-NO2 groups received 2% DSS (w/v) in the drinking water for 7 days. OA and OA-NO2 (0.5 or 5 mg/kg/day) were given subcutaneously using Alzet osmotic minipumps (Model 1007D; Durect Corp., Cupertino, CA, USA) from the first day of the experiment. Control animals received tap water only.
Mice were examined daily by a blinded investigator to determine the disease activity index (DAI). The DAI was achieved by scoring body weight loss, stool consistency, and bleeding as described [19]. At the end of the study period mice were anesthetized with isoflurane, and blood was collected by heart puncture, followed by cervical dislocation. The colon was removed and its length was recorded. Tissues and plasma samples were frozen and kept at −80°C until analysis. Levels of the NF-κB subunit p65 were assessed by immunohistochemical staining of colon biopsies, as described but with slight modifications [20]. In this case, the primary antibody was the NF-κB p65 monoclonal antibody (sc-8008; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and the secondary antibody was the biotinylated anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA, USA). The intensity of the staining was scored (0–2) by a blinded investigator.
Measurements of nitro-fatty acids
Acetonitrile extraction was used to isolate lipids from murine intestine and plasma stored at −80°C until processing. Quantitative analysis of free OA-NO2 levels was conducted by high-performance liquid chromatography–electrospray ionization mass spectrometry using a triple-quadrupole mass spectrometer (API 5000; Applied Biosystems/MDS Sciex, Framingham, MA, USA) [21,22]. Samples were resolved by high-performance liquid chromatography (HPLC) using a 150×2-mm C18 Luna reversed-phase column (particle size 3 mm; Phenomenex, Belmont, CA, USA) at a flow rate of 0.25 ml/min and a linear gradient of acetonitrile (+0.1% acetic acid) in water (45–80% in 45 min). Mass spectrometric detection of OA-NO2 was performed using multiple reaction monitoring (MRM) mode by monitoring the mass transition m/z 326/46 for the native compound and m/z 344/46 for the 13C-labeled internal standard. Quantitative analysis of protein-adducted molecules was performed in the MRM scan mode. Separately, β-mercaptoethanol (BME) was utilized to capture net free and protein-adducted OA-NO2, and then the data were calculated for the pools of free and adducted species. Lipid extracts were treated with 500 mM BME for 30 min at 37°C and analyzed by HPLC-ESI-MS/MS. HPLC separations were performed on a C18 reverse-phase column (150×2 mm, 3-μm particle size) with a mobile phase consisting of A (0.1% acetic acid in H2O) and B (0.1% acetic acid in acetonitrile). Products were resolved from the column and compared with standards using the following gradient program: 45% B for 1 min, 45–80% B over 44 min, 80–100% B over 1 min, 100% B for 7 min, 100–45% B over 6 s, and 45% B for 10 min to reequilibrate the column by monitoring for molecules that undergo an M−/[M–BME]− transition. The transition used for BME–OA-NO2 was m/z 404.4/326.3 and for BME–[13C18]OA-NO2, m/z 422.4/344.3. The declustering potentials were −50 V for BME adducts and collision energies were set at −17 for BME adducts. Zero-grade air was used as source gas, and nitrogen was used in the collision chamber. Data were acquired and analyzed using Analyst 1.4.2 software (Applied Biosystems). Quantification was achieved by comparing peak area ratios between analytes and their corresponding internal standards and then calculating analyte concentration using an internal standard curve. Concentrations were normalized to plasma volume or wet tissue weight.
Cell culture assays
Colonocyte cell lines (SW480) were kindly donated by V. Arulampalam. Cultures were maintained in DMEM supplemented with 1% penicillin/streptomycin and 10% FBS in a humidified incubator at 37°C and 0.5% CO2.
The regulation of PPARγ by OA and/or OA-NO2 was evaluated as follows: cells were treated with OA, OA-NO2 (1 μM, 24 h), or 5-ASA (3 μM–30 mM, 16 h), in the absence or presence of the PPARγ inhibitor GW9662 (1 μM, 1 h pretreatment). 5-ASA (common treatment for IBD) was used as a control. The mRNA levels of PPARγ were further analyzed by quantitative RT-PCR. In continuation, the cells were transiently transfected with a plasmid containing the luciferase gene under the control of two tandem PPARγ response elements (2× Cyp-luc, kindly donated by V. Arulampalam), using the Fugene transfection reagent. The cells were further treated with OA, OA-NO2 (1 μM), or 5-ASA (300 μM) in the presence or absence of GW9662 (1 μM). Transcription levels of PPARγ were measured by luciferase assay using the Dual-Luciferase Reporter Assay System. The transfection efficiency was normalized to the Renilla luciferase activity expressed by the pRL-TK plasmid (Promega).
RNA isolation and quantitative RT-PCR
Total RNA was extracted from cells and frozen tissues using a power homogenizer (KEBO-Lab, Stockholm, Sweden) and Trizol reagent. One microgram of total RNA was reverse transcribed with M-MLV reverse transcriptase. The relative expression levels of PPARγ, STAT-1, and fatty acid binding protein 2 (FABP2) were determined by real-time PCR in a 7900 sequence detection system (Applied Biosystems). Primers were designed with Primer Express software (Applied Biosystems) and amplification was carried out with SYBR green master mix. Sequences of the primers used were hPPARγ forward, 5′-CCTGATAGGCCCCACTGTGT-3′, and reverse, 5′-CAGGTGGGAGTGGAACAAT-3′; mPPARγ forward, 5′-TCACAAGAGCTGACCCAATGG-3′, and reverse, 5′-GATCGCACTTTGGTATTCTTGGA-3′; mSTAT-1 forward, 5′-CTTATTCCATGGACAAGGTTTTG-3′, and reverse, 5′-GGTGCTTCTTAATGAGCTCTAGG-3′; mFABP2 forward, 5′-AAATGGGTGTTAATATAGTGAAAA-3′, and reverse, 5′-CCTTCTTGTGTAATTGTCAGCTTC-3′; and β-actin forward, 5′-GCTCCTCCTGAGCGCAAGT-3′, and reverse, 5′-GTGGACAGTGAGGCCAGGAT-3′. Thermal conditions were 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C, 30 s at 55°C, and 1 min at 72°C.
Statistics
Data are presented as means±SEM. Statistical analysis was performed using Student’s t test, the Mann–Whitney test, or one-way ANOVA. P < 0.05 was considered significant (GraphPad Software, Inc).
Results
Detection of OA-NO2 in blood and colon tissue
OA-NO2 measurements revealed that after 7 days of treatment, OA-NO2 is present in both plasma and colon samples (Table 1) from the OA-NO2 groups, while remaining undetectable in the untreated groups (control, DSS, and DSS + OA). The concentration of total OA-NO2 in plasma was 14 nM after administration of 0.5 mg/kg/day and 270 nM with the higher dose of OA-NO2 (5 mg/kg/day). Free OA-NO2 in plasma was 8 nM after administration of 5 mg/kg/day OA-NO2 and again undetectable in the other groups. Colon samples also revealed an increase in the concentration of total OA-NO2 in the treated group compared with the untreated groups.
Table 1.
Nitrated oleic acid concentration in plasma and colon samples
| OA-NO2 | Free OA-NO2 in plasma (nM) | Total OA-NO2 in plasma (nM) | Total OA-NO2 in colon (ng/g tissue) |
|---|---|---|---|
| Untreated | 0 | 0 | 0 |
| 0.5 mg/kg/day | 0.64±0.10 | 13.85±4.49 | 0.59**±0.13 |
| 5 mg/kg/day | 8.06*±1.31 | 269.8±29.35 | nm |
Nitrated oleic acid concentration in plasma and colon samples from the OA-NO2-treated groups (0.5 and 5 mg/kg/day) vs the untreated groups. nm, not measured.
P < 0.001, one-way ANOVA, post hoc Tukey.
P < 0.01, Student t test.
Attenuation of DSS-induced colitis by administration of nitrated oleic acid
Shortening of the large intestine is a typical feature of experimental colitis, and colon length is considered a robust marker of the severity of inflammation [23]. As shown in Fig. 1, DSS administration caused a 30% reduction in colon length compared with the control group. Coadministration of OA-NO2 dose-dependently prevented the DSS-induced colon shortening (Fig. 1C). With the higher dose of OA-NO2 (5 mg/kg/day) the colon length did not differ significantly from the controls not receiving DSS (Fig. 1B), demonstrating a protective effect of nitrated OA. The lower dose of OA (0.5 mg/kg/day, Fig. 1A) did not prevent colon shortening, whereas the higher dose had some protective effect.
Fig. 1.

Colon length measurements after administration of DSS, in the absence or presence of OA or OA-NO2. (A) 0.5 mg/kg/day and (B) 5 mg/kg/day. (C) DSS-induced reduction in colon length compared to the control group (0 mg/kg is the DSS group). Mann–Whitney test, ***P < 0.0001; **P < 0.004; *P < 0.02.
To assess the symptoms associated with DSS-induced colitis, we recorded the DAI, which is achieved by scoring body weight loss, stool consistency, and rectal bleeding. The DSS group showed an increase in the DAI compared to the control group (Fig. 2), and this parameter was clearly reduced after OA-NO2 coadministration. OA treatment did not cause any significant improvement in DAI.
Fig. 2.

General assessment of colitis based on the DAI. DAI was reduced after treatment with OA-NO2 (5 mg/kg/day), compared with the DSS group. Mann–Whitney test; *P < 0.02.
Finally, colonic inflammation was also evaluated by measuring p65 (an NF-κB subunit) levels in colon samples. As shown in Fig. 3, the expression of p65 was increased in the colonic mucosa of the DSS group and this increase was almost completely prevented in animals treated with 5 mg/kg/day OA-NO2. Native oleic acid did not significantly attenuate the increase in DSS-induced p65 levels.
Fig. 3.

The DSS-induced increase in the expression of p65 is attenuated by OA-NO2 (5 mg/kg/day). (A) Immunohistochemistry staining and (B) quantification of the staining (a.u., arbitrary units). Mann–Whitney test; **P < 0.004.
Activation of PPARγ by nitrated oleic acid
A role for PPARγ in the OA-NO2 protection against DSS-induced colitis was hypothesized; thus treatment with OA-NO2 was evaluated for an impact on this nuclear receptor.
OA-NO2 increased the transcriptional expression of PPARγ in colon homogenates from the DSS + OA-NO2 group, whereas native oleic acid (DSS + OA) had no effect (Fig. 4). Moreover, the mRNA levels of two target genes of PPARγ, STAT-1 and FABP2, were induced after OA-NO2 (but not OA) administration, in the colon samples (Fig. 5). The mRNA expression of PPARγ was also induced in SW480 cells after OA-NO2 administration (Fig. 6), whereas OA had no effect. The addition of the PPARγ inhibitor GW9662 completely reverted the increase in PPARγ expression.
Fig. 4.
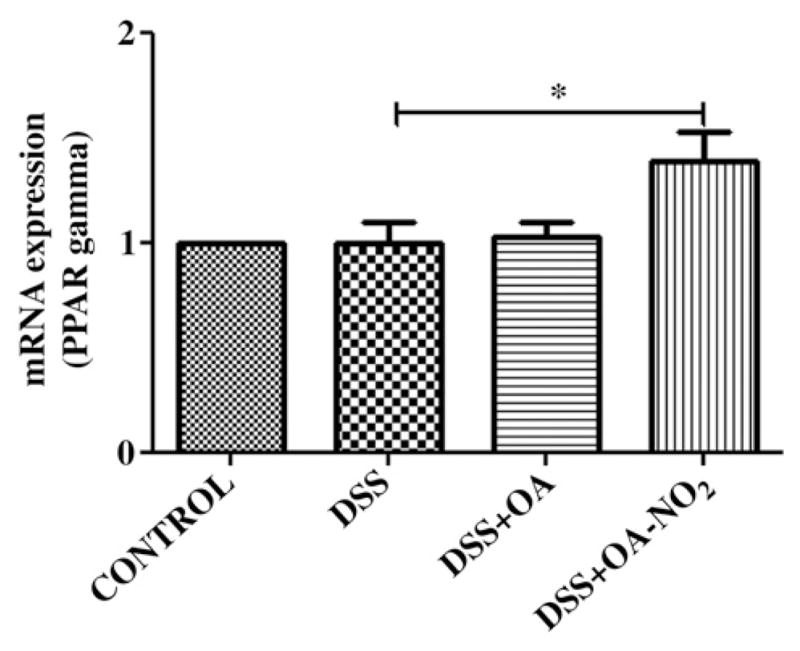
Nitrated oleic acid up-regulates PPAR expression in vivo. PPAR gene expression was measured by quantitative RT-PCR in the colon samples from the four experimental groups (OA-NO2: 5 mg/kg/day). Student t test; *P < 0.01.
Fig. 5.

Analysis of PPARγ target genes. Gene expression of STAT-1 and FABP2 was measured in the colon samples from the four experimental groups (OA-NO2: 5 mg/kg/day) by quantitative RT-PCR. Student t test; *P < 0.01.
Fig. 6.

Nitrated oleic acid up-regulates PPARγ expression in vitro. SW480 cells were treated with OA or OA-NO2 (1 μM), in the absence or presence of GW9662 (1 μM). PPAR gene expression was measured by quantitative RT-PCR. Student t test; ***P < 0.001.
5-ASA is commonly used in the treatment of IBD and has recently been suggested to act via activation of PPARγ [6]. Therefore we next analyzed the effect of 5-ASA on this nuclear receptor and compared it with the activation caused by OA-NO2. SW480 cells were treated with increasing doses (3 μM–30 mM) of 5-ASA, and only the dose of 300 μM significantly increased the expression of PPARγ (Table 2). This up-regulation of PPARγ was abolished by GW9662 pretreatment. Interestingly, PPARγ up-regulation was higher after the administration of 1 μM OA-NO2 than after 300 μM 5-ASA (Fig. 7), demonstrating that OA-NO2 is a considerably more potent activator of PPARγ than 5-ASA.
Table 2.
5-ASA up-regulates PPARγ expression
| Experiment | PPARγ expression (fold) |
|---|---|
| Expt 1 | |
| 5-ASA (0 μM) | 1 |
| 5-ASA (3 μM) | 1.21 |
| 5-ASA (30 μM) | 0.93 |
| 5-ASA (300 μM) | 2.7* |
| 5-ASA (3 mM) | 0.91 |
| 5-ASA (30 mM) | 1.44 |
| Expt 2 | |
| 5-ASA (300 μM) | 2.7 |
| GW (1 μM) + 5-ASA (300 μM) | 0.4* |
Expt 1: SW480 cells were treated with increasing doses of 5-ASA. PPARγ expression was significantly higher with the 300 μM dose. Expt 2: SW480 cells were treated with 5-ASA in the absence or presence of GW9662. PPARγ gene expression was measured by quantitative RT-PCR.
P < 0.01, Student t test.
Fig. 7.

OA-NO2 vs 5-ASA in the regulation of PPARγ expression. SW480 cells were treated with OA (1 μM), OA-NO2 (1 μM), or 5-ASA (300 μM). PPAR gene expression was measured by quantitative RT-PCR. Student t test; ***P < 0.001.
To confirm the involvement of OA-NO2 in the regulation of PPARγ, SW480 cells were transiently transfected with the PPARγ response element and treated with OA-NO2 and OA in the presence or absence of GW9662 (Fig. 8). OA-NO2 increased the transcriptional activity of PPARγ 2.5-fold, whereas OA or vehicle had no effect. Pretreatment with GW9662 prevented the up-regulation of PPARγ in the presence of OA-NO2. Taken together, these results show that OA-NO2 is a potent activator of PPARγ in the colon. Such activation may explain the potent anti-inflammatory effects of OA-NO2 observed in this study.
Fig. 8.

PPARγ is transcriptionally activated by OA-NO2 but not by OA in vitro. SW480 cells were transiently transfected with the PPARγ response element and treated with OA, OA-NO2 (1 μM), or 5-ASA (300 μM) in the presence or absence of GW9662 (1 μM). Expression levels of PPARγ were measured by luciferase assay. Student t test; *P < 0.01.
Discussion
Diverse nitration products of unsaturated fatty acids (nitroalkenes) are formed endogenously via NO-dependent oxidative inflammatory reactions [22,24]. Studies in cell cultures [15] have demonstrated pluripotent anti-inflammatory signaling actions of nitrated fatty acids (NO2-FAs), prompting the evaluation of the capability of these compounds to suppress inflammation in vivo.
Here we show that systemic administration of OA-NO2 attenuates DSS-induced colitis in mice. This protective effect is associated with in vivo activation of colonic PPARγ and concomitant down-regulation of NF-κB, a key mediator of the proinflammatory response. Although activation of PPARγ by OA-NO2 may represent a central mechanism for the observed protection, interactions with other anti-inflammatory pathways might also contribute [15]. These findings are concordant with recent studies suggesting that activation of PPARγ is associated with anti-inflammatory effects [2,5,25]. Indeed, activation of PPARγ, e.g., by thiazolidinedione derivatives, inhibits the inflammatory response in experimental colitis [5,6,8,26]. Moreover, recent data have suggested that 5-ASA also acts through the activation of PPARγ. 5-ASA is also able to serve as a PPARγ ligand and promote its translocation from the cytoplasm to the nucleus, perhaps in a positive feedback loop [6]. Our data confirm that 5-ASA activates PPARγ expression, and this effect is almost abolished in the presence of the PPARγ antagonist GW9662. However, our results clearly demonstrate that nitrated oleic acid is a considerably more potent activator of PPARγ than 5-ASA.
Although NO2-FAs may release some NO under certain circumstances, current data indicate that NO and cGMP do not significantly transduce NO2-FA signaling actions. Rather, the strongly electron-withdrawing nitro (NO2) group renders nitroalkene fatty acid derivatives electrophilic. This supports a kinetically rapid and reversible covalent adduction of nucleophilic amino acids, primarily cysteine [27]. As a consequence, NO2-FAs can posttranslationally modify critical redox-reactive thiols of various signaling mediators and transcription factors to alter their function [15]. The activation of PPARγ is a consequence of such electrophilic protein adduction by NO2-FAs [17,28].
Herein, native oleic acid administration as a control for OA-NO2 failed to significantly affect DAI or colonic expression of NF-κB and PPARγ. However, at a higher dose (5 mg/kg/day), OA had some protective effect, as revealed by the prevention of colon shortening. At present, the mechanism behind this effect is unclear. Recently, Chen and colleagues [29] demonstrated protective effects of dietary OA in the same DSS-induced model of colitis. They found that DSS caused an inhibition of hepatic stearoyl-CoA desaturase 1, thereby decreasing oleic acid biogenesis. Apparently, this inhibition exacerbated proinflammatory responses to DSS challenge, an effect limited by dietary oleic acid supplementation. It is possible that a greater protective effect of OA would have been revealed if we had increased the dose. Indeed, the total dose of OA in the study by Chen and colleagues was close to 100 mg per day, i.e., ~1000-fold higher than the dose used in the present study. It is also plausible that the effects of OA and the previously observed protective effects of monounsaturated lipids [30] at some stage involve endogenous nitration of the fatty acids. Conditions for such reactions seem particularly favorable when using the oral administration route. Thus, in the acidic stomach salivary-derived nitrite ( ) continuously meets gastric acid, thereby forming potent nitrating species including the nitrogen dioxide radical ( ) [31–33]. Thus, the weak protective effect of OA could possibly be explained by some degree of endogenous nitration.
Consumption of monounsaturated fat, such as in the olive-oil-rich Mediterranean diet, is related to benefits for diseases associated with chronic inflammation, including cardiovascular disease, rheumatoid arthritis, and gastrointestinal inflammation [34]. Oleic acid is also the major constituent of olive oil. Future studies will reveal whether dietary interventions can be used to increase the circulating levels of NO2-FAs and if this would offer any long-term protection against chronic inflammatory disorders, including IBD.
We conclude that administration of nitrated oleic acid attenuates inflammation in experimental inflammatory bowel disease. This protection involves activation of colonic PPARγ.
Acknowledgments
We thank M. Stensdotter for expert technical assistance. This project was supported by The Eli and Edythe Broad Foundation (J.O. L.); Vinnova (CIDaT, J.O.L.); the EU (Eicosanox LSMHCT-2004-005033 and Flaviola, J.O.L.); National Institutes of Health Grants R01 HL58115 (B.A.F.), R01 HL64937 (B.A.F.), and T32DK007052-34 (M.P.C.); and the Hartwell Foundation (M.P.C.). B.A.F. acknowledges a financial interest in Complexa, Inc.
Abbreviations
- DSS
dextran sulfate sodium
- IBD
inflammatory bowel disease
- NO2-FA
nitrated fatty acid
- OA-NO2
nitrated oleic acid
- OA
native oleic acid
- DAI
disease activity index
- PPARγ
peroxisome proliferator-activated receptor γ
- 5-ASA
5-aminosalicylic acid
References
- 1.Lichtenstein GR, Abreu MT, Cohen R, Tremaine W. American Gastroenter-ological Association Institute technical review on corticosteroids, immunomodulators, and infliximab in inflammatory bowel disease. Gastroenterology. 2006;130:940–987. doi: 10.1053/j.gastro.2006.01.048. [DOI] [PubMed] [Google Scholar]
- 2.Dubuquoy L, Rousseaux C, Thuru X, Peyrin-Biroulet L, Romano O, Chavatte P, Chamaillard M, Desreumaux P. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut. 2006;55:1341–1349. doi: 10.1136/gut.2006.093484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogler G. Significance of anti-inflammatory effects of PPARgamma agonists? Gut. 2006;55:1067–1069. doi: 10.1136/gut.2005.089946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saubermann LJ, Nakajima A, Wada K, Zhao S, Terauchi Y, Kadowaki T, Aburatani H, Matsuhashi N, Nagai R, Blumberg RS. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine response and prevent acute colitis. Inflamm Bowel Dis. 2002;8:330–339. doi: 10.1097/00054725-200209000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Katayama K, Wada K, Nakajima A, Mizuguchi H, Hayakawa T, Nakagawa S, Kadowaki T, Nagai R, Kamisaki Y, Blumberg RS, Mayumi T. A novel PPAR gamma gene therapy to control inflammation associated with inflammatory bowel disease in a murine model. Gastroenterology. 2003;124:1315–1324. doi: 10.1016/s0016-5085(03)00262-2. [DOI] [PubMed] [Google Scholar]
- 6.Rousseaux C, Lefebvre B, Dubuquoy L, Lefebvre P, Romano O, Auwerx J, Metzger D, Wahli W, Desvergne B, Naccari GC, Chavatte P, Farce A, Bulois P, Cortot A, Colombel JF, Desreumaux P. Intestinal antiinflammatory effect of 5-aminosalicylic acid is dependent on peroxisome proliferator-activated receptor-gamma. J Exp Med. 2005;201:1205–1215. doi: 10.1084/jem.20041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramakers JD, Verstege MI, Thuijls G, Te Velde AA, Mensink RP, Plat J. The PPARgamma agonist rosiglitazone impairs colonic inflammation in mice with experimental colitis. J Clin Immunol. 2007;27:275–283. doi: 10.1007/s10875-007-9074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J Clin Invest. 1999;104:383–389. doi: 10.1172/JCI7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lewis JD, Lichtenstein GR, Stein RB, Deren JJ, Judge TA, Fogt F, Furth EE, Demissie EJ, Hurd LB, Su CG, Keilbaugh SA, Lazar MA, Wu GD. An open-label trial of the PPAR-gamma ligand rosiglitazone for active ulcerative colitis. Am J Gastroenterol. 2001;96:3323–3328. doi: 10.1111/j.1572-0241.2001.05333.x. [DOI] [PubMed] [Google Scholar]
- 10.Lewis JD, Lichtenstein GR, Deren JJ, Sands BE, Hanauer SB, Katz JA, Lashner B, Present DH, Chuai S, Ellenberg JH, Nessel L, Wu GD. Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology. 2008;134:688–695. doi: 10.1053/j.gastro.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lundberg JO, Lundberg JM, Alving K, Weitzberg E. Nitric oxide and inflammation: the answer is blowing in the wind. Nat Med. 1997;3:30–31. doi: 10.1038/nm0197-30. [DOI] [PubMed] [Google Scholar]
- 12.Lundberg JO, Hellstrom PM, Lundberg JM, Alving K. Greatly increased luminal nitric oxide in ulcerative colitis. Lancet. 1994;344:1673–1674. doi: 10.1016/s0140-6736(94)90460-x. [DOI] [PubMed] [Google Scholar]
- 13.Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation: formation of novel nitrogen-containing oxidized lipid derivatives. J Biol Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- 14.Lima ES, Di Mascio P, Rubbo H, Abdalla DS. Characterization of linoleic acid nitration in human blood plasma by mass spectrometry. Biochemistry. 2002;41:10717–10722. doi: 10.1021/bi025504j. [DOI] [PubMed] [Google Scholar]
- 15.Cui T, Schopfer FJ, Zhang J, Chen K, Ichikawa T, Baker PR, Batthyany C, Chacko BK, Feng X, Patel RP, Agarwal A, Freeman BA, Chen YE. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J Biol Chem. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker PR, Lin Y, Schopfer FJ, Woodcock SR, Groeger AL, Batthyany C, Sweeney S, Long MH, Iles KE, Baker LM, Branchaud BP, Chen YE, Freeman BA. Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J Biol Chem. 2005;280:42464–42475. doi: 10.1074/jbc.M504212200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schopfer FJ, Lin Y, Baker PR, Cui T, Garcia-Barrio M, Zhang J, Chen K, Chen YE, Freeman BA. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor gamma ligand. Proc Natl Acad Sci U S A. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nettles KW. Insights into PPARgamma from structures with endogenous and covalently bound ligands. Nat Struct Mol Biol. 2008;15:893–895. doi: 10.1038/nsmb0908-893. [DOI] [PubMed] [Google Scholar]
- 19.Ito R, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, Imanishi J, Kita M, Ueda Y, Iwakura Y, Kataoka K, Okanoue T, Mazda O. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol. 2006;146:330–338. doi: 10.1111/j.1365-2249.2006.03214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jansson EA, Petersson J, Reinders C, Sobko T, Bjorne H, Phillipson M, Weitzberg E, Holm L, Lundberg JO. Protection from nonsteroidal anti-inflammatory drug (NSAID)-induced gastric ulcers by dietary nitrate. Free Radic Biol Med. 2007;42:510–518. doi: 10.1016/j.freeradbiomed.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 21.Schopfer FJ, Batthyany C, Baker PR, Bonacci G, Cole MP, Rudolph V, Groeger AL, Rudolph TK, Nadtochiy S, Brookes PS, Freeman BA. Detection and quantification of protein adduction by electrophilic fatty acids: mitochondrial generation of fatty acid nitroalkene derivatives. Free Radic Biol Med. 2009;46:1250–1259. doi: 10.1016/j.freeradbiomed.2008.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolph V, Schopfer FJ, Khoo NK, Rudolph TK, Cole MP, Woodcock SR, Bonacci G, Groeger AL, Golin-Bisello F, Chen CS, Baker PR, Freeman BA. Nitro-fatty acid metabolome: saturation, desaturation, beta-oxidation, and protein adduction. J Biol Chem. 2009;284:1461–1473. doi: 10.1074/jbc.M802298200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diaz-Granados N, Howe K, Lu J, McKay DM. Dextran sulfate sodium-induced colonic histopathology, but not altered epithelial ion transport, is reduced by inhibition of phosphodiesterase activity. Am J Pathol. 2000;156:2169–2177. doi: 10.1016/S0002-9440(10)65087-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker PR, Schopfer FJ, Sweeney S, Freeman BA. Red cell membrane and plasma linoleic acid nitration products: synthesis, clinical identification, and quantitation. Proc Natl Acad Sci U S A. 2004;101:11577–11582. doi: 10.1073/pnas.0402587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, Gumucio DL, Marin HE, Peters JM, Young HA, Gonzalez FJ. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut. 2006;55:1104–1113. doi: 10.1136/gut.2005.081745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Desreumaux P, Dubuquoy L, Nutten S, Peuchmaur M, Englaro W, Schoonjans K, Derijard B, Desvergne B, Wahli W, Chambon P, Leibowitz MD, Colombel JF, Auwerx J. Attenuation of colon inflammation through activators of the retinoid X receptor (RXR)/peroxisome proliferator-activated receptor gamma (PPARgamma) heterodimer: a basis for new therapeutic strategies. J Exp Med. 2001;193:827–838. doi: 10.1084/jem.193.7.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker LM, Baker PR, Golin-Bisello F, Schopfer FJ, Fink M, Woodcock SR, Branchaud BP, Radi R, Freeman BA. Nitro-fatty acid reaction with glutathione and cysteine: kinetic analysis of thiol alkylation by a Michael addition reaction. J Biol Chem. 2007;282:31085–31093. doi: 10.1074/jbc.M704085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yong L, Zhang J, Schopfer FJ, Martynowski D, Garcia-Barrio MT, Kovach A, Suino-Powell K, Baker PRS, Freeman BA, Chen YE, Xu HE. Molecular recognition of nitrated fatty acids by PPARγ. Nat Struct Mol Biol. 2008;15:865–867. doi: 10.1038/nsmb.1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen C, Shah YM, Morimura K, Krausz KW, Miyazaki M, Richardson TA, Morgan ET, Ntambi JM, Idle JR, Gonzalez FJ. Metabolomics reveals that hepatic stearoyl-CoA desaturase 1 downregulation exacerbates inflammation and acute colitis. Cell Metab. 2008;7:135–147. doi: 10.1016/j.cmet.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bassaganya-Riera J, Reynolds K, Martino-Catt S, Cui Y, Hennighausen L, Gonzalez F, Rohrer J, Benninghoff AU, Hontecillas R. Activation of PPAR gamma and delta by conjugated linoleic acid mediates protection from experimental inflammatory bowel disease. Gastroenterology. 2004;127:777–791. doi: 10.1053/j.gastro.2004.06.049. [DOI] [PubMed] [Google Scholar]
- 31.Lundberg JO, Weitzberg E, Lundberg JM, Alving K. Intragastric nitric oxide production in humans: measurements in expelled air. Gut. 1994;35:1543–1546. doi: 10.1136/gut.35.11.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lundberg JO, Weitzberg E, Gladwin MT. The nitrate–nitrite–nitric oxide pathway in physiology and therapeutics. Nat Rev Drug Discov. 2008;7:156–167. doi: 10.1038/nrd2466. [DOI] [PubMed] [Google Scholar]
- 33.Lundberg JO, Weitzberg E, Cole JA, Benjamin N. Nitrate, bacteria and human health. Nat Rev Microbiol. 2004;2:593–602. doi: 10.1038/nrmicro929. [DOI] [PubMed] [Google Scholar]
- 34.Alarcon de la Lastra C, Barranco MD, Motilva V, Herrerias JM. Mediterranean diet and health: biological importance of olive oil. Curr Pharm Des. 2001;7:933–950. doi: 10.2174/1381612013397654. [DOI] [PubMed] [Google Scholar]