

Abstract
Stimulation of the fimbria in rat hippocampal slices evoked an extracellular negativity in the granule cell layer and a small depolarization in granule cells at their resting potentials. The intracellular potentials appeared to be GABAA receptor-mediated IPSPs because they reversed at −69.1 ± 1.0 mV (mean ± S.E.M.. n = 14) and were blocked by the GABAA receptor antagonist bicuculline (10–50 μM, n = 14). However, during the first few minutes of perfusion with bicuculline. IPSPs transiently and paradoxically increased in amplitude. As IPSPs increased, the reversal potential and latency to onset remained the same. These effects were reversible, and during the wash period IPSPs first increased and then stabilized at a smaller amplitude, similar to IPSPs evoked in control conditions. As the GABAA receptor-mediated IPSP decreased, it was followed by a second hyperpolarization. This late hyperpolarization appeared to be a GABAB receptor-mediated IPSP, because it reversed near the equilibrium potential for potassium (mean −81.8 ± 2.3 mV, n = 12, [K+]o = 5 mM) and was blocked by the GABAB receptor antagonist 2-hydroxy saclofen (250–500 μM, n = 5). The results suggest that GABAA and GABAB receptor-mediated IPSPs evoked in granule cells by fimbria stimulation are normally inhibited by activation of GABAA receptors. The inhibition by GABAA receptors is strong enough that, in control conditions, the GABAA IPSPs are barely detectable and the GABAB IPSPs are undetectable. The relevant GABAA receptors could be located presynaptically, on the nerve terminals of inhibitory interneurons that innervate granule cells, or on the dendrites and somata of the interneurons, where they may be affected by GABAergic inputs activated by fimbria stimulation. These data demonstrate the strength and complexity of pathways utilizing GABAA receptors and GABAB receptors to inhibit dentate granule cells.
Keywords: IPSP, GABA, Presynaptic inhibition, GABAA receptor, GABAB receptor, Dentate gyrus, Septohippocampal projection, Interneuron
The control of hippocampal inhibition is of great importance, given the evidence that inhibition controls hippocampal excitability [8,17,29,32] and affects synaptic plasticity [7,21,34]. Inhibition in the dentate gyrus may be particularly important in controlling the spread of epileptic discharges through the limbic system [25]. Numerous studies have examined the nature and control of hippocampal inhibition, and a complex set of mechanisms has emerged (for review see ref. 1). One source of control is exerted by presynaptic GABAB receptors that suppress the release of GABA in hippocampus, [3,6,9,11,12,33] as well as the dentate gyrus [4,18,20,22]. In addition, some studies have indicated that presynaptic GABAA receptors, i.e., GABAA receptors on GABAergic interneurons, may modulate GABAA receptor-mediated inhibition. For example, it has been reported that the GABAA receptor antagonist picrotoxin blocks frequency-dependent depression of IPSPs in area CA1, [2] and that the GABAA agonist muscimol disinhibits immature CA1 pyramidal cells [5]. There is a precedent for presynaptic inhibition by GABAA receptors, since it has been shown in rat frontal cortex that GABAA receptors are responsible for presynaptic inhibition of GABA release [19]. The results presented below describe (1) a GABAA and GABAB receptor-mediated IPSP evoked in granule cells by fimbria stimulation, and (2) strong inhibition of that IPSP that is mediated by GABAA receptors.
Transverse rat hippocampal slices were prepared as previously described [26]. Animals were treated in accordance with guidelines set by the National Institutes of Health and the New York State Department of Health. Briefly, adult female Sprague–Dawley rats were anesthetized with ether and decapitated. The brain was immediately removed and the hippocampus was isolated within a block of tissue including adjacent structures. The hippocampus was cut in 400 μm thick slices with a vibratome while immersed in 4°C buffer (in mM: 126.0 NaCl, 5.0 KC1, 2.0 CaCl2, 2.0 MgCl2, 26.0 NaHCO3, 1.25 NaH2PO4, and 10.0 d-glucose; pH 7.4). Slices were immediately transferred to a recording chamber (Fine Science Tools), where they were perfused with warm (33–34°C), oxygenated (95% O2, 5% CO2) buffer.
Recordings were made with glass microelectrodes (borosilicate glass containing a capillary fiber, A&M Systems) that were filled with 1 M potassium acetate (80–150 MΩ), using an intracellular amplifier with a bridge circuit (Axoclamp 2A, Axon Instruments); bridge balance was monitored continuously. For stimulation of the fimbria, a twisted metal bipolar electrode was placed in the white matter of the fimbria (Fig. 1). Fimbria stimulation was set at the level required to evoke a submax-imal response of CA3b pyramidal cells in the same slice; any slice without at least a 5 mV antidromic population spike in CA3b was not used. Data were recorded on an oscilloscope (Nicolet) and taped (Neurodata Instruments) for analysis offline. Bicuculline methiodide (10 mM in 0.9% NaCl, Sigma), 2-hydroxy saclofen (10 mM in 10 mM NaOH, Tocris Neuramin), atropine meth-ylbromide (10 mM in 0.9% NaCl, Sigma), and mecam-ylamine (10 mM in 0.9% NaCl, Sigma) were stored in concentrated aliquots and dissolved in buffer to reach the desired final concentration immediately before use.
Fig. 1.
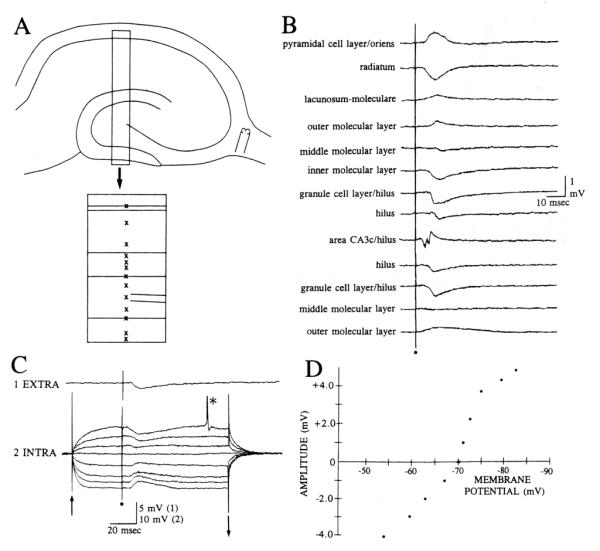
Fimbria stimulation evokes a negativity in the granule cell layer by extracellular recording that corresponds to a hyperpolarization of granule cells. A: a diagram of the slice shows where the stimulating electrode was placed in the fimbria and the area where recordings shown in B were made is boxed. The box is enlarged below, showing specific sites (marked by x’s) for extracellular recording. B: responses to stimulation of the fimbria are shown for several sites in the slice, as illustrated in A. The same fimbria stimulus was used to elicit all responses. Stimulation occurred at the dot. C: simultaneous intracellular and extracellular recording of granule cell responses to fimbria stimulation. 1. Extra: extracellular recording from the granule cell layer. The recording site was close to the cell shown in part C2. 2. Intra: intracellular recordings from a granule cell. Eight superimposed responses of a granule cell to the same fimbria stimulus are shown. Intracellular responses were recorded during depolarizing or hyperpolarizing current steps except for the central trace, which was evoked without a current step. The start and end of the steps are marked by arrows. The action potential at the asterisk is truncated. Stimulation occurred at the dot. Membrane potential, −72 mV; resting potential, −79 mV. D: amplitudes of granule cell responses to fimbria stimulation are plotted as a function of membrane potential for the cell shown in C.
This study was based on intracellular recordings from 38 granule cells and extracellular recordings from 45 slices. Fimbria stimulation was used to evoke an IPSP in dentate granule cells without concomitant orthodromic or antidromic excitation, as occurs when IPSPs are elicited by perforant path input or mossy fiber stimulation. Fimbria-evoked IPSPs were detected both extracellularly as well as intracellularly. The extracellular correlate to the IPSP was a small negativity recorded in the granule cell layer (mean amplitude 0.5 ± 0.1 mV, n = 45) with a latency to onset of 9.6 ± 0.4 ms (Fig. 1). This negativity became smaller as the recording site was moved either towards the hilus or the molecular layer, and correlated with a positivity in the outer molecular layer (Fig. 1). It is unlikely that the positivity was passively conducted from stratum lacunosum-moleculare of area CA1, where a positivity was evoked by the same fimbria stimulus, because similar positivities in the molecular layer were recorded in the upper and lower blades (Fig. 1 A,B). The negativity in the granule cell layer was unaffected by bath-application of the cholinergic muscarinic antagonist atropine co-applied with the nicotinic antagonist me-camylamine (10 μM each, n = 4; 25 μM, n = 3; 50 μM, n = 2; data not shown).
The intracellularly-recorded IPSP was a small depolarization of granule cells at their resting potential, and a hyperpolarization at more depolarized potentials (Fig. 1). The hyperpolarizations were small (mean maximal hyperpolarization 2.9 ± 0.3 mV, n = 14; Fig. 1) and began at a mean latency of 10.1 ± 0.6 ms after the fimbria stimulus. The mean latencies of the intracellular and extracellular potentials were not significantly different (t-test, P > 0.05), supporting the premise that they were due to similar mechanisms. The reversal potentials of hyperpolarizations were close to the equilibrium potential for chloride (mean reversal potential, −69.1 ±1.0 mV, n = 14), indicating that they were IPSPs mediated by GABA acting at GABAA receptors. Consistent with that possibility, bicuculline blocked the IPSP in every cell where it was tested (Fig. 2, 10 μM, n = 2; 25 μM, n = 7; 50 μM, n = 5). Atropine and mecamylamine did not affect the IPSP (n = 9; data not shown).
Fig. 2.
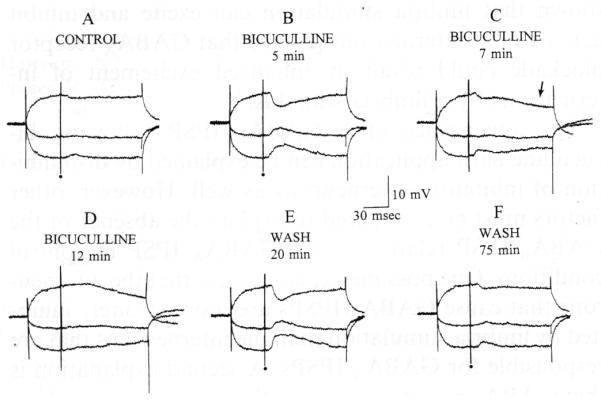
Bicuculline increased and then blocked the hyperpolarization evoked by fimbria stimulation. A: 3 responses to fimbria stimulation, triggered during hyperpolarizing or depolarizing current steps, are shown superimposed. Stimulation occurred at the dot. Membrane potential, −66 mV; resting potential, −82 mV. B: responses to the same fimbria stimulus are shown 5 min after 25 μM bicuculline was added to the buffer. Note the increase in IPSP amplitude. C: 7 min after bicuculline was added to the buffer, the IPSP was reduced and a second, later hyperpolarization was evident (arrow). D: at 12 min, the short latency IPSP was blocked, but the second hyperpolarization was not. E: 20 min after perfusion with drug-free buffer was resumed, stimulation produced a very large early IPSP but no late hyperpolarization. F: following prolonged perfusion with drug-free buffer, the response to fimbria stimulation was similar to control conditions.
During the first few minutes of bicuculline application, a transient increase in the amplitude of the IPSP occurred in all experiments (n= 14; Fig. 2). At the point during bicuculline application when IPSPs were largest, maximum IPSP amplitude was 7.0 ± 0.5 mV, 241% of the mean amplitude of control IPSPs. The latencies to onset of the IPSPs did not change during bicuculline bath-application (post bicuculline, 10.0 ±0.5 ms, n = 14, paired t-test, P > 0.05). The reversal potential of the increased IPSP was not different from the IPSP evoked in control conditions (mean Erev post bicuculline = −69.5 ± 0.8 mV, n = 14, paired t-test, P > 0.05). Input resistance did not change significantly (control, 65 ± 3.4 MΩ; post bicuculline, 72 ± 5.1 Ω, n = 14, paired t-test, P > 0.05). The effects of bicuculline were completely reversible. During reversal, the GABAA IPSP first became quite large and subsequently stabilized at the small amplitude of control IPSPs (Fig. 2).
During the blockade of the GABAA IPSP, a second distinct hyperpolarization emerged at a longer latency from the stimulus (Figs. 2 and 3). After the initial GABAA IPSP was blocked, only the second hyperpolarization was evident (Fig. 2). This hyperpolarization was large (mean maximum amplitude, 8.7 ± 1.0 mV, n = 12), long lasting (mean duration 986 ± 14 ms, peak at 150–280 ms after the stimulus) and reversed at −81.8 ± 2.3 mV (n = 12). Application of the GABAB receptor antagonist 2-hydroxy saclofen (250–500 μM, n = 5) blocked the late hyperpolarization reversibly (Fig. 3). A small depolarization remained after saclofen application in some cases (Fig. 3); investigation of this apparent EPSP is currently underway.
Fig. 3.

The fimbria-evoked late hyperpolarization, revealed after bicuculline application, was blocked by the GABAB receptor antagonist 2-hydroxy saclofen. A–D: 3 responses to an identical fimbria stimulus are shown superimposed, triggered during current steps (top and bottom traces) or without any current injection (central trace). In control (A), a small IPSP was recorded. Following reduction of the GABAA IPSP (B), a late hyperpolarization was evoked after the residual GABAA IPSP. The residual GABAA IPSP is marked by an arrow, the late hyperpolarization is marked by an asterisk. The late hyperpolarization was blocked by 500 μM 2-hydroxy saclofen (C) reversibly (D). Stimulation occurred at the dot. Action potentials are truncated. Membrane potential, −71 mV; resting potential, −85 mV.
The results demonstrate that in control conditions fimbria stimulation evokes very small IPSPs in granule cells that are mediated by GABAA receptors. The GABAergic septohippocampal pathway could mediate this IPSP. However, the long latency to onset of the IPSP, and the evidence that GABAergic septohippocampal neurons innervate interneurons preferentially [10,15], suggest that another circuit is also possible. One possibility is that fimbria stimulation excited pyramidal cells that subsequently excited dentate interneurons, and the interneurons were responsible for the granule cell IPSPs. This pathway is suggested by drawings of intracellularly-la-belled CA3 pyramidal cells, which have axon collaterals in the hilus; [13,16] these collaterals could innervate dentate interneurons. Either hilar interneurons or interneu-rons in the granule cell layer could be responsible for granule cell IPSPs, since both have dendrites in the hilus and both inhibit granule cells [28]. This pathway is supported by several lines of evidence. First, a large antidromic (and hence short latency) pyramidal cell population spike was evoked by the same stimulus that evoked IPSPs in granule cells. Second, pyramidal cells are thought to use an excitatory amino acid as a neurotransmitter [23], and fimbria-evoked IPSPs were insensitive to cholinergic antagonists yet sensitive to the glutamatergic antagonist CNQX. In five experiments bath-application of 5 μM CNQX blocked fimbria-evoked IPSPs completely (data not shown). Blockade of fimbria-evoked IPSPs by CNQX was completely reversible in three of five granule cells where impalements were maintained for over 30 minutes after returning to CNQX-free buffer. In the other two cells blockade of the IPSP was only partly reversed. One variant of this pathway involves the ability of hilar ‘mossy’ cells to substitute for the pyramidal cells. Thus, hilar mossy cells could be excited by fimbria stimulation and in turn excite inhibitory interneurons of the dentate gyrus. In support of the latter possibility, fimbria stimulation in slices can excite hilar mossy cells by a CNQX-sensitive mechanism [27], mossy cells are thought to use glutamate as a neurotransmitter [31], and it has been argued that mossy cells innervate inhibitory interneurons [30]. Thus either pyramidal cells or mossy cells, or both, could be responsible for the excitation of GABAergic neurons that caused fimbria-evoked IPSPs in granule cells.
That an IPSP evoked in a hippocampal principal cell is mediated by GABAA receptors is not surprising, given that IPSPs in the hippocampus and dentate gyrus involve, either partially or exclusively, GABAA receptors [1,9,14]. It also is not surprising that the extracellularly-recorded IPSP was maximal in the granule cell layer and reversed in the molecular layer, since the site of the inhibitory ‘basket’ cell plexus is in the granule cell layer/inner molecular layer [24]. However, it is notable that blockade of GABAA receptors can increase a GABAA receptor-mediated IPSP and uncover large GABAB receptor-mediated IPSPs.
There are several possible mechanisms that could explain how GABAA receptors inhibit GABAA receptor-mediated granule cell IPSPs. First, GABA may normally act to inhibit its own release by GABAA autoreceptors. If this were the case, bicuculline might have impaired presynaptic inhibition at a time when postsynaptic GABAA receptors were incompletely blocked, resulting in a transient increase in the GABAA IPSP of granule cells. Another possibility is that bicuculline disinhibited interneurons at a time when GABAA receptors on granule cells were only partially blocked. Indeed, it has been shown that fimbria stimulation can excite and inhibit dentate hilar interneurons [27], so that GABAA receptor blockade could result in enhanced excitement of interneurons by a fimbria stimulus.
The appearance of a GABAB IPSP following bicuculline bath-application can be explained by disinhibi-tion of inhibitory interneurons as well. However, other factors must be considered to explain the absence of the GABAB IPSP relative to the GABAA IPSP in control conditions. One possible explanation is that the interneurons that cause GABAB IPSPs are more strongly inhibited by fimbria stimulation than the interneurons that are responsible for GABAA IPSPs. A second explanation is that GABAB receptors occur at the same synapses where GABAA receptors exist, but because GABAB receptors are further from the active zone they require greater release of GABA for their activation. Finally, GABAB synapses may be located electrically distal to the intraso-matic electrode, a location that could make GABAB IPSPs undetected until large amounts of GABA were released. Further experiments will be necessary to differentiate among these possibilities, but regardless of the underlying mechanisms (s), the results underscore the strength and complexity of the inhibitory network in the hippocampus and dentate gyrus.
I thank Dr. Robert S. Sloviter and Dr. Daniel H. Lowenstein for their comments and discussion, Dr. Simon Neubort for statistical calculations, and Mrs. Annmarie Curcio for secretarial assistance. This study was supported by NS 30831.
References
- [1].Alger BE. Gating of GABAergic inhibition in hippocampal pyramidal cells. Ann. N.Y. Acad. Sci. 1991;627:249–263. doi: 10.1111/j.1749-6632.1991.tb25929.x. [DOI] [PubMed] [Google Scholar]
- [2].Benardo LS. GABAA receptor-mediated mechanisms contribute to frequency-dependent depression of IPSPs in the hippocampus. Brain Res. 1993;607:81–88. doi: 10.1016/0006-8993(93)91491-a. [DOI] [PubMed] [Google Scholar]
- [3].Blaxter TJ, Carlen PL. Pre- and postsynaptic effects of baclofen in the rat hippocampal slice. Brain Res. 1985;341:195–199. doi: 10.1016/0006-8993(85)91489-1. [DOI] [PubMed] [Google Scholar]
- [4].Burgard EC, Sarvey JS. Long-lasting potentiation and epileptiform activity produced by GABAB receptor activation in the dentate gyrus of rat hippocampal slice. J. Neurosci. 1991;11:1198–1209. doi: 10.1523/JNEUROSCI.11-05-01198.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chesnut TJ, Swann JW. Disinhibitory actions of the GABAA agonist muscimol in immature hippocampus. Brain Res. 1989;502:365–374. doi: 10.1016/0006-8993(89)90633-1. [DOI] [PubMed] [Google Scholar]
- [6].Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J. Physiol. 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Davies CH, Starkey SJ, Pozza MF, Collingridge GL. GABAB autoreceptors regulate the induction of LTP. Nature. 1991;349:609–611. doi: 10.1038/349609a0. [DOI] [PubMed] [Google Scholar]
- [8].Dingledine R, Gjerstad L. Reduced inhibition during epileptiform activity in the in vitro hippocampal slice. J. Physiol. 1980;305:297–313. doi: 10.1113/jphysiol.1980.sp013364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dutar P, Nicoll RA. Pre- and postsynaptic GABAB receptors in the hippocampus have different pharmacological properties. Neuron. 1988;1:585–591. doi: 10.1016/0896-6273(88)90108-0. [DOI] [PubMed] [Google Scholar]
- [10].Freund TF, Antal M. GABA-containing neurons in the septum control inhibitory interneurons in the hippocampus. Nature. 1988;336:170–174. doi: 10.1038/336170a0. [DOI] [PubMed] [Google Scholar]
- [11].Harrison NL, Lovinger DM, Lambert NA, Teyler TJ, Prager R, Ong J, Kerr DIB. The actions of 2-hydroxy-saclofen at presynaptic GABAB receptors in the rat hippocampus. Neurosci. Lett. 1990;119:272–276. doi: 10.1016/0304-3940(90)90851-y. [DOI] [PubMed] [Google Scholar]
- [12].Inoue M, Matsuo T, Ogata N. Characterization of pre- and postsynaptic actions of (-)-baclofen in the guinea-pig hippocampus in vitro. Br. J. Pharmacol. 1985;84:843–851. doi: 10.1111/j.1476-5381.1985.tb17378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ishizuka N, Weber J, Amar^l DG. Organization of in-trahippocampal projections originating from CA3 pyramidal cells in the rat. J. Comp. Neurol. 1990;295:580–623. doi: 10.1002/cne.902950407. [DOI] [PubMed] [Google Scholar]
- [14].Knowles WD, Schneiderman JH, Wheal HV, Stafstrom CE, Schwartzkroin PA. Hyperpolarizing potentials in guinea pig hippocampal CA3 neurons. Cell. Mol. Neurobiol. 1984;4:207–230. doi: 10.1007/BF00733586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kohler C, Chan-Palay V, Wu J-Y. Septal neurons containing glutamic acid decarboxylase immunoreactivity project to the hippocampal region in the rat brain. Anat. EmbryoL. 1984;169:41–44. doi: 10.1007/BF00300585. [DOI] [PubMed] [Google Scholar]
- [16].Li X-G, Ylinen A, Tepper JM, Jando G, Buzsaki G. Axon arborization of CA3 pyramidal cells in vivo: an intracellular labeling study. Soc. Neurosci. Abstr. 1992;18:1418. [Google Scholar]
- [17].Miles R, Wong RKS. Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J. Physiol. 1987;388:611–629. doi: 10.1113/jphysiol.1987.sp016634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Misgeld U, Klee MR, Zeise ML. Differences in baclofen-sensitivity between CA3 neurons and granule cells of the guinea pig hippocampus in vitro. Neurosci. Lett. 1984;47:307–311. doi: 10.1016/0304-3940(84)90531-7. [DOI] [PubMed] [Google Scholar]
- [19].Mitchell PR, Martin IL. Is GABA release modulated by presynaptic receptors? Nature. 1978;274:904–905. doi: 10.1038/274904a0. [DOI] [PubMed] [Google Scholar]
- [20].Mott DD, Bragdon AC, Lewis DV, Wilson WA. Baclofen has a proepileptic effect in the rat dentate gyrus. J. Pharmacol. Exp. Ther. 1989;9:721–725. [PubMed] [Google Scholar]
- [21].Mott DD, Lewis DV. Facilitation of the induction of long-term potentiation by GABAB receptors. Science. 1991;252:1718–1720. doi: 10.1126/science.1675489. [DOI] [PubMed] [Google Scholar]
- [22].Otis TS, Mody I. Differential activation of GABAA and GABAB receptors by spontaneously released transmitter. J. Neu-rophysiol. 1992;67:227–235. doi: 10.1152/jn.1992.67.1.227. [DOI] [PubMed] [Google Scholar]
- [23].Ottersen O, Storm-Mathisen J. Excitatory and inhibitory amino acids in the hippocampus. In: Chan-Palay V, Kohler C, editors. The Hippocampus. New Vistas, Liss; New York: 1989. pp. 97–117. [Google Scholar]
- [24].Ramon y, Cajal S. Histologie du Systeme Nerveux de 1’Homme et des Vertebres. A. Maloine; Paris: 1911. [Google Scholar]
- [25].Ribak CE, Gall CM, Mody I. The Dentate Gyrus and Its Role in Seizures. Elsevier; Amsterdam: 1992. p. 317. [Google Scholar]
- [26].Scharfman HE. Dentate hilar cells with dendrites in the molecular layer have lower thresholds for synaptic activation by perforant path than granule cells. J. Neurosci. 1991;11:1660–1673. doi: 10.1523/JNEUROSCI.11-06-01660.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Scharfman HE. Activation of dentate hilar neurons by stimulation of the fimbria in rat hippocampal slices. Neurosci. Lett. 1993;156:61–66. doi: 10.1016/0304-3940(93)90440-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Scharfman HE, Kunkel DD, Schwartzkroin PA. Synaptic connections of dentate granule cells and hilar neurons: results of paired intracellular recordings and intracellular horseradish peroxidase injections. Neuroscience. 1990;37:693–707. doi: 10.1016/0306-4522(90)90100-i. [DOI] [PubMed] [Google Scholar]
- [29].Schwartzkroin PA. Hippocampal slices in experimental human epilepsy. In: Delgado-Escueta AV, Ward AA, Woodbury DM, Porter RJ, editors. Basic Mechanisms of the Epilepsies, Advances in Neurology. Vol. 44. Raven; New York: 1986. pp. 991–1010. [PubMed] [Google Scholar]
- [30].Sloviter RS. Permanently altered hippocampal structure, excitability and inhibition after experimental status epilepticus in the rat: the ‘dormant basket cell’ hypothesis and its possible relevance to temporal lobe epilepsy. Hippocampus. 1991;1:41–66. doi: 10.1002/hipo.450010106. [DOI] [PubMed] [Google Scholar]
- [31].Soriano E, Frotscher M. Hippocampus. Mossy cells of the rat fascia dentata are glutamate-immunoreactive. in press. [DOI] [PubMed] [Google Scholar]
- [32].Stelzer A. GABAA receptor populations control the excitability of neuronal populations. Int. Rev. Neurobiol. 1992;33:195–287. doi: 10.1016/s0074-7742(08)60693-5. [DOI] [PubMed] [Google Scholar]
- [33].Thompson SM, Gahwiler B. Comparison of the actions of baclofen at pre- and postsynaptic receptors in the rat hippocampus in vitro. J. Physiol. 1992;451:329–345. doi: 10.1113/jphysiol.1992.sp019167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wigstrom H, Gustafsson B. Facilitated induction of hippocampal long-lasting potentiation during blockade of inhibition. Nature. 1983;301:603–604. doi: 10.1038/301603a0. [DOI] [PubMed] [Google Scholar]