

Abstract
Pulmonary arterial hypertension (PAH) and cancer share elements of pathophysiology. This provides an opportunity for the cross-development of anticancer agents that can be used in improving PAH care. The adaptation of new drugs across these disease populations warrants a structured approach. This study was a 16-week, phase Ib, single-center, open-label trial of the multikinase/angiogenesis inhibitor sorafenib. In order to assess the safety of sorafenib in PAH, patients with advanced but stable disease on parenteral prostanoids (with or without oral sildenafil) were initiated on treatment at the lowest active dosage administered to cancer patients: 200 mg daily. Patients underwent weekly clinical evaluations and monthly functional testing and dose escalations to a final dosage of 400 mg twice daily. Among 12 patients (10 of them women), sorafenib was well tolerated at 200 mg twice daily. The most common adverse events were moderate skin reactions on the hands and feet and alopecia. Our conclusion was therefore that this is a tolerable dosing regimen for testing the therapeutic activity of sorafenib in PAH patients.
Pulmonary arterial hypertension (PAH) is a fatal disease characterized by vasoconstriction, endothelial dysfunction, excessive smooth muscle cell proliferation, and in situ thrombosis of pulmonary arterioles.1–4 The incidence has been estimated to be 5–25 cases per million individuals per year.5 Efforts to develop new molecular entities for uncommon diseases are limited by the capacity to recruit sufficient numbers of patients and subsequently to recoup the investment in the development effort. The number of new molecular entities in development for cancer indications dwarfs the number of those in development for many other diseases, including PAH.6 Although the discovery methods leading to the development of these compounds justifies developing them as anticancer agents, a better characterization of their pharmacology can justify cross-developing them for noncancer indications7 such as PAH.
PAH and cancer share elements of pathophysiology. PAH is an angioproliferative disease.3,8–12 As part of the pulmonary vascular remodeling, endothelial cells can form plexiform lesions1,13 that express angiogenic vascular endothelial growth factor (VEGF) and VEGF receptors (VEGF-Rs).3 These endothelial cells seem to proliferate by monoclonal expansion, become resistant to apoptosis, and contribute to microvascular obstruction.14,15 This process is akin to cancer pathophysiology16–18 Targeting endothelial and smooth muscle cell proliferation with agents that interfere with tumor angiogenesis is a major shift from the current vasodilatory treatment approach for PAH19 and presents an unusual opportunity that warrants a structured approach.
In addition to the large number of agents developed for cancer indications, an advantage is that the development of these as anticancer agents means that the requisite testing for safe human use has already been completed and that clinical experience with cancer patients informs the assessment of drug-related adverse events and management of drug-related toxicity. However, the dosage required for shrinking or stalling the growth of tumors might be very different from that required for controlling aberrant pulmonary microvascular proliferation. Rather than proceeding immediately to a randomized trial, as has been done in the case of one anticancer agent (imatinib, NCT00477269), we undertook a phase Ib trial with sorafenib to determine short-term safety and dosage and to identify potential drug–drug interactions relevant to PAH patients that may not have been seen in the oncology cohorts.
Sorafenib (Nexavar; Bayer/Onyx Pharmaceuticals, West Haven, CT) is an inhibitor of multiple kinases, including Raf-1, VEGF-R2, and PDGFR-β.20,21 These kinases intracellularly mediate signaling by growth factors that are important to abnormal proliferation and migration of smooth muscle and endothelial cells. Sorafenib is indicated for advanced renal22 and hepatocellular cancers.23 On the basis of the monoclonal endothelial cell proliferation within plexiform lesions in advanced PAH, our group hypothesized that sorafenib may have therapeutic potential in more advanced stages of PAH. We, and others, have demonstrated that sorafenib prevents, reduces, and reverses pulmonary artery remodeling and the associated harmful consequences in three rodent pulmonary hypertension models.24,25
Balancing scientific concerns about an appropriate target population with the capacity for rapid accrual at a single center, we limited enrollment in our study to PAH patients who were the most likely to have plexiform lesions contributing to their disease physiology, namely, those receiving continuous infusion of prostacyclins. We performed an open-label study of sorafenib with 12 patients over 16 weeks with weekly clinical safety evaluations and monthly objective function evaluations guiding individual dose escalation. In order to ensure that patients could complete the study and withstand unexpected toxicities, we excluded patients in World Health Organization functional class (FC) IV.
Results
Patient characteristics
A total of 22 subjects were screened (eight declined to be enrolled; two failed screening). Table 1 shows the selected baseline demographics and disease characteristics. At baseline, the median age for the 12 patients enrolled was 49 years (range 28–77 years), the median time on epoprostenol was 94 months (range 33–156 months; median dosage 30 ng/kg/min (range 24–48 ng/kg/min)), and median time on subcutaneous treprostinil was 34 months (range 24–56 months; median dosage 88 ng/kg/min (range 70–100 ng/kg/min)); one subject was on intravenous treprostinil for 75 months at a dosage of 142 ng/kg/min. Six subjects were also on background sildenafil at study enrollment (in addition to background prostanoid). The median time on sildenafil treatment was 7 months (range 29–61 months) at a median dosage of 60 mg daily (range 20–120 mg).
Table 1.
Baseline demographics
| Patient | Age (years) | Gender | Etiology | PAH medication(s) | WHO FC |
|---|---|---|---|---|---|
| 1 | 57 | F | IPAH | EPO/SIL | III |
| 2 | 58 | M | IPAH | TRE (IV)/SIL | II |
| 3 | 48 | M | HPAH | EPO | III |
| 4 | 41 | F | CTD | EPO | II |
| 5 | 37 | F | IPAH | TRE (SC)/SIL | I |
| 6 | 49 | F | HPAH | EPO | III |
| 7 | 60 | F | Anorexigen | TRE (SC) | III |
| 8 | 57 | F | IPAH | EPO/SIL | III |
| 9 | 28 | F | HPAH | EPO/SIL | III |
| 10 | 47 | F | IPAH | TRE (SC)/SIL | III |
| 11 | 77 | F | IPAH | EPO | III |
| 12 | 46 | F | IPAH | TRE (SC)/SIL | II |
CTD, connective-tissue disease; EPO, epoprostenol; F, female; FC functional class; HPAH, heritable pulmonary arterial hypertension; IPAH, idiopathic pulmonary arterial hypertension; IV, intravenous; M, male; PAH, pulmonary arterial hypertension; SC subcutaneous; SIL, sildenafil; TRE, treprostinil; WHO, World Health Organization.
Tolerability
Of the 12 subjects, 11 completed all study evaluations, with 10 continuing on sorafenib after week 16. One subject did not complete the study. After 2 days of sorafenib at 200 mg daily, the subject abruptly developed an atypical moderate (CTCAE grade 2) rash on the face that extended to the upper torso. After evaluation by the study dermatologist and review by the safety committee, the subject was withdrawn from the study. The rash resolved in 1 week. Of the remaining 11 subjects, 7 completed week 16 on 200 mg twice daily, 1 on 200 mg once daily, 1 on 200 mg three times daily, and 2 on 400 mg twice daily (Table 2). At the monthly safety visits, none of the subjects met the criteria for withdrawal from the study. The 6 subjects on sildenafil were on the following dosages of sorafenib at the end of the study: 1 on 200 mg daily, 3 on 200 mg twice daily, 1 on 200 mg three times daily, and 1 on 400 mg twice daily. During the 16-week trial, only one serious adverse event, unrelated to the study drug, was observed.
Table 2.
Subject-wise sorafenib dose escalation
| Subject | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Maximum dosage (mg/day) | 800 | 800 | 800 | 800 | 800 | 800 | 800 | 400 | 600 | 600 | 600 | 200 |
| Final dosage (mg/day) | 400 | 400 | 800 | 400 | 400 | 800 | 400 | 200 | 600 | 400 | 400 | 200 |
| Time on maximum dosage (weeks) | 4 | 6 | 8 | 2 | 4 | 6 | 3 | 8 | 11a | 2 | 1 | 0.3 |
Visit delayed 4 weeks because of severe respiratory viral infection.
Sorafenib toxicity
During the 16-week study, all the subjects experienced at least one sorafenib-related adverse event. All these events were manageable and not severe (Table 3). The most frequent adverse events were diarrhea, hand–foot syndrome, rash, and alopecia. The typical hand–foot syndrome consists of an initial irritation with minimal skin changes without pain (grade 1) that can progress to peeling, callus, and edema interfering with activities and warranting intervention (grade 2). All dose reductions were caused by grade 2 hand–foot syndrome and grade 1 alopecia. Grade 2 hand–foot syndrome adverse events were managed with topical therapies (emollients or 10% urea cream), temporary drug interruption (range 7–10 days), and/or down-titration of the sorafenib dose per protocol. Scalp alopecia developed in seven patients.
Table 3.
Sorafenib-related adverse events
| Adverse event | No. of subjects experiencing events | No. of subjects with grade 2 events |
|---|---|---|
| Hand/foot reaction | 7 | 6 |
| Diarrhea | 4 | 2 |
| Rash | 8 | 2 |
| Alopecia | 7 | 0 |
| Hypophosphatemia | 5 | 1 |
| Dry skin | 4 | 0 |
| Anorexia | 4 | 0 |
Pharmacodynamics and prostacyclin-related adverse events
Two elements of sorafenib activity were unique to PAH patients. Five subjects in this study who received treatment with epoprostenol experienced attenuation or resolution of flushing and rash. This occurred within as little as 2 weeks (Figure 1) and at the lowest dose of sorafenib administered. Subjects on treprostinil did not have any significant changes in prostacyclin-related adverse events. The subjects taking epoprostenol (four of seven subjects) as well as those taking treprostinil (one of four subjects) had increases in episodes of diarrhea, which were judged to be caused by sorafenib in combination with the prostacyclin; all these events were manageable (via treatment with loperamide). In contrast to cancer patients, no elevation in mean systolic or diastolic blood pressure was detected in patients with PAH (Figure 2).26
Figure 1.

Improvement in characteristic erythematous epoprostenol rash in subject 4. At baseline (left) and after 119 days (16 weeks) of sorafenib administration (right).
Figure 2.
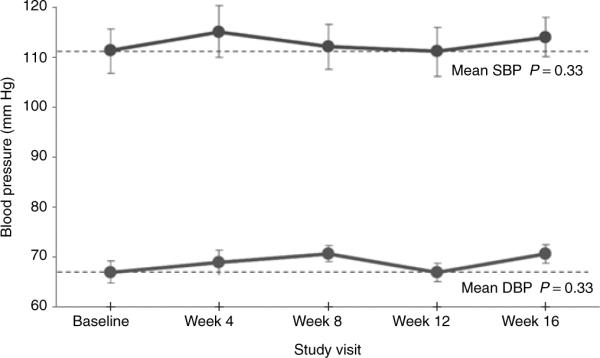
Systemic blood pressure changes. Changes in mean systolic (upper curve) and mean diastolic (lower curve) blood pressure during 16 weeks of therapy with sorafenib. Dotted lines are drawn at baseline values for clarity. DBP, diastolic blood pressure; SBP, systolic blood pressure. Error bars are SEM.
Exercise capacity and FC
From baseline to week 16, treadmill metabolic equivalents for the patients increased (P = 0.02; Table 4, Figure 3), but there was no significant median increase in 6-min walk distance (6MWD) (P = 0.37). Consistent with the results from our previous study,27 exercise treadmill test detected significant improvement in 75% of the patients with good pretreatment exercise tolerance (patient 5 was at the maximum level for both tests). Meanwhile, patients whose baseline 6MWD was <450 m had a median 48-m improvement in 6MWD (range 23–61 m; P = 0.02; not a prespecified analysis). Patient 11 showed an overall decline, and patient 5 showed no objective improvement. Three subjects improved from World Health Organization FC III to II; the FCs of the other eight subjects remained unchanged.
Table 4.
Baseline and week 16 values and changes in exploratory efficacy parameters
| Pt ID | BL | Week 16 | Δ | BL | Week 16 | Δ | BL | Week 16 | Δ |
|---|---|---|---|---|---|---|---|---|---|
| 6-min Walk distance (m) |
Exercise treadmill test (metabolic equivalents) |
Right ventricular ejection fraction (3-dimensional) (%) |
|||||||
| 1 | 411 | 472 | 61 | 6.1 | 6.4 | 0.3 | 23 | 17 | −6 |
|
| |||||||||
| 2 | 585 | 541 | −44 | 6.3 | 7.8 | 1.5 | 28 | 38 | 10 |
|
| |||||||||
| 3 | 481 | 495 | 14 | 7.3 | 8.9 | 1.6 | 27 | 36 | 9 |
|
| |||||||||
| 4 | 492 | 421 | −71 | 4.6 | 6.4 | 1.8 | 33 | 46 | 13 |
|
| |||||||||
| 5 | 666 | 682 | 16 | 11.0 | 11.0 | 0.0 | 34 | 36 | 2 |
|
| |||||||||
| 6 | 398 | 445 | 47 | 6.1 | 6.8 | 0.7 | 22 | 31 | 9 |
|
| |||||||||
| 7 | 349 | 389 | 40 | 5.2 | 5.3 | 0.1 | 16 | 30 | 14 |
|
| |||||||||
| 8 | 265 | 288 | 23 | 4.1 | 4.4 | 0.3 | 41 | 43 | 2 |
|
| |||||||||
| 9a | 262 | 296 | 34 | 3.3 | 3.7 | 0.4 | 19 | 20 | 1 |
|
| |||||||||
| 10 | 363 | 448 | 85 | 6.3 | 8.9 | 2.6 | 18 | 16 | −2 |
|
| |||||||||
| 11 | 473 | 437 | −36 | 5.6 | 4.8 | −0.8 | 17 | 13 | −4 |
|
| |||||||||
| Median | 411 | 445 | 23 | 6.1 | 6.4 | 0.4 | 23 | 31 | 2 |
|
| |||||||||
| Min | 262 | 296 | −71 | 3.3 | 3.7 | −0.8 | 16 | 13 | −6 |
|
| |||||||||
| Max | 666 | 682 | 85 | 11.0 | 11.0 | 2.6 | 41 | 46 | 14 |
|
| |||||||||
| P value | 0.33 | 0.02 | 0.09 | ||||||
| Mean pulmonary arterial pressure (mm Hg) |
Cardiac output/cardiac index (l/min)/(l/min/m2) |
Pulmonary vascular resistance index (Units × m2) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 53 | 61 | 8 | 3.4/1.9 | 2.8/1.6 | −0.6/−0.3 | 22 | 34 | 12 |
|
| |||||||||
| 2 | 63 | 59 | −4 | 4.6/2.4 | 4.3/2.3 | −0.3/−0.1 | 21 | 21 | 0.0 |
|
| |||||||||
| 3 | 55 | 47 | −8 | 4.8/2.4 | 4.6/2.3 | −0.2/−0.1 | 17 | 17 | 0.0 |
|
| |||||||||
| 4 | 48 | 40 | −8 | 7.1/4.2 | 6.4/3.8 | −0.7/−0.4 | 9 | 8 | −1.0 |
|
| |||||||||
| 5 | 36 | 35 | −1 | 7.6/3.6 | 7.0/3.4 | −0.6/−0.2 | 7 | 7 | 0.0 |
|
| |||||||||
| 6 | 45 | 43 | −2 | 4.6/3.1 | 3.2/2.2 | −1.4/−0.9 | 12 | 16 | 4 |
|
| |||||||||
| 7 | 51 | 36 | −15 | 6.5/3.5 | 5.5/2.9 | −1.0/−0.6 | 11 | 8 | −3.0 |
|
| |||||||||
| 8 | 41 | 35 | −6 | 9.3/5.0 | 6.5/3.5 | −2.8/−1.5 | 5 | 9 | 4 |
|
| |||||||||
| 9a | 48 | 53 | 5 | 3.7/2.3 | 3.6/2.2 | −0.1/−0.1 | 20 | 18a | −2.0 |
|
| |||||||||
| 10 | 58 | 55 | −3 | 2.6/1.9 | 2.4/1.7 | −0.2/−0.2 | 25 | 24 | −1.0 |
|
| |||||||||
| 11 | 64 | 64 | 0 | 2.7/1.7 | 2.2/1.5 | −0.5/−0.2 | 31 | 35 | 4.0 |
|
| |||||||||
| Median | 51 | 47 | −3 | 4.6/2.4 | 4.3/2.3 | −0.6/−0.2 | 17 | 17 | 0.0 |
|
| |||||||||
| Min | 36 | 35 | −15 | 2.6/1.7 | 2.2/1.5 | −2.8/−1.5 | 5 | 7 | 0.0 |
|
| |||||||||
| Max | 64 | 64 | 8 | 9.3/5.0 | 7.0/3.8 | 0.1/−0.1 | 31 | 35 | 12 |
|
| |||||||||
| P value | 0.11 | <0.01b | 0.62 | ||||||
Δ, change; BL, baseline.
Tests delayed 4 weeks because of severe respiratory infection.
Applies to both cardiac output and cardiac index.
Figure 3.
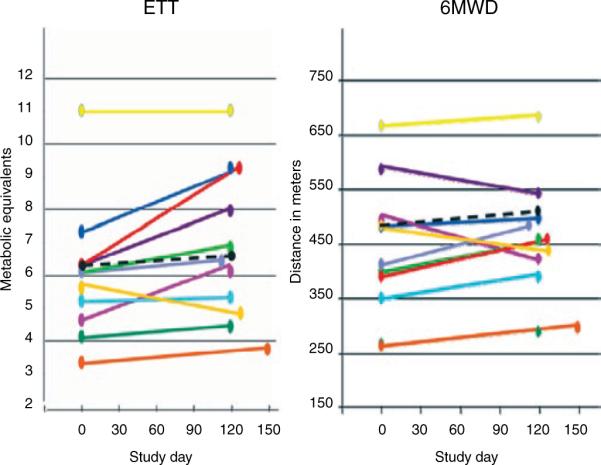
Exercise capacity: results for individual patients. 6MWD, 6-min walk distance; ETT, exercise treadmill test.
Hemodynamics
There was a modest, statistically significant decrease in cardiac output (CO)/cardiac index (CI) (P < 0.01); however, CI remained within normal range at the end of the study in subjects who started at normal or supranormal levels (Table 4). Patients who had a low CI had measurable but clinically insignificant decreases. Four patients (3, 4, 7, and 8) had significant (>5 mm Hg) decreases in pulmonary arterial pressure, and three of these patients (3, 4, and 7) had significant increases in right ventricular ejection fraction (RVEF) and exercise capacity (patients 3 and 4 in exercise treadmill test and patient 7 in 6MWD).
Echocardiography
At baseline, all the subjects had a reduced RVEF as measured by three-dimensional echocardiography. Five subjects had a tricuspid annular plane systolic excursion ≤1.8 cm, with four of these five having less-than-normal CI by right heart catheterization. There were no statistically significant changes in tricuspid annular plane systolic excursion (P = 0.09), free wall tissue Doppler (P = 0.07), or left ventricular ejection fraction (P = 0.78). At week 16, the median change in the three-dimensional echocardiography RVEF was 2% (range −6 to 14%; P = 0.09) (Table 4). Some of the patients showed marked improvement in RV measures.
Pulmonary function testing
At baseline, the subjects had either pulmonary function test results consistent with mild restrictive lung disease or normal lung volumes (Supplementary Tables S1 and S2 online). Although no subject had any clinically apparent decrease in pulmonary function, subjects 2 and 9 had declines in diffusing capacity for carbon monoxide: from 89 to 67% and from 60 to 45%, respectively. Subject 2 showed a decline in forced vital capacity but maintained the ratio of forced vital capacity to forced expiratory volume in 1 second.
Electrocardiography and N-terminal basic natriuretic peptide
There was no significant change in QTc (range −64 to 29 ms). All changes in N-terminal basic natriuretic peptide (NT-proBNP) values were within the known range of intraindividual biologic variability (serial measurements of NT-proBNP in healthy subjects vary by ≤90%) (Supplementary Tables S1 and S2 online).28
DISCUSSION
This report is the first to detail the safety, tolerability, and exploratory efficacy evaluation of sorafenib in patients with PAH, a uniformly fatal condition. Considerations included an appropriate specific patient population, tolerability concerns in PAH patients distinct from those in cancer patients, and potential phase II end points. We exclusively enrolled PAH patients who were on aggressive medical therapy (i.e., on parenteral prostanoids) so as to enrich for a population that we hypothesized would most likely have plexiform lesions with advanced disease. For safety and uniformity in testing combination treatments, patients were eligible only if the second agent was sildenafil.29,30 The minimum 6MWD of >150 m excluded World Health Organization FC IV patients in whom the potential risks of decompensation from drug-related toxicities outweighed the potential benefits.
In order to ensure patient safety and optimize interpretation of clinical data, patient evaluations included both the pulmonary hypertension management team and the oncology drug development team. In this early stage of testing a new agent that was unfamiliar to the cardiology team, the oncologists discerned direct, indirect, and unrelated toxicities of sorafenib, guided the supportive care for common toxicities of sorafenib, and aided the team with decisions on dose titration. In our study, we found that PAH patients, unwilling to contend with the common dermatologic adverse events that were acceptable to cancer patients, were unable to tolerate sorafenib at the FDA-approved dosage of 400 mg twice daily.
In this study, the drug was administered only on a continuous basis; although higher doses administered intermittently might have been tolerable, this was not explored. However, this is now recognized as a relevant issue that could be addressed in a randomized phase II study. Because no patient remained on 400 mg twice daily after 16 weeks and sorafenib is envisioned to be administered continuously for disease stabilization, we recommend 200 mg twice daily as the starting dosage for at least one arm of a phase II trial in this PAH patient population. Some patients concluded the study on 200 mg daily, which is equivalent to the minimum dosage indicated for the treatment of renal cell carcinoma.
The purpose of outcome measures in this trial included (i) safety monitoring for the individual patient, (ii) determination of whether to continue the individual subjects' therapy, (iii) exploratory development of new biomarkers for subsequent phases of study, and (iv) exploratory assessment for preliminary evidence of therapeutic activity. Many subjects showed improvement in their exercise capacity and RVEF during sorafenib treatment. In the future, placebo-controlled testing is necessary in order to establish whether this was a true sorafenib effect or merely a placebo effect. For those with poor exercise capacity (6MWD ≤450 m), this effect could be demonstrated by change in 6MWD; but for those with reasonably good exercise capacity (6MWD ≥450 m at baseline), improvement in exercise capacity was more readily detected by the exercise treadmill test. Similar to our previous results,27 we found that less clinically compromised patients showed inconsistent changes with respect to the 6MWD31 but consistently showed improvement on the exercise treadmill test. Researchers conducting a placebo-controlled phase II trial should consider evaluating the exercise treadmill test as a secondary/exploratory end point.
The potential direct effects of kinase inhibitors on cardiac function raise safety concerns for use in pulmonary hypertension. Sunitinib and imatinib have been associated with left ventricular dysfunction.32,33 We required patients to have normal left ventricular ejection fraction at baseline, and we performed serial echocardiography throughout the study. Left ventricular ejection fraction did not change significantly over the study period. Unexpectedly, we identified a small decline in median CI. In most of the subjects, the declines in CO/CI coincided with functional improvements. Even in those with the most obvious and consistent improvements in RVEF, with declines in mean pulmonary arterial pressure and increases in exercise capacity, the CI did not improve (for example, in subject 7). As the clinical significance and underlying mechanism remain ill defined, we assert that this is a safety concern that should be attentively addressed by close observation in a placebo-controlled phase II trial.
There are three leading hypotheses that are consistent with these data. First, although CO/CI measurements by thermodilution are considered accurate in PAH,34 systematic measurement variances attributable to the effects of unmeasured variables such as tricuspid regurgitation, heart rate, and equipment settings might account for the small changes. Second, it is a matter for concern, that sorafenib might have a direct effect on myocardial function, as is suspected to be the case with imatinib and as has been seen with sunitinib. However, the clinical validation of the effects of imatinib on left ventricular function remains controversial.35,36 The reproducibly measurable cardiac changes with sunitinib are not subtle, are typically accompanied by symptoms, and are presumed to represent a secondary response to systemic pressure elevations.37 In the PAH patients in this study, all of whom were receiving background prostanoid therapy, no elevations in systemic pressures were identified, symptoms of diminished CO were not apparent, and the measured changes were typically small. A third hypothesis—the one with which these findings are most consistent—is that declines in CO/CI are due to reduction in the blood volume distributed to the skin vasculature in the setting of concurrent prostanoid therapy.
There are two precedents for the sorafenib-associated decline in CO being attributed to a shift in blood volume away from the skin. First, high CO has been observed in patients with chronic erythematous skin disease, and symptoms of high-output failure improved with resolution of the erythema.38 Second, Rowell performed a series of physiologic studies of the effects of exercise and skin temperature changes on CO.39 At rest, the fraction of CO to skin has been estimated to be ~4%. It has been estimated to increase to as much as 30% under conditions of high temperature. Although obviously not as extreme, the changes observed in patients who had sorafenib added to prostacyclin therapy (diminished erythema and improved exercise tolerance but decreased CO) are reminiscent of the results reviewed by Rowell.39 In these experiments, after subjects had exercised at high ambient temperatures, the skin was cooled to enable assessment of the magnitude of blood volume shifts between skin and skeletal muscle. The observed pattern was one of diminished skin blood flow and increased exercise tolerance, along with a subtle reduction in the measured CO. Validation of this hypothesis for the observed findings in PAH patients receiving prostacyclins would require a prospective study with intensive monitoring. This might not be tested in the context of future development of kinase inhibitors as therapy for PAH; such an observation could happen only through a dedicated cross-development trial. This observation might influence the interpretation of studies on molecular mechanisms of thermal vasoregulation of skin to include prostacyclin and the VEGF-signaling pathway.40,41
This single-center, open-label, phase Ib study is subject to the potential biases associated with the study design.42 We may have underdosed some subjects, especially those who showed less improvement, as a result of the lenient down-titration adopted in the study. Validated assessments of quality of life such as the US version of the Cambridge Pulmonary Hypertension Outcome Review could have reduced the subjectivity of reporting and provided assessment of another potential end point for a future phase II study.43,44 On the basis of this study, it is unclear whether sorafenib should be used in PAH as a short-term supplement or as a chronic treatment; this requires further investigation. Finally, because all the subjects were receiving parenteral prostanoid therapy, we cannot extrapolate this dosing regimen or safety evaluation to PAH cohorts on other background PAH therapies or to treatment-naive patients.
In summary, this phase Ib trial suggests that, for PAH patients receiving parenteral prostacyclins (with or without oral sildenafil), sorafenib can be added safely at a starting dosage of 200 mg twice daily. To our knowledge, this is the first phase Ib trial of an anti-cancer kinase inhibitor performed specifically to cross-develop the agent for a noncancer indication. Through this investigation, we have identified biomarkers that might serve as useful end points in a phase II study. We observed evidence of pharmacodynamic interactions—apparent sorafenib-induced resolution of common epoprostenol-associated skin flushing and the absence of systemic blood pressure elevations associated with inhibition of the VEGF-signaling pathway—that provide new clues to understanding the mechanism of action of sorafenib in both diseases. Finally, we do not recommend “compassionate” use of this drug. A more complete understanding of the short- and long-term risks and benefits of sorafenib therapy is necessary to justify treatment of PAH with this multikinase inhibitor outside of a clinical trial.
METHODS
Study design
This was an investigator-initiated, 16-week, open-label phase Ib trial with an optional extension. The protocol included weekly safety visits and monthly assessments of FC and exercise capacity (6MWD45 and Naughton-Balke exercise treadmill test27,46) as well as laboratory testing, including measurements of NT-proBNP and serum inorganic phosphorus. At baseline and at the week-16 visit, subjects underwent right heart catheterization, electrocardiogram, pulmonary function testing, and two- and three-dimensional echocardiography (complete echocardiography was also performed at week 8).
This study was conducted under IND no. 75,684, held by the University of Chicago. Bayer, Inc., permitted cross-referencing of its data on file at the Food and Drug Administration and provided sorafenib but did not sponsor the study. The protocol was approved by the University of Chicago institutional review board; the justification for undertaking the study, in the context of the risks/benefits involved, was based on the poor long-term prognosis of the enrolled population of PAH patients, the routine safety surveillance conducted jointly by experts in these patients, the fact that sorafenib is used in oncology patients, and the close surveillance by the pulmonary hypertension team of PAH patients already familiar with their disease and individualized titration of their PAH therapeutics (prostacyclins).
Patients
Patients who had PAH that was of idiopathic or heritable etiology or associated with connective-tissue disease or anorexigen use,4 and were in FC I–III, of age >18 years, and on stable doses (≥30 days) of monotherapy with intravenous epoprostenol or subcutaneous/intravenous treprostinil or on combination therapy with oral sildenafil, were enrolled (from April 2007 to June 2008). Right heart catheterization and pulmonary function tests were performed within the 30 days prior to initiation of sorafenib. All the subjects had 6MWD >150 m (no upper limit was set).
Treatment
Subjects were started on a dosage of 200 mg daily, with monthly uptitration to a maximum of 400 mg twice daily or 200 mg three times daily. Dose-limiting toxicity was defined as an intolerable adverse event from prostanoid, sorafenib, and/or phosphodiesterase inhibition. If dose-limiting toxicity occurred, dose escalation was terminated and the dose being received at that time point was determined to be the maximum tolerated dose. Toxicities were graded according to the National Cancer Institute's “Common Terminology Criteria for Adverse Events” (http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf). Prostanoid and sildenafil doses remained unchanged during the study. Adherence was confirmed by subject interviews, residual pill counts, and records in subjects' diaries.
Withdrawal criteria
Subjects were withdrawn for (i) therapy interruption >10 days, (ii) inability to tolerate the 200 mg once-daily dosage, (iii) decrease in 6MWD by ≥20% from baseline or a decrease of ≥30 m with subjective or clinical signs/symptoms of progression (verified with a repeat test after 2 weeks), or (iv) an unresolved or recurrent grade 3/4 adverse event.
Sorafenib dose modifications
Subjects were examined weekly; supportive treatment for adverse events was in accordance with standard oncology protocols22 and was guided by oncologist co-investigators. Doses were reduced or scheduled dose increases delayed for clinically significant toxicities that were judged by the treating physician and safety committee to be at least possibly related to the drug. Additional dose delays or modifications were allowed if ≤10 days had elapsed between doses and ≥200 mg daily was the dosage. Subjects with thinning/patchy alopecia or rash associated with discomfort were referred to a dermatologist familiar with the effects of sorafenib for evaluation and management.
Outcome measures and statistical analysis
Baseline demographics, safety, and outcome measures were reported as median values and range (minimum, maximum). However, systemic blood pressure was reported as mean values with SEM, based on conventional reporting. Exploratory measures of drug effect were analyzed using the paired Wilcoxon rank sum test and included changes from baseline to week 16 in the following parameters: 6MWD, exercise treadmill test, hemodynamics, pulmonary function tests, NT-proBNP, and results of two-dimensional echocardiography/three-dimensional echocardiography. This was a pilot study on a small number of patients and was designed to assess safety and tolerability rather than efficacy.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge horst Olschewski for helpful interpretation of the data and review of the manuscript. M.G.-M. was supported by a Doris Duke Clinical Scientist Development award. S.L.A. is supported by NIH-RO1-HL071115. M.L.M. is supported by NIH-K23-CA124802. Sorafenib was provided by Bayer, Inc.
Footnotes
SUPPLEMENTARY MATERIAL is linked to the online version of the paper at http://www.nature.com/cpt
CONFLICT OF INTEREST M.G.-M., M.L.M., and M.J.R. are co-inventors on a pending patent (“Compositions and Methods for Treating Pulmonary Hypertension” Mardi Gomberg-Maitland et al. WO/2007/087575). M.G.-M. has received research grant support from Actelion, Gilead, Lilly/Icos, Pfizer, and United Therapeutics and has served as a consultant and/or on advisory boards for Biomarin, Gilead, Medtronic, Millennium Pharmaceuticals, and Pfizer Inc. M.J.R. has been a consultant for Onyx Pharmaceuticals. M.L.M. has been a consultant for Astellas, Abbott Laboratories, Millennium Pharmaceuticals, Takeda Pharmaceuticals, and UCB Pharma and received research support from Bayer, Inc. R.J.B. has received research grants from Actelion, Eli Lilly, Gilead, Novartis, Pfizer, and United Therapeutics and payment for speakers' bureau appointments from Gilead; he has also been a consultant/adviser for Actelion, Bayer, Eli Lilly, Pfizer, and Gilead. M.E.L. has received lecture fees from, has participated in advisory boards for, has consulted for, and has stock ownership in Bayer, Inc., and Onyx Pharmaceuticals. L.S., L.B., S.C., and T.J.P. declared no conflict of interest.
References
- 1.Wagenvoort C, Wagenvoort N. Pathology of Pulmonary Hypertension. 2nd edn. John Wiley and Sons; New York: 1977. [Google Scholar]
- 2.Wagenvoort CA, Mulder PG. Thrombotic lesions in primary plexogenic arteriopathy. Similar pathogenesis or complication? Chest. 1993;103:844–849. doi: 10.1378/chest.103.3.844. [DOI] [PubMed] [Google Scholar]
- 3.Tuder RM, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J. Pathol. 2001;195:367–374. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 4.Chin KM, Rubin LJ. Pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2008;51:1527–1538. doi: 10.1016/j.jacc.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 5.Humbert M, et al. Pulmonary arterial hypertension in France: results from a national registry. Am. J. Respir. Crit. Care Med. 2006;173:1023–1030. doi: 10.1164/rccm.200510-1668OC. [DOI] [PubMed] [Google Scholar]
- 6.Medical Advertising News. 2004. Product Pipeline; p. 35074. [Google Scholar]
- 7.Maitland ML, Ratain MJ. Terminal ballistics of kinase inhibitors: there are no magic bullets. Ann. Intern. Med. 2006;145:702–703. doi: 10.7326/0003-4819-145-9-200611070-00015. [DOI] [PubMed] [Google Scholar]
- 8.Bonnet S, et al. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation. 2006;113:2630–2641. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 9.Giaid a., Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1995;333:214–221. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 10.Giaid A, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1993;328:1732–1739. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 11.Rabinovitch M. Molecular pathogenesis of pulmonary arterial hypertension. J. Clin. Invest. 2008;118:2372–2379. doi: 10.1172/JCI33452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tuder RM, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999;159:1925–1932. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 13.Tuder R. Plexiform lesions in primary pulmonary hypertension may represent an abnormal form of angiogenesis. Semin. Respir. Crit. Care Med. 1994;15:207–214. [Google Scholar]
- 14.Lee SD, Shroyer KR, Markham NE, cool c.D., Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J. Clin. Invest. 1998;101:927–934. doi: 10.1172/JCI1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michelakis ED. Spatio-temporal diversity of apoptosis within the vascular wall in pulmonary arterial hypertension: heterogeneous BMP signaling may have therapeutic implications. Circ. Res. 2006;98:172–175. doi: 10.1161/01.RES.0000204572.65400.a5. [DOI] [PubMed] [Google Scholar]
- 16.Adnot S. Lessons learned from cancer may help in the treatment of pulmonary hypertension. J. Clin. Invest. 2005;115:1461–1463. doi: 10.1172/JCI25399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adnot S, Eddahibi S. Lessons from oncology to understand and treat pulmonary hypertension. Int. J. Clin. Pract. 2007;Suppl. 16:19–25. doi: 10.1111/j.1742-1241.2007.01618.x. [DOI] [PubMed] [Google Scholar]
- 18.Humbert M, Hoeper MM. Severe pulmonary arterial hypertension: a forme fruste of cancer? Am. J. Respir. Crit. Care Med. 2008;178:551–552. doi: 10.1164/rccm.200806-867ED. [DOI] [PubMed] [Google Scholar]
- 19.Rai PR, et al. The cancer paradigm of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008;178:558–564. doi: 10.1164/rccm.200709-1369PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strumberg D, et al. Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43-9006 in patients with advanced refractory solid tumors. J. Clin. Oncol. 2005;23:965–972. doi: 10.1200/JCO.2005.06.124. [DOI] [PubMed] [Google Scholar]
- 21.Wilhelm SM, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–7109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 22.Escudier B, et al. TARGET Study Group Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 23.Llovet JM, et al. SHARP Investigators Study Group Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 24.Klein M, et al. Combined tyrosine and serine/threonine kinase inhibition by sorafenib prevents progression of experimental pulmonary hypertension and myocardial remodeling. Circulation. 2008;118:2081–2090. doi: 10.1161/CIRCULATIONAHA.108.779751. [DOI] [PubMed] [Google Scholar]
- 25.Moreno-Vinasco L, et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol. Genomics. 2008;33:278–291. doi: 10.1152/physiolgenomics.00169.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veronese ML, et al. Mechanisms of hypertension associated with BAY 43-9006. J. Clin. Oncol. 2006;24:1363–1369. doi: 10.1200/JCO.2005.02.0503. [DOI] [PubMed] [Google Scholar]
- 27.Gomberg-Maitland M, Huo D, Benza RL, McLaughlin VV, tapson VF, Barst RJ. Creation of a model comparing 6-minute walk test to metabolic equivalent in evaluating treatment effects in pulmonary arterial hypertension. J. Heart Lung Transplant. 2007;26:732–738. doi: 10.1016/j.healun.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 28.Wu AH. Biological variation for N-terminal pro- and B-type natriuretic peptides and implications for therapeutic monitoring of patients with congestive heart failure. Am. J. Cardiol. 2003;92:628–631. doi: 10.1016/s0002-9149(03)00741-0. [DOI] [PubMed] [Google Scholar]
- 29.Gomberg-Maitland M, McLaughlin V, Gulati M, Rich S. Efficacy and safety of sildenafil added to treprostinil in pulmonary hypertension. Am. J. Cardiol. 2005;96:1334–1336. doi: 10.1016/j.amjcard.2005.06.083. [DOI] [PubMed] [Google Scholar]
- 30.Simonneau G, et al. PACES Study Group Addition of sildenafil to long-term intravenous epoprostenol therapy in patients with pulmonary arterial hypertension: a randomized trial. Ann. Intern. Med. 2008;149:521–530. doi: 10.7326/0003-4819-149-8-200810210-00004. [DOI] [PubMed] [Google Scholar]
- 31.Frost AE, et al. The 6-min walk test (6MW) as an efficacy endpoint in pulmonary arterial hypertension clinical trials: demonstration of a ceiling effect. Vascul. Pharmacol. 2005;43:36–39. doi: 10.1016/j.vph.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Chu TF, et al. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007;370:2011–2019. doi: 10.1016/S0140-6736(07)61865-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerkelä R, et al. Cardiotoxicity of the cancer therapeutic agent imatinib mesylate. Nat. Med. 2006;12:908–916. doi: 10.1038/nm1446. [DOI] [PubMed] [Google Scholar]
- 34.hoeper MM, et al. Determination of cardiac output by the Fick method, thermodilution, and acetylene rebreathing in pulmonary hypertension. Am. J. Respir. Crit. Care Med. 1999;160:535–541. doi: 10.1164/ajrccm.160.2.9811062. [DOI] [PubMed] [Google Scholar]
- 35.Rosti G, Martinelli G, Baccarani M. In reply to `Cardiotoxicity of the cancer therapeutic agent imatinib mesylate'. Nat. Med. 2007;13:15. doi: 10.1038/nm0107-15a. author reply 15–15; author reply 16. [DOI] [PubMed] [Google Scholar]
- 36.Verweij J, et al. Imatinib does not induce cardiac left ventricular failure in gastrointestinal stromal tumours patients: analysis of EORTC-ISG-AGITG study 62005. Eur. J. Cancer. 2007;43:974–978. doi: 10.1016/j.ejca.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 37.Khakoo AY, et al. Heart failure associated with sunitinib malate: a multitargeted receptor tyrosine kinase inhibitor. Cancer. 2008;112:2500–2508. doi: 10.1002/cncr.23460. [DOI] [PubMed] [Google Scholar]
- 38.Shuster S. High-output cardiac failure from skin disease. Lancet. 1963;1:1338–1340. doi: 10.1016/s0140-6736(63)91921-4. [DOI] [PubMed] [Google Scholar]
- 39.Rowell LB. Human Cardiovascular Control. Oxford University Press; New York, Oxford: 1993. Control of regional blood flow during dynamic exercise. [Google Scholar]
- 40.Kellogg DL, Jr, Zhao JL, Wu Y. Endothelial nitric oxide synthase control mechanisms in the cutaneous vasculature of humans in vivo. Am. J. Physiol. Heart Circ. Physiol. 2008;295:H123–H129. doi: 10.1152/ajpheart.00082.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sigaudo-Roussel D, Fromy B, Saumet JL. In vivo vasodilating mechanisms: who's NOS involved? J. Physiol. (Lond.) 2008;586:689–690. doi: 10.1113/jphysiol.2007.149658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gomberg-Maitland M. Traditional and alternative designs for pulmonary arterial hypertension trials. Proc. Am. Thorac. Soc. 2008;5:610–616. doi: 10.1513/pats.200803-024SK. [DOI] [PubMed] [Google Scholar]
- 43.Gomberg-Maitland M, Thenappan T, Rizvi K, Chandra S, Meads DM, McKenna SP. United States validation of the cambridge Pulmonary Hypertension Outcome Review (CAMPHOR) J. Heart Lung Transplant. 2008;27:124–130. doi: 10.1016/j.healun.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 44.McKenna SP, Doughty N, Meads DM, Doward LC, Pepke-Zaba J. The Cambridge Pulmonary Hypertension Outcome Review (CAMPHOR): a measure of health-related quality of life and quality of life for patients with pulmonary hypertension. Qual. Life Res. 2006;15:103–115. doi: 10.1007/s11136-005-3513-4. [DOI] [PubMed] [Google Scholar]
- 45.ATS statement: guidelines for the six-minute walk test. Am. J. Respir. Crit. Care Med. 2002;166:111–117. doi: 10.1164/ajrccm.166.1.at1102. [DOI] [PubMed] [Google Scholar]
- 46.Patterson JA, Naughton J, Pietras RJ, Gunnar RM. Treadmill exercise in assessment of the functional capacity of patients with cardiac disease. Am. J. Cardiol. 1972;30:757–762. doi: 10.1016/0002-9149(72)90151-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.