

Abstract
Aims
Circulating endogenous, dietary, and foreign chemicals can contribute to vascular dysfunction. The mechanism by which the vasculature protects itself from these chemicals is unknown. This study investigates whether the pregnane X receptor (PXR), the major transcriptional regulator of hepatic drug metabolism and transport that responds to such xenobiotics, mediates vascular protection by co-ordinating a defence gene programme in the vasculature.
Methods and results
PXR was detected in primary human and rat aortic endothelial and smooth muscle cells (SMC) and blood vessels including the human and rat aorta. Metabolic PXR target genes cytochrome P450 3A, 2B, 2C, and glutathione S-transferase mRNA and activity were induced by PXR ligands in rodent and human vascular cells and absent in the aortas from PXR-null mice stimulated in vivo or in rat aortic SMC expressing dominant-negative PXR. Activation of aortic PXR by classical agonists had several protective effects: increased xenobiotic metabolism demonstrated by bioactivation of the pro-drug clopidogrel, which reduced adenosine diphosphate-induced platelet aggregation; increased expression of multidrug resistance protein 1, mediating chemical efflux from the vasculature; and protection from reactive oxygen species-mediated cell death.
Conclusion
PXR co-ordinately up-regulates drug metabolism, transport, and antioxidant genes to protect the vasculature from endogenous and exogenous insults, thus representing a novel gatekeeper for vascular defence.
Keywords: Pregnane X receptor, Nuclear receptors, Vascular endothelium, Vascular smooth muscle cells, Cytochrome P450
1. Introduction
The vascular wall serves as a barrier controlling the movement of solutes, fluids, and cells from the vascular space to the tissues and as such is exposed to circulating chemicals from both endogenous and foreign sources, including drugs and dietary and environmental contaminants.1 These chemicals can contribute to vascular dysfunction and the development of cardiovascular disease.2 Yet, little consideration has been given to how the vasculature regulates and protects itself and therefore all organs from such insults.
The body defends itself from chemical insults by biotransforming chemicals and eliminating the metabolites using a system of enzymes and transport proteins in the liver and intestine. The xenosensing pregnane X receptor (PXR; NR1I2) is central to this process.3 PXR, a member of the nuclear receptor superfamily of ligand-dependent transcription factors, can be activated by a variety of structurally distinct ligands, including drugs (e.g. dihydropyridine calcium channel blockers),3 environmental contaminants (e.g. polychlorinated biphenyls),4 and endogenous compounds such as bile acids, oxysterols, and steroid hormones3,5 and in response regulates the enterohepatic defence system at a transcriptional level. Activated PXR binds to response elements in the promoters and up-regulates the transcription of Phase I and II drug-metabolizing enzymes, e.g. cytochrome P450 (CYP)s and glutathione S-transferases (GSTs), and transporters, e.g. multidrug resistance protein 1 (MDR1).3,6
Numerous CYPs are expressed in vascular cells, where they produce endogenous mediators such as epoxyeicosatrienoic acids (EETs).7 This includes CYPs potentially regulated by PXR; CYP3A, 2B, and 2C.8–10 However, neither the roles of any of these CYPs in drug metabolism within the vasculature nor the contribution of PXR-regulated xenosensing to the protective homeostatic barrier role of the vasculature has been investigated. Vascular expression of PXR mRNA has been shown previously in rat and Psammomys obesus gerbil thoracic aortic smooth muscle cells (SMC), rat, pig, and human brain capillary endothelial cells, and mouse mesenteric arteries.11–18 The latter was indicated in regulating vasodilation during pregnancy via up-regulation of CYP epoxygenases.18 Here, we show that PXR is expressed in the vasculature across species, where it co-ordinates a gene programme of Phase I and II drug-metabolizing enzymes and transporters, providing a mechanism by which the vasculature can protect itself and the underlying tissues from circulating xenobiotic and endobiotic insults.
2. Methods
For detailed methods and reagent sources, see Supplementary material online.
2.1. Immunohistochemistry
Immunohistochemistry was performed on Ambion Human LandMarkTM LD cardiovascular tissue microarrays as described previously.19 Dewaxed sections were blocked with 10% goat preimmune serum, incubated with 1:50 dilution of rabbit anti-PXR antibody (Santa Cruz Biotechnology, CA, USA) overnight and processed according to the avidin–biotin complex method (Vector Labs, Peterborough, UK) using a 1:100 dilution of Vector goat anti-rabbit IgG biotinylated secondary antibody. Control sections were treated as above but incubated overnight in the absence of primary antibody. The slides were counterstained with haematoxylin.
2.2. Cell culture
Rat aortic SMC (RASMC; WKY3m-22), Huh-7, and HepG2 cells were grown and maintained in Dulbecco's modified Eagle's medium containing 10% foetal bovine serum (DMEM) as described previously.20 For dominant-negative experiments, RASMC were transfected with a 2:5 complex of dominant-negative PXR (PXR-DN)21 or empty vector pcDNA 3.1 V5 His plasmid DNA and LipofectamineTM 2000 (Invitrogen, Paisley, UK) as previously described22 for 24 h prior to treatment. Primary human aortic endothelial cells (HAEC) and human aortic SMC (HASMC) were obtained from Promocell (Heidelberg, Germany) and cultured according to Promocell recommendations. Due to species differences, pregnenolone 16α-carbonitrile (PCN; 10 µmol/L) and rifampicin (10 μmol/L) were used to activate rodent and human PXR, respectively.
2.3. Organ culture
Wistar rat aorta organ culture was performed essentially as described previously.23 Animals were cared for in accordance with the Home Office Guidance in the Operation of the Animals (Scientific Procedures) Act 1986. Animals were administered buprenorphine analgesic (0.03 mg/kg subcutaneously) prior to anaesthesia with sodium thiopentone (85 mg/kg ip), approved in Home office project number PPL 70/7055, protocol 4. Anaesthesia was monitored by reaction to foot pinching at 15 min intervals and respiration monitoring and maintained by supplementary infusions of sodium thiopentone. The animal was euthanized prior to aorta removal by removal of the heart under deep anaesthetic. Aortas were cleaned and equilibrated for an hour in DMEM. Aortas were treated with vehicle [dimethyl sulfoxide (DMSO)] or 10 μmol/L PCN for 24 h with or without 100 μmol/L (±)-clopidogrel hydrochloride for the final hour of PCN treatment. Ten microlitres of this conditioned media was incubated with 100 μL of human platelet-rich plasma (PRP) for 30 min at 37°C for platelet aggregation studies. Human blood was collected by venepuncture into tri-sodium citrate (3.2% w/v final) and centrifuged (175 g, 15 min) to obtain PRP. Aggregation of the PRP in response to 0.1 μmol/L adenosine diphosphate (ADP) was measured by a modified 96-well plate light transmission method.24 The platelet study was approved by the St Thomas's Hospital Research Ethics Committee (Ref. 07/Q0702/24), conducted according to the Declaration of Helsinki and all volunteers gave written informed consent prior to providing blood samples. The aortas were used for reverse transcription–polymerase chain reaction (RT–PCR) analysis.
2.4. RT–PCR analysis
RT–PCR was performed using standard techniques as described in Supplementary material online. Copy DNA (cDNA) was synthesized from 2 μg total RNA and extracted from cultured cells or rat aorta using TRIzol reagent (Invitrogen). PCR was performed using GoTaq® Flexi DNA polymerase according to the manufacturer's protocol (Promega). PCR products were size fractionated and visualized with a 1.5% agarose gel containing ethidium bromide. For quantification, bands were analysed using ImageJ (NIH, Bethesda, MD, USA) and presented as a ratio to β-actin.
2.5. Real-time PCR analysis
cDNA was prepared from 1 to 5 μg total RNA extracted from cultured vascular cells, mouse aorta, positive control mouse, and rat liver and Huh-7 (human hepatoma) cells with TRIzol reagent. Real-time PCR was performed using the primers, probes, and reagents listed in Supplementary material online using glyceraldehyde-3-phosphate dehydrogenase as an internal control. For PXR detection, absolute quantification was performed using a genomic DNA (Promega) standard curve.
2.6. PXR knockout mice
Mouse care and procedures were approved by the Animal Ethics Committee, National Institute of Environmental Health Sciences, and conformed to the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health. Genetically matched wild-type (PXR+/+) and null (PXR–/–) mice in 129sv/C57BL6 mixed background,25 which had been backcrossed with C57BL6 six times, were randomly divided into two groups (n = 3/group) and were treated with PCN (20 mg/kg body weight ip) or vehicle (DMSO) for 24 h. The mice were killed by CO2 inhalation as described in NIEHS Animal Resource SOP 8.602 and the aortas removed.
2.7. Non-lytic P450-GloTM assay for CYP3A4
CYP3A activity was measured using a substrate from Promega that luminesces after metabolism. RASMC were cultured in 96-well plates and treated with PCN (10 μmol/L) for 48 h, changing the media daily. Cells were incubated at 37°C for 4 h with the CYP3A4 substrate luciferin-PFBE (1:40 dilution) in the presence of 10 μmol/L ketoconazole or a range of clopidogrel concentrations. Luminescence in the conditioned media was measured after 20 min room temperature incubation with an equal volume of luciferin detection reagent.
2.8. Cell viability
Cell viability was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.26 RASMC treated with PCN (10 μmol/L) for 24 h, followed by 3 h hydrogen peroxide (1 mmol/L) treatment, were incubated for 30 min at 37°C in DMEM containing 0.2 mg/mL MTT. The medium was removed and the formazan precipitate dissolved in DMSO. Absorbance was measured at 550 nm. The effect of PXR-DN on hydrogen peroxide-induced cell death in living cells was measured indirectly using green fluorescent protein (GFP) as a transfection marker, as described previously; a 9:1 mixture of PXR-DN and GFP, totalling 0.6 μg plasmid per well, was used.27 Twenty-four hour post-transfection cells were treated as above. Fluorescent microscopy images were captured from three (×200) magnification fields for each sample, which were prepared in triplicate, and the number of GFP cells was counted.
2.9. Efflux assay
MDR1-mediated efflux was measured by rhodamine 123 fluorescent dye as described previously.28 HAEC were cultured in 96-well plates and treated with either vehicle (0.1% DMSO) or 10 μmol/L rifampicin for 24 h. Cells were loaded with 1 μmol/L rhodamine 123 at 37°C in the presence of 100 μmol/L verapamil (MDR1 inhibitor) for 10 min. After washing on ice, the cells were incubated for 6 min in 200 μL Hanks' buffered salt solution at room temperature to allow rhodamine 123 efflux. Fluorescence in the conditioned Hanks' buffered salt solution was measured at 485 nm excitation and 535 nm emission.
2.10. Cellular GST and glutathione peroxidase assays
Cellular GST and glutathione peroxidase (GPx) activity were measured as described previously.29 Protein extracts were prepared by sonication and centrifugation from RASMC treated for 24 h with either vehicle (0.1% DMSO) or 10 μmol/L PCN. The protein concentration of the supernatants was determined by BioRad protein assay (Hemel-Hempstead, UK). GST activity was measured by the change in absorbance at 340 nm of 1-chloro-2,4-dinitrobenzene (CDNB), due to enzyme-dependent formation of CDNB–glutathione conjugate, at 30 s intervals for 5 min at room temperature. GST activity was calculated using the extinction coefficient 9.6 mmol/L cm and the results were expressed as nmol CDNB–glutathione conjugate formed per minute per milligram cellular protein. GPx activity was measured by the change in absorbance at 340 nm due to enzyme-dependent NADPH consumption at 30 s intervals for 5 min at 37°C. This measurement was based on the consumption of NADPH by glutathione reductase in the reaction mix during the reduction in GPx-generated glutathione disulfide. GPx activity was calculated using extinction coefficient 6.22 mmol/L cm and the results were expressed as nmol NADPH consumed per minute per milligram cellular protein.
3. Results
3.1. PXR is expressed in human vasculature
PXR, the master regulator of enterohepatic drug metabolism and transport,3 was detected in vascular SMC of the human aorta (Figure 1A1a), in the cardiac muscle (Figure 1A2a), and in small vessels in the heart (Figure 1A2c). PXR was expressed in positive control tissues, liver (Figure 1A3a) and small intestine (Figure 1A4a) and was absent in negative control tissue, spleen (Figure 1A5a). No positive staining was observed when the primary antibody was omitted (Figure 1A1–5b, controls).
Figure 1.
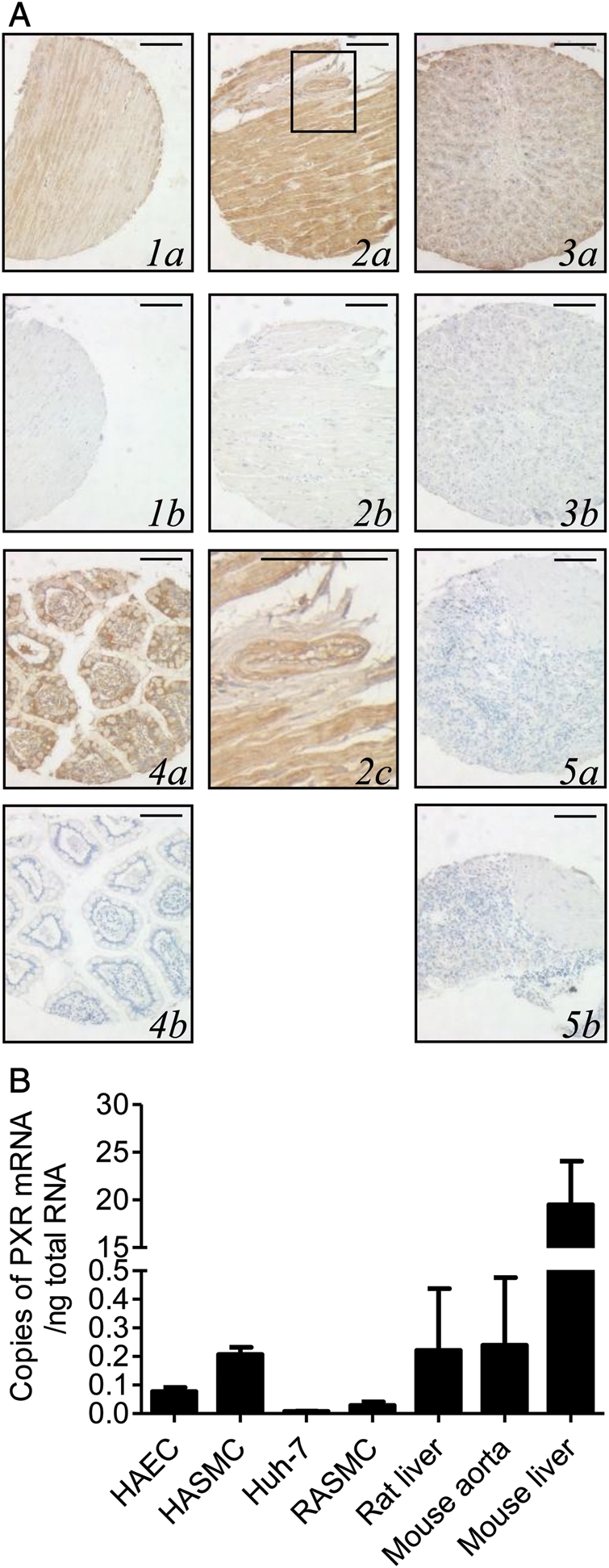
PXR is expressed in cardiovascular tissue. (A) Immunoreactive PXR protein (brown immunoperoxidase) in the aorta (1a), heart (2a) including vessels in the highlighted area of the heart (2c), liver (3a), and small intestine (4a) but not the spleen (5a) or when primary antibody was omitted (1–5b). Magnification ×200. Scale bar represents 100 μm. (B) PXR mRNA in the rat liver (n = 2), RASMC (n = 20), Huh-7 (n = 3), HAEC (n = 13), HASMC (n = 2), mouse aorta (n = 4), and mouse liver (n = 3). Data represent the number of copies of PXR mRNA/ng total RNA ±SE.
3.2. PXR is expressed in vascular cells in vitro
PXR mRNA was detected by real-time PCR in the PXR-positive controls human hepatoma cell line Huh-7 and rat and mouse liver (Figure 1B). PXR mRNA was also present in RASMC, HASMC, HAEC, and in the freshly harvested mouse and rat aorta (Figure 1B; see Supplementary material online, Figure S1). PXR protein expression was confirmed in HAEC, HASMC, and RASMC by western blotting (see Supplementary material online, Figure S1).
3.3. PXR regulates Phase I drug metabolism in vascular cells
PXR is considered a xenosensing receptor that co-ordinately regulates the transcription of genes involved in drug metabolism and transport in the liver and intestine.3 We hypothesized that PXR regulated a similar defence gene programme in the vasculature, as summarized in Figure 2.
Figure 2.

PXR protects the vasculature against chemical and oxidative insults. Activation of PXR in endothelial cells and SMC induces transcription of Phase I and II drug-metabolizing enzymes and transporters, e.g. CYP3A and GSTs, consequently altering metabolic handling and enhancing endogenous homeostatic mediator production, e.g. EETs in the vasculature. Oxidative stress response genes, e.g. GPx, are also up-regulated, which with GSTs can protect against oxidative damage.
PXR is known to regulate Phase I enzyme subfamilies CYP3A, CYP2B, and CYP3A in the human and rodent liver.3 Similarly, CYP3A23, CYP2B2, and CYP2C6 were significantly induced in RASMC and in ex vivo cultured rat aorta in response to 10 μmol/L PCN (Figure 3A). The equivalent human CYP isoforms CYP3A4, CYP2B6, and CYP2C8 were induced by 10 μmol/L rifampicin in HAEC and HASMC (see Supplementary material online, Table S2). Consistent with known variations in human expression and human hepatocyte primary cell culture for CYPs, there was a great variation in basal expression of CYPs in human primary vascular cells; however, there was a consistent induction of at least one CYP by PXR in the majority of cultures.30,31 CYP3A23 mRNA induction was matched by a similar induction of CYP3A activity after PCN treatment of RASMC, which was inhibited by CYP3A inhibitor ketoconazole (Figure 3B). However, when RASMC were transiently transfected with either PXR-DN or control empty vector (pcDNA3.1), CYP3A23 mRNA was significantly induced after PCN treatment in the control RASMC but not in those transfected with PXR-DN (Figure 3C). To further confirm PXR-dependent regulation of CYPs in the vasculature, in vivo CYP induction in the PXR wild-type and knockout mouse aorta was examined 24 h after ip PCN administration. Cyp2b10, the mouse equivalent of human CYP2B6, mRNA was significantly induced in the aorta from wild-type mice but not PXR-null mice after treatment with PCN (Figure 3D).
Figure 3.

PXR-dependent cytochrome P450 induction. Data represent fold change from vehicle (DMSO)-treated samples ±SE due to 24 h treatment with PCN (10 μmol/L in vitro; 20 mg/kg body weight ip in vivo). (A) Densitometric analysis of semi-quantitative RT–PCR for CYP3A23 (n = 3), CYP2B2 (nRASMC= 6, nAorta= 7), and CYP2C6 (n = 3) in RASMC or ex vivo cultured Wistar rat aorta normalized to β-actin control. (B) CYP3A activity in RASMC was assessed as the conversion of CYP3A luminogenic substrate over 4 h post-PCN treatment, with or without CYP3A inhibitor ketoconazole (10 µmol/L). n = 4 triplicate samples. (C) Real-time PCR for CYP3A23 (n = 3) in RASMC transfected with either control vector or PXR-DN plasmid normalized to GAPDH control. (D) Real-time RT–PCR for Cyp2b10 (n = 3) on in vivo treated wild-type and PXR-null mice aorta, normalized to GAPDH control. *P < 0.05 vs. vehicle (A, B, one-sample t-test; D, Mann–Whitney non-parametric test). **P < 0.01 two-way ANOVA with Bonferroni's multiple comparison test.
3.4. PXR activators induce functional clopidogrel pro-drug conversion to the active P2Y12 antagonist in the rat aorta
Clopidogrel is a commonly used anti-platelet drug, which inhibits ADP responses by blocking the P2Y12 ADP receptor in platelets. Clopidogrel is also a pro-drug that requires bioactivation by CYP1A2 and PXR target genes CYP3A, CYP2B6, CYP2C9, and CYP2C19 to produce its active P2Y12 ADP receptor inhibitor.3,32,33 The CYP3A induced by PCN in RASMC was able to accept clopidogrel as a substrate, as clopidogrel competed with the luciferin-PFBE substrate in the CYP3A assay (Figure 4A).
Figure 4.
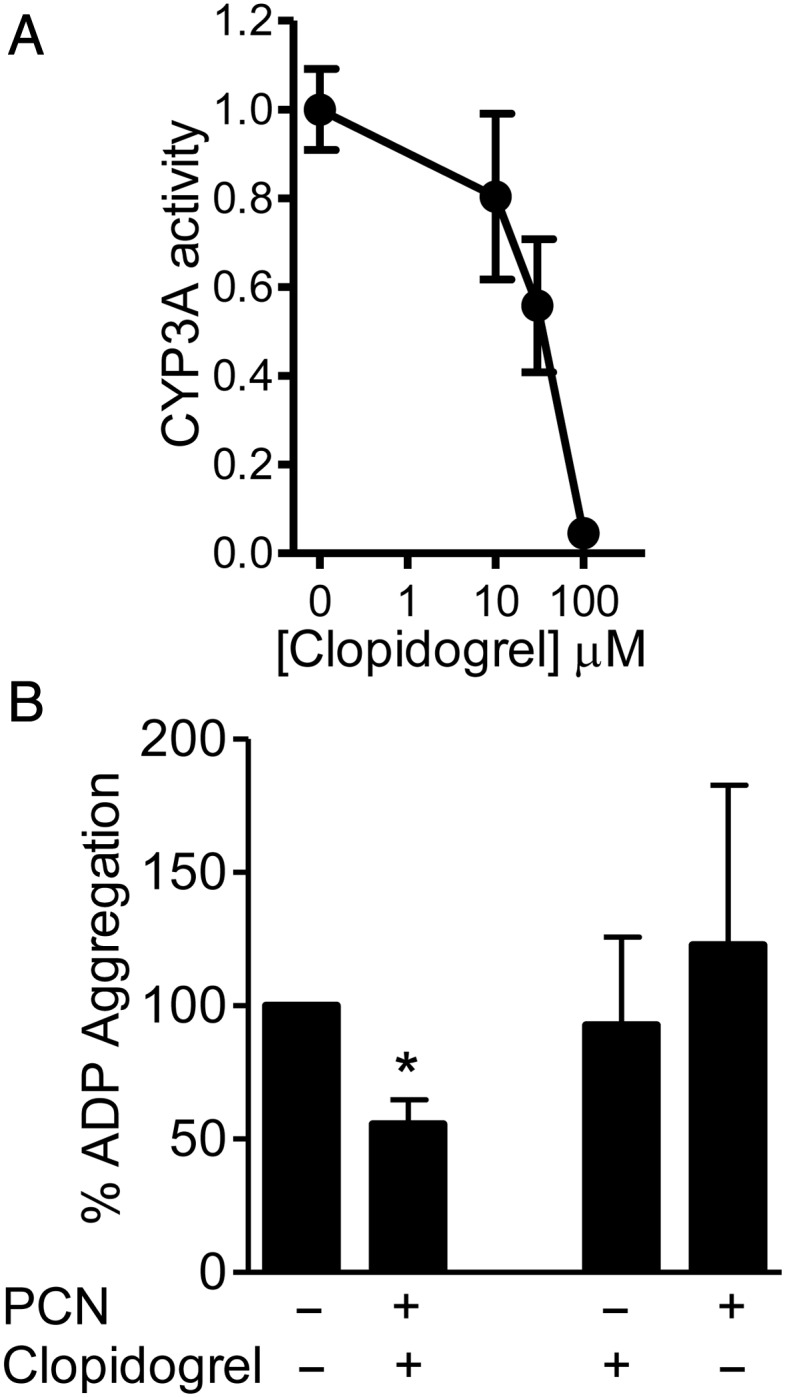
(A) Clopidogrel competes with CYP3A luminogenic substrate for metabolism in RASMC. Data represent fold change (n = 3 triplicate samples) from vehicle ±SE. (B) PXR activation in the rat aorta by PCN (10 μmol/L 24 h) enhances the anti-platelet aggregation effects of clopidogrel. Ten microlitres of conditioned media, from ex vivo cultured rat aorta incubated with clopidogrel (100 μmol/L 1 h post-PCN), were incubated with PRP for 30 min prior to 1 μmol/L ADP stimulation. Data represent aggregation/mg aortic protein as a percentage of control ±SE, n = 4, duplicate samples; *P < 0.05 vs. vehicle control determined by one-sample t-test.
CYP3A, 2B, and 2C mRNA was significantly induced in ex vivo cultured rat aorta in response to PCN (Figure 3A). Conditioned media from rat aorta pre-incubated with PCN for 24 h and then treated with clopidogrel (100 μmol/L) for 1 h was tested to see whether a functional conversion of clopidogrel pro-drug had occurred. Human PRP was stimulated with 1 μmol/L ADP (Figure 4B) to induce sub-maximal aggregation (∼50% maximum aggregation). When conditioned media was added to the PRP, ADP-induced aggregation was significantly reduced (Figure 4B). In contrast, conditioned media from the rat aorta incubated with either clopidogrel or PCN alone did not alter ADP-induced aggregation.
3.5. PXR ligands regulate transport in vascular cells
MDR1 [ATP-binding cassette subfamily B (ABCB1)], a PXR-regulated efflux transporter actively translocates substrates across biological membranes.6 Expression of MDR1 mRNA and efflux of Rhodamine 123, an MDR1-specific fluorescent substrate, was induced after PXR ligand treatment of HAEC (Figure 5).
Figure 5.

PXR activation induces multidrug resistance protein 1 expression and activity. HAEC were treated with rifampicin (10 μmol/L; 24 h) (A) Real-time PCR of MDR1 (n = 6) normalized to GAPDH control, expressed as the fold change from vehicle (0.1% DMSO)-treated cells ±SE. (B) MDR1-mediated efflux of fluorescent substrate rhodamine 123, represented as a percentage of vehicle control efflux ±SE, n = 8. *P < 0.05 vs. vehicle determined by paired Student's t-test.
3.6. PXR ligands decrease oxidative stress in vascular cells
PXR transcriptionally regulates the GST family of Phase II drug-metabolizing enzymes, which protect against toxicity and oxidative stress by conjugating glutathione to their substrates and by their GPx activity.34,35 GSTM1 mRNA expression and GST and GPx activities (Figure 6A) were significantly induced after PCN treatment of RASMC. To confirm whether this protected vascular cells against oxidative damage, RASMC death was induced in a concentration-dependent manner by H2O2 (data not shown). Pre-treatment of the cells for 24 h with PCN reduced the H2O2-induced cell death at similar levels as the GPx mimetic ebselen, whereas the non-peroxidase antioxidant apocynin did not prevent the cell death (Figure 6B and C; see Supplementary material online, Figure S2). Transfection of RASMC with PXR-DN blocked the ability of PCN to reduce H2O2-induced cell death (Figure 6D).
Figure 6.

PXR activation induces GST activity (n = 5), GPx activity (n = 6), and GSTM1 mRNA expression (n = 6) in RASMC treated with PCN (10 μmol/L 24 h) (A). Data represent fold change in enzyme activity or GSTM1:GAPDH ratio for real-time PCR from vehicle (0.1% DMSO)-treated cells ±SE. (B and C) PXR activation (10 μmol/L PCN, 24 h) protects RASMC from hydrogen peroxide (1 mmol/L, 3 h post-PCN)-induced cell death. (B) Representative photomicrographs of RASMC morphology post-treatment. Magnification ×100. Scale bar represents 200 μm. (C) Data represent viability as a percentage of control ±SE (n = 5 triplicate samples). (D) Expression of PXR-DN prevents PXR activator-mediated protection of RASMC viability. RASMC transfected with control vector (pcDNA 3.1) or PXR-DN plasmid in the presence of GFP expression vector (pEGFPN-1) at a ratio of 9:1 were treated 24 h post-transfection with 10 μmol/L PCN for 24 h and then 1 mmol/L hydrogen peroxide for 3 h before GFP cells were counted by fluorescent microscopy. Data represent the mean from three (×200) magnification fields per triplicate, from n = 3 experiments ±SE. (A) *P < 0.05 vs. vehicle determined by one-sample t-test. (C) *P < 0.05, (A) **P < 0.01 vs. control/vehicle determined by paired Student's t-test. (D) *P < 0.05, ***P < 0.001 determined by two-way ANOVA with Bonferroni's post hoc test.
4. Discussion
Exposure to chemicals from endogenous, dietary, and environmental sources has been shown to contribute to the development of cardiovascular diseases.2 There is an established enterohepatic system responsible for protecting the body from these toxic insults, by metabolizing the majority of these chemicals. Expression of PXR, a master regulator of enterohepatic drug metabolism and transport,3 was detected in the human aorta and heart tissue sections, HAEC, HASMC, RASMC, and rat and mouse aorta. This expression of PXR in vascular cells contrast with a previous finding of no PXR in the aorta and heart from C57/BL6 and 129 × 1/SvJ mice.36 However, our data in combination with the detection of PXR mRNA in SMC from obese Zucker rat and P. obesus gerbil thoracic aorta and in wild-type mouse mesenteric arteries indicate that arterial PXR expression is common across species and that the mRNA is translated into a functional protein.11,12,18
The vascular endothelium expresses known hepatic drug-metabolizing enzymes, such as CYPs, which produce endogenous mediators by metabolism of compounds, such as arachidonic acid.7,9 To confirm PXR-regulated vascular Phase I metabolism, CYP induction was examined in primary RASMC. CYP3A23, CYP2B2, and CYP2C6 were significantly induced in RASMC by PXR activation. In addition, the up-regulation of CYP3A23 mRNA corresponded to an induction of similar magnitude of CYP3A functional activity. Focused cDNA microarrays showed that Phase I and II drug-metabolizing enzymes and transporters were also expressed in and up-regulated in a concerted manner by PXR activation in human vascular cells (see Supplementary material online, Table S3), similar to the patterns observed previously in human hepatocytes, rat liver, and wild-type but not PXR knockout mouse liver.37,38 The induction of cytochrome P450s from the 3A, 2B, and 2C subfamilies by PXR activation in HAEC and HASMC was confirmed by RT–PCR. There was, however, variability in basal CYP expression in the primary human vascular cells, similar to observations in primary hepatocyte cultures, where CYP expression and function decline with culture time and are altered by disease.31
To establish whether PXR regulation of vascular drug-metabolizing enzymes has a functional impact on the metabolism and therefore efficacy of cardiovascular drugs directly in the vessel wall, the anti-platelet aggregation drug clopidogrel was selected for testing. Clopidogrel is a pro-drug that requires metabolism by CYP1A2 and PXR target genes CYP3A, CYP2B6, CYP2C9, and CYP2C19 to form an active metabolite that inhibits ADP-induced platelet activation by irreversibly binding to the P2Y12 ADP platelet receptor.3,32,33 Enhanced CYP transcription, e.g. allelic variant CYP2C19*17, increases the bioactivation and patient responsiveness to clopidogrel.39 Clopidogrel metabolic conversion is considered to occur exclusively in the liver. Yet, in a cultured rat aorta system, PXR activation up-regulated the transcription of clopidogrel-metabolizing CYPs and induced clopidogrel bioactivation, generating an active P2Y12 antagonist that inhibited platelet aggregation. This corresponds to the clinical observations that PXR activation enhanced responsiveness to clopidogrel, whereas low polymorphic PXR expression correlates with clopidogrel non-responsiveness,33 suggesting that PXR represents a target to overcome resistance to clopidogrel treatment. This is important as 25% of the patients with acute myocardial infarction undergoing coronary artery stenting are hyporesponsive or resistant to clopidogrel.40 Our results indicate for the first time the drug metabolism capability of vascular cells, the systemic relevance of which should be considered. Indeed, PCN-induced CYP3A in RASMC accepted clopidogrel and simvastatin (see Supplementary material online, Figure S3A) as substrates, in competition with the CYP3 Aactivity assay substrate luciferin-PFBE. Simvastatin is a widely used 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase inhibitor, metabolized by CYP3A.41 In addition to its cholesterol-reducing effects, HMG-CoA reductase inhibition, and thus high levels of simvastatin, directly induces vascular SMC apoptosis, via reduced RhoA prenylation activating a TGFβ-Smad apoptotic pathway.42,43 When RASMC were pre-treated with PCN, the cells were protected from simvastatin cytotoxicity (see Supplementary material online, Figure S3B). Given the PXR dependence of the protection (see Supplementary material online, Figure S3C), increased metabolism and clearance of simvastatin by PXR-up-regulated vascular CYP3A may play an important role in determining local simvastatin efficacy. Overall, the induction of these key drug-metabolizing enzymes in vascular cells, especially CYP3A4, which is responsible for the oxidative metabolism of over 50% of pharmaceutical drugs consumed by man, shows that the vasculature, a system of greater mass than the liver,44 represents an important site for drug metabolism.
The CYPs are not just involved in xenobiotic metabolism, and CYP2C enzymes are the major endothelial epoxygenases, producing EETs (important vascular homeostasis and anti-inflammatory signalling molecules) from arachidonic acid.9 CYP3A and CYP2B can also contribute to EET production.10,45 Hagedorn et al.18 indicated that hormonal activation of PXR during pregnancy initiates mesenteric artery vasorelaxation by inducing CYP epoxygenases. Although PXR mRNA was detected in mouse mesenteric arteries and vasorelaxation could be inhibited by a CYP epoxygenase inhibitor, no direct induction of CYPs in the artery was shown.18 The local up-regulation of CYPs in the vasculature we observed provides this missing link and suggests that generally PXR may protect the vasculature from disruptions to vascular homeostasis and inflammation and regulate tone. This is in contrast to a recent report, which attributed CYP2C induction by PXR ligands to the other xenosensing receptor, constitutive androstane receptor (CAR), as human saphenous vein endothelial cells and umbilical vein endothelial cell line ECV304 did not express PXR.3,46 This may highlight a difference between venous- and arterial-derived endothelial cells, because in addition to HAEC, we have detected PXR expression in human coronary artery endothelial cells but could not detect PXR in human umbilical vein endothelial cells (unpublished data). CAR is detected in both human HAEC and HASMC on the cDNA arrays and in RASMC by RT–PCR (unpublished data).
As endothelial cells form a barrier between the circulation and the underlying tissues, we examined whether MDR1 actively transported drugs across the endothelial barrier and was regulated by PXR as in the liver.6 MDR1 mRNA and activity was induced after PXR ligand rifampicin treatment, indicating that vascular PXR-mediated transport is not limited to blood–brain barrier endothelial cells as previously thought,13,17 but that similar to the liver, PXR in vascular cells can co-ordinately regulate transport as well as drug metabolism, which may impact on the bioavailability of cardiovascular drugs.47 In addition, MDR1 transports free cholesterol into and out of cells and therefore changes in MDR1 could potentially be harnessed to reduce cholesterol accumulation in atherosclerotic lesions.48
Data from the cDNA arrays indicated that many Phase II conjugating drug-metabolizing enzymes from the GST and SULT families were up-regulated by PXR (see Supplementary material online, Table S3). These enzymes play an important role in protection from toxic and oxidative stress. GSTs can protect against oxidative damage by conjugating glutathione to their substrates (xenobiotics or products of oxidative stress) and by selenium-independent GPx activity.34,35 In endothelial cells, GSTA4 protects against hydrogen peroxide-induced apoptosis in vitro,35 similar to the PXR-dependent protection we observed here in RASMC after PXR-mediated induction of GST activity. The cDNA arrays showed GSTM1 up-regulation in HAEC and HASMC by PXR, which was confirmed by real-time PCR in RASMC. Recent work by Yang et al.49 has linked reduced GSTM1 expression to decreased clearance of superoxide and thus increased oxidative stress in RASMC from atherosclerosis susceptible C57BL/6 mice. In addition, PXR activation-induced GPx activity, which given the protection against hydrogen peroxide-induced cell death by PCN was the same as a GPx mimetic, indicates a contribution of induced peroxidase activity to the protective effect. Here, for the first time in any tissue, PXR is shown to protect against oxidative stress, co-ordinating an antioxidant response in vascular cells, inducing total GST and GPx activity, and protecting against oxidative stress-induced cytotoxicity.
In summary, PXR is expressed in the vasculature, where it co-ordinately regulates the expression and activity of Phase I and II drug-metabolizing enzymes, transporters, and oxidative stress response genes (Figure 2). Consequently, PXR activation alters the efficacy of well-known cardiovascular drugs clopidogrel and simvastatin in vascular cells. This demonstrates the importance of considering the vasculature and PXR in the metabolic handling and tissue distribution of cardiovascular drugs, nutritional components, and xenobiotics, with implications in the understanding of adverse reactions, drug–drug interactions, and individual patient responses. Importantly, this study has not only confirmed that PXR regulates the same chemical defence system in the vasculature as in the liver, but expanded that defence system to include protection against oxidative stress, making PXR a novel gatekeeper for vascular defence.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by a Barts and the London Charitable Trust, research and advisory board non-clinical research fellowship (427/438) to K.E.S.; the Wellcome Trust (074361/Z/04/Z) to D.B.-B.; European Community FP6 funding (LSHM-CT-2004-0050333) to T.D.W.; the British Heart Foundation (BS/02/002) to D.B.-B.; and the Intramural Research Program of the National Institutes of Health and National Institute of Environmental Health Sciences to M.N.
Supplementary Material
References
- 1.Pries AR, Kuebler WM. Normal endothelium. Handb Exp Pharmacol. 2006;176(Pt 1):1–40. doi: 10.1007/3-540-32967-6_1. [DOI] [PubMed] [Google Scholar]
- 2.Hennig B, Reiterer G, Majkova Z, Oesterling E, Meerarani P, Toborek M. Modification of environmental toxicity by nutrients: implications in atherosclerosis. Cardiovasc Toxicol. 2005;5:153–160. doi: 10.1385/ct:5:2:153. [DOI] [PubMed] [Google Scholar]
- 3.Kliewer SA, Goodwin B, Willson TM. The nuclear pregnane X receptor: a key regulator of xenobiotic metabolism. Endocr Rev. 2002;23:687–702. doi: 10.1210/er.2001-0038. doi:10.1038/mt.2008.257. [DOI] [PubMed] [Google Scholar]
- 4.Schuetz EG, Brimer C, Schuetz JD. Environmental xenobiotics and the antihormones cyproterone acetate and spironolactone use the nuclear hormone pregnenolone X receptor to activate the CYP3A23 hormone response element. Mol Pharmacol. 1998;54:1113–1117. doi: 10.1124/mol.54.6.1113. doi:10.1111/j.1530-0277.2002.tb02626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shenoy SD, Spencer TA, Mercer-Haines NA, Abdolalipour M, Wurster WL, Runge-Morris M, et al. Induction of CYP3A by 2,3-oxidosqualene:lanosterol cyclase inhibitors is mediated by an endogenous squalene metabolite in primary cultured rat hepatocytes. Mol Pharmacol. 2004;65:1302–1312. doi: 10.1124/mol.65.5.1302. doi:10.1086/302540. [DOI] [PubMed] [Google Scholar]
- 6.Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14581–14587. doi: 10.1074/jbc.M010173200. doi:10.1002/ijc.22199. [DOI] [PubMed] [Google Scholar]
- 7.Elbekai RH, El-Kadi AO. Cytochrome P450 enzymes: central players in cardiovascular health and disease. Pharmacol Ther. 2006;112:564–587. doi: 10.1016/j.pharmthera.2005.05.011. doi:10.1016/j.cbi.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 8.Fisslthaler B, Hinsch N, Chataigneau T, Popp R, Kiss L, Busse R, et al. Nifedipine increases cytochrome P4502C expression and endothelium-derived hyperpolarizing factor-mediated responses in coronary arteries. Hypertension. 2000;36:270–275. doi: 10.1161/01.hyp.36.2.270. doi:10.1097/FPC.0b013e32832ecf2e. [DOI] [PubMed] [Google Scholar]
- 9.Fleming I. Cytochrome p450 and vascular homeostasis. Circ Res. 2001;89:753–762. doi: 10.1161/hh2101.099268. doi:10.1111/j.1360-0443.2008.02203.x. [DOI] [PubMed] [Google Scholar]
- 10.Hoebel BG, Steyrer E, Graier WF. Origin and function of epoxyeicosatrienoic acids in vascular endothelial cells: more than just endothelium-derived hyperpolarizing factor? Clin Exp Pharmacol Physiol. 1998;25:826–830. doi: 10.1111/j.1440-1681.1998.tb02162.x. doi:10.1111/j.1360-0443.2004.00597.x. [DOI] [PubMed] [Google Scholar]
- 11.Hamlat N, Forcheron F, Negazzi S, del Carmine P, Feugier P, Bricca G, et al. Lipogenesis in arterial wall and vascular smooth muscular cells: regulation and abnormalities in insulin-resistance. Cardiovasc Diabetol. 2009;8:64. doi: 10.1186/1475-2840-8-64. doi:10.1084/jem.194.5.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamlat N, Negazzi S, Forcheron F, Bricca G, Beylot M, Aouichat-Bouquerra S. Lipogenesis in arterial wall and vascular smooth muscle cells of Psammomys obesus: its regulation and abnormalities in diabetes. Diabetes Metab. 2010;36:221–228. doi: 10.1016/j.diabet.2010.01.003. doi:10.1073/pnas.95.5.2509. [DOI] [PubMed] [Google Scholar]
- 13.Bauer B, Hartz AMS, Fricker G, Miller DS. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood–brain barrier. Molecular Pharmacology. 2004;66:413–419. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 14.Narang VS, Fraga C, Kumar N, Shen J, Throm S, Stewart CF, et al. Dexamethasone increases expression and activity of multidrug resistance transporters at the rat blood–brain barrier. Am J Physiol Cell Physiol. 2008;295:C440–C450. doi: 10.1152/ajpcell.00491.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ott M, Fricker G, Bauer B. Pregnane X receptor (PXR) regulates P-glycoprotein at the blood–brain barrier: functional similarities between pig and human PXR. J Pharmacol Exp Ther. 2009;329:141–149. doi: 10.1124/jpet.108.149690. [DOI] [PubMed] [Google Scholar]
- 16.Zastre JA, Chan GN, Ronaldson PT, Ramaswamy M, Couraud PO, Romero IA, et al. Up-regulation of P-glycoprotein by HIV protease inhibitors in a human brain microvessel endothelial cell line. J Neurosci Res. 2009;87:1023–1036. doi: 10.1002/jnr.21898. doi:10.1073/pnas.88.18.8149. [DOI] [PubMed] [Google Scholar]
- 17.Chan GN, Hoque MT, Cummins CL, Bendayan R. Regulation of P-glycoprotein by orphan nuclear receptors in human brain microvessel endothelial cells. J Neurochem. 2011;118:163–175. doi: 10.1111/j.1471-4159.2011.07288.x. doi:10.1111/j.1530-0277.2010.01403.x. [DOI] [PubMed] [Google Scholar]
- 18.Hagedorn KA, Cooke CL, Falck JR, Mitchell BF, Davidge ST. Regulation of vascular tone during pregnancy: a novel role for the pregnane X receptor. Hypertension. 2007;49:328–333. doi: 10.1161/01.HYP.0000253478.51950.27. doi:10.1016/j.bcp.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Bishop-Bailey D, Walsh DT, Warner TD. Expression and activation of the farnesoid X receptor in the vasculature. Proc Natl Acad Sci USA. 2004;101:3668–3673. doi: 10.1073/pnas.0400046101. doi:10.1097/01.alc.0000174909.91034.7c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bishop-Bailey D, Hla T, Warner TD. Intimal smooth muscle cells as a target for peroxisome proliferator-activated receptor-gamma ligand therapy. Circ Res. 2002;91:210–217. doi: 10.1161/01.res.0000029080.15742.85. doi:10.1111/j.1530-0277.2011.01439.x. [DOI] [PubMed] [Google Scholar]
- 21.Kocarek TA, Shenoy SD, Mercer-Haines NA, Runge-Morris M. Use of dominant negative nuclear receptors to study xenobiotic-inducible gene expression in primary cultured hepatocytes. J Pharmacol Toxicol Methods. 2002;47:177–187. doi: 10.1016/S1056-8719(03)00002-9. doi:10.1073/pnas.241506298. [DOI] [PubMed] [Google Scholar]
- 22.Li YT, Swales KE, Thomas GJ, Warner TD, Bishop-Bailey D. Farnesoid x receptor ligands inhibit vascular smooth muscle cell inflammation and migration. Arterioscler Thromb Vasc Biol. 2007;27:2606–2611. doi: 10.1161/ATVBAHA.107.152694. [DOI] [PubMed] [Google Scholar]
- 23.Bishop-Bailey D, Larkin SW, Warner TD, Chen G, Mitchell JA. Characterization of the induction of nitric oxide synthase and cyclo-oxygenase in rat aorta in organ culture. Br J Pharmacol. 1997;121:125–133. doi: 10.1038/sj.bjp.0701100. doi:10.1111/j.1530-0277.2001.tb02356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Armstrong PC, Truss NJ, Ali FY, Dhanji AA, Vojnovic I, Zain ZN, et al. Aspirin and the in vitro linear relationship between thromboxane A2-mediated platelet aggregation and platelet production of thromboxane A2. J Thromb Haemost. 2008;6:1933–1943. doi: 10.1111/j.1538-7836.2008.03133.x. doi:10.1089/10430340152712665. [DOI] [PubMed] [Google Scholar]
- 25.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 27.Bishop-Bailey D, Hla T. Endothelial cell apoptosis induced by the peroxisome proliferator-activated receptor (PPAR) ligand 15-deoxy-Delta 12, 14-prostaglandin J2. J Biol Chem. 1999;274:17042–17048. doi: 10.1074/jbc.274.24.17042. [DOI] [PubMed] [Google Scholar]
- 28.Bogman K, Peyer AK, Torok M, Kusters E, Drewe J. HMG-CoA reductase inhibitors and P-glycoprotein modulation. Br J Pharmacol. 2001;132:1183–1192. doi: 10.1038/sj.bjp.0703920. doi:10.1093/toxsci/65.2.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Cao Z, Zhu H. Upregulation of endogenous antioxidants and phase 2 enzymes by the red wine polyphenol, resveratrol in cultured aortic smooth muscle cells leads to cytoprotection against oxidative and electrophilic stress. Pharmacol Res. 2006;53:6–15. doi: 10.1016/j.phrs.2005.08.002. doi:10.1111/j.1530-0277.2007.00553.x. [DOI] [PubMed] [Google Scholar]
- 30.George J, Goodwin B, Liddle C, Tapner M, Farrell GC. Time-dependent expression of cytochrome P450 genes in primary cultures of well-differentiated human hepatocytes. J Lab Clin Med. 1997;129:638–648. doi: 10.1016/s0022-2143(97)90199-2. doi:10.1097/FPC.0b013e3282609e67. [DOI] [PubMed] [Google Scholar]
- 31.Gomez-Lechon MJ, Castell JV, Donato MT. Hepatocytes—the choice to investigate drug metabolism and toxicity in man: in vitro variability as a reflection of in vivo. Chem Biol Interact. 2007;168:30–50. doi: 10.1016/j.cbi.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 32.Kazui M, Nishiya Y, Ishizuka T, Hagihara K, Farid NA, Okazaki O, et al. Identification of the human cytochrome P450 enzymes involved in the two oxidative steps in the bioactivation of clopidogrel to its pharmacologically active metabolite. Drug Metab Dispos. 2010;38:92–99. doi: 10.1124/dmd.109.029132. doi:10.1073/pnas.89.7.2581. [DOI] [PubMed] [Google Scholar]
- 33.Lau WC, Gurbel PA, Watkins PB, Neer CJ, Hopp AS, Carville DG, et al. Contribution of hepatic cytochrome P450 3A4 metabolic activity to the phenomenon of clopidogrel resistance. Circulation. 2004;109:166–171. doi: 10.1161/01.CIR.0000112378.09325.F9. doi:10.1111/j.1369-1600.2006.00030.x. [DOI] [PubMed] [Google Scholar]
- 34.Hayes JD, Pulford DJ. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. doi:10.1152/ajpendo.00187.2007. [DOI] [PubMed] [Google Scholar]
- 35.Yang Y, Yang Y, Trent MB, He N, Lick SD, Zimniak P, et al. Glutathione-S-transferase A4-4 modulates oxidative stress in endothelium: possible role in human atherosclerosis. Atherosclerosis. 2004;173:211–221. doi: 10.1016/j.atherosclerosis.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 36.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. doi:10.1016/j.ymgme.2003.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guzelian J, Barwick JL, Hunter L, Phang TL, Quattrochi LC, Guzelian PS. Identification of genes controlled by the pregnane X receptor by microarray analysis of mRNAs from pregnenolone 16alpha-carbonitrile-treated rats. Toxicol Sci. 2006;94:379–387. doi: 10.1093/toxsci/kfl116. doi:10.1006/mthe.2001.0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maglich JM, Stoltz CM, Goodwin B, Hawkins-Brown D, Moore JT, Kliewer SA. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol Pharmacol. 2002;62:638–646. doi: 10.1124/mol.62.3.638. doi:10.1080/09595230600944453. [DOI] [PubMed] [Google Scholar]
- 39.Sibbing D, Koch W, Gebhard D, Schuster T, Braun S, Stegherr J, et al. Cytochrome 2C19*17 allelic variant, platelet aggregation, bleeding events, and stent thrombosis in clopidogrel-treated patients with coronary stent placement. Circulation. 2010;121:512–518. doi: 10.1161/CIRCULATIONAHA.109.885194. doi:10.1096/fj.09-132563. [DOI] [PubMed] [Google Scholar]
- 40.Sharma RK, Reddy HK, Singh VN, Sharma R, Voelker DJ, Bhatt G. Aspirin and clopidogrel hyporesponsiveness and nonresponsiveness in patients with coronary artery stenting. Vasc Health Risk Manag. 2009;5:965–972. doi: 10.2147/vhrm.s6787. doi:10.1038/sj.gt.3302149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prueksaritanont T, Gorham LM, Ma B, Liu L, Yu X, Zhao JJ, et al. In vitro metabolism of simvastatin in humans [SBT]identification of metabolizing enzymes and effect of the drug on hepatic P450s. Drug Metab Dispos. 1997;25:1191–1199. doi:10.1111/j.1530-0277.2008.00658.x. [PubMed] [Google Scholar]
- 42.Guijarro C, Blanco-Colio LM, Massy ZA, O'Donnell MP, Kasiske BL, Keane WF, et al. Lipophilic statins induce apoptosis of human vascular smooth muscle cells. Kidney Int Suppl. 1999;71:S88–S91. [PubMed] [Google Scholar]
- 43.Rodriguez-Vita J, Sanchez-Galan E, Santamaria B, Sanchez-Lopez E, Rodrigues-Diez R, Blanco-Colio LM, et al. Essential role of TGF-beta/Smad pathway on statin dependent vascular smooth muscle cell regulation. PLoS One. 2008;3:e3959. doi: 10.1371/journal.pone.0003959. doi:10.1073/pnas.0500930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gerlach E, Nees S, Becker BF. The vascular endothelium: a survey of some newly evolving biochemical and physiological features. Basic Res Cardiol. 1985;80:459–474. doi: 10.1007/BF01907911. doi:10.1007/BF02197242. [DOI] [PubMed] [Google Scholar]
- 45.Imaoka S, Hashizume T, Funae Y. Localization of rat cytochrome P450 in various tissues and comparison of arachidonic acid metabolism by rat P450 with that by human P450 orthologs. Drug Metab Pharmacokinet. 2005;20:478–484. doi: 10.2133/dmpk.20.478. [DOI] [PubMed] [Google Scholar]
- 46.Bertrand-Thiebault C, Masson C, Siest G, Batt AM, Visvikis-Siest S. Effect of HMGCoA reductase inhibitors on cytochrome P450 expression in endothelial cell line. J Cardiovasc Pharmacol. 2007;49:306–315. doi: 10.1097/FJC.0b013e31803e8756. doi:10.1073/pnas.91.10.4407. [DOI] [PubMed] [Google Scholar]
- 47.Taubert D, von BN, Grimberg G, Lazar A, Jung N, Goeser T, et al. Impact of Pglycoprotein on clopidogrel absorption. Clin Pharmacol Ther. 2006;80:486–501. doi: 10.1016/j.clpt.2006.07.007. doi:10.1002/hep.21023. [DOI] [PubMed] [Google Scholar]
- 48.Petruzzo P, Cappai A, Brotzu G, Batetta B, Putzolu M, Mulas MF, et al. Lipid metabolism and molecular changes in normal and atherosclerotic vessels. Eur J Vasc Endovasc Surg. 2001;22:31–36. doi: 10.1053/ejvs.2001.1378. [DOI] [PubMed] [Google Scholar]
- 49.Yang Y, Parsons KK, Chi L, Malakauskas SM, Le TH. Glutathione S-transferase-micro1 regulates vascular smooth muscle cell proliferation, migration, and oxidative stress. Hypertension. 2009;54:1360–1368. doi: 10.1161/HYPERTENSIONAHA.109.139428. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.