

Abstract
O-GlcNAcylation is the covalent attachment of β-D-N-acetylglucosamine (GlcNAc) sugars to serine or threonine residues of nuclear and cytoplasmic proteins, and it is involved in extensive crosstalk with other post-translational modifications, such as phosphorylation. O-GlcNAcylation is becoming increasing realized as having important roles in cancer-relevant processes, such as cell signalling, transcription, cell division, metabolism and cytoskeletal regulation. However, currently little is known about the specific roles of aberrant O-GlcNAcylation in cancer. In this Opinion article, we summarize the current understanding of O-GlcNAcylation in cancer and its emerging functions in transcriptional regulation at the level of chromatin and transcription factors.
Glycosylation is a generic term that is used to define the covalent attachment of glycans to proteins. However, this term does not reflect the enormous complexity of different types of glycan modifications in biology. Alterations in complex cell surface glycan structures are found in many diseases, and have long been known to have a direct role in cancer aetiology1. A commonly cited estimate suggests that over 50% of all proteins are glycosylated, but even this estimate is far too low, as it does not consider that many, if not most, proteins within the nucleus and cytoplasm are dynamically modified by the attachment of β-D-N-acetylglucosamine (GlcNAc) moieties to the hydroxyl group of serine or threonine residues1,2.
This modification is not usually extended into larger, more complex oligosaccharide structures nor is it found to any extent at the cell surface or in the extracellular space; thus, O-GlcNAc is a unique type of intracellular glycan attachment. The vast majority of O-GlcNAc-modified proteins are soluble nuclear or cytoplasmic proteins that are modified in response to cellular or environmental cues, such as growth factors, signalling molecules, glucose and other nutrient fluxes, and stress3,4. O-GlcNAc-modified proteins have diverse cellular roles in signalling, metabolism, transcriptional regulation, cell cycle control, protein trafficking and the regulation of cellular structure. These roles are discussed further in recent reviews5,6. Because many of these processes are perturbed during tumorigenesis, alterations in O-GlcNAcylation are likely to have a role in cancer. This Opinion article focuses on O-GlcNAc, and discusses our current restricted knowledge of the ontology of O-GlcNAc in cancer and some of the biological consequences of O-GlcNAcylation, particularly regarding the emerging transcriptional roles of O-GlcNAc at the level of chromatin and transcription factor regulation.
O-GlcNAc in cancer cell biology
O-GlcNAcylation is catalysed by a single enzyme, termed O-linked N-acetylglucosamine transferase (OGT), which transfers N-acetylglucosamine from uridine diphosphate N-acetylglucosamine (UDP-GlcNAc) to protein substrates7. Furthermore, a single enzyme known as N-acetyl-β-glucosaminidase (OGA; also known as O-GlcNAcase), which is encoded by the MGEA5 gene, rapidly removes the O-GlcNAc modification8 (BOX 1). Together, these enzymes dynamically alter the post-translational state and function of proteins in response to cellular signals.
The clinical importance of alterations in O-GlcNAc signalling to the onset, progression and metastasis of cancer is largely unknown. When comparing solid tumours with matched normal tissue no clear pattern has emerged. Two studies on a small set of paired tissues from breast or thyroid cancers showed an increase in OGA enzymatic activity, although information on OGA protein expression was not available9,10. Western blot methods showed decreased O-GlcNAc levels in some tumour samples compared with controls9. However, other studies suggested that O-GlcNAc levels increase in some cancers. Histological sections from breast, lung and colon tumours demonstrated increased O-GlcNAcylation compared with matched adjacent tissue. In the case of the lung and colon tissue, both OGT and OGA expression seemed to increase11,12. Similarly, in patients with chronic lymphocytic leukaemia, the O-GlcNAcylation of proteins was increased when compared with normal lymphocytes, and both OGT and OGA protein levels were also increased13. Strikingly, the patients whose lymphocytes displayed the highest levels of O-GlcNAcylated proteins had a better prognosis, as determined by standard prognostic markers13. These studies suggested that even though leukaemic lymphocytes had higher levels of O-GlcNAcylation than normal lymphocytes, when O-GlcNAcylation reached very high levels, it blocked signalling pathways that are key to rapid leukaemia cell proliferation14.
There are potential sources of confusion from these studies. Crucial tumorigenic roles of specific O-GlcNAcylated proteins might be masked when analysing only global cellular O-GlcNAcylation. In addition, cellular levels of O-GlcNAc, OGT and OGA have a tight relationship with multiple cellular activities. For example, chromatin immunoprecipitation followed by microarray (ChIP–chip) analysis using an O-GlcNAc-specific antibody demonstrated that the chromatin O-GlcNAcylation of hundreds of genes was altered after the loss of either OGT or OGA activity in Caenorhabditis elegans15. Not unexpectedly, gene expression significantly increased in about 50% of the genes and decreased in the other 50%, paralleling the multiple roles of O-GlcNAc in gene regulation15. Loss of OGA activity led to changes in the expression of genes that are involved in lifespan and ageing, stress response, carbohydrate metabolism, membrane transport and chromatin remodelling. Indeed, chromatin immunoprecipitation, using antibodies against OGA, shows the presence of OGA at multiple genes16. The loss of OGT activity caused alterations in the expression of genes that are involved in amino acid metabolism, mitochondrial function, reproduction, stress response and ageing15. Pharmacological or genetic methods that increase O-GlcNAcylation also rapidly increase the expression of OGA14. In addition, in a recent study using an inducible Ogt-knockout mouse embryonic fibroblast (MEF) cell line, the loss of OGT expression caused a parallel loss in OGA expression17. These data suggest that, in the case of the solid tumour samples, the increased OGA activity and expression might be a direct response to alterations in O-GlcNAc levels.
Despite conflicting data in primary tumour samples, increased O-GlcNAcylation does seem to be a general characteristic of cancer cells. For example, numerous breast cancer cell lines show higher levels of O-GlcNAcylation, and OGT protein expression is increased compared with less aggressive breast cancer cell lines18. When OGT was knocked down in these cells using RNA interference (RNAi), the cells stopped growing and formed fewer colonies in soft agar18. The RNAi-mediated knockdown of OGT was followed by an increase in p27 (also known as KIP1) levels, and p27 is an inhibitor of G1 cell cycleprogression18. The increase in p27 levels is correlated with decreased fork-head box protein M1 (FOXM1) protein levels, and FOXM1 is a transcription factor that is known to repress p27 expression18. Proteins of the FOXO family are substrates of OGT, and their increased O-GlcNAcylation correlates with increased FOXO target gene transcription19–21. Furthermore, breast cancer cells in which OGT was knocked down that were transplanted into nude mice formed fewer tumours compared with controls18. These results are interesting, but are probably due to the essential functions of OGT in cells. The alterations in FOXM1 are especially interesting, as many crucial genes that are involved in mitotic progression are regulated by this transcription factor22. O-GlcNAcylation regulates multiple pathways during cell cycle progression and might have a particular influence at M phase6 (BOX 2).
Elevations in O-GlcNAc levels have also been implicated in epithelial–mesenchymal transition through the regulation of the transcription repressor SNAIL1 (also known as SNAI1)23. Normal cells are held tightly together through the interactions of the extracellular domains of cell adhesion proteins, such as E-cadherin. SNAIL1 negatively regulates E-cadherin gene expression; however, under normal conditions, SNAIL1 is phosphorylated by the kinases caesin kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β), which promotes the ubiquitylation and the degradation of SNAIL1 by the proteasome23. SNAIL1 is O-GlcNAcylated at Ser112, and the O-GlcNAc-modified form of SNAIL1 blocks phosphorylation by GSK3β23. Elevations in O-GlcNAc levels promote SNAIL1 stabilization and in turn lead to increased SNAIL1 expression and repression of E-cadherin23. The regulation of transcriptional mediators that are associated with tumori genesis and the modification of chromatin might also explain why the deregulation of O-GlcNAcylation is evident in cancer cells.
Chromatin and O-GlcNAc
Shortly after the first paper to characterize O-GlcNAc was published, a Drosophila melanogaster gene named super sex combs (sxc) was identified24, which was much later found to be the D. melanogaster homologue of OGT25,26. Although homozygote sxc−/− embryos died during embryogenesis, sxc+/− animals survived until adulthood, but possessed several body segments that had been homeotically transformed (such as, legs in place of antenna)24. The sxc gene is part of the Polycomb gene regulatory complex, which is involved in gene silencing by histone methylation, resulting in histone 3 trimethylated on lysine 27 (H3K27me3)27. Many of the gene targets of the Polycomb complex are HOX genes, which are members of a family of homeotic transcription factors that are important for proper development and differentiation28. Importantly, aberrant HOX gene expression is linked to numerous cancers28.
Early studies on D. melanogaster polytene chromosomes demonstrated intense staining of O-GlcNAc in areas of condensed chromatin, with the transcriptionally active areas (or puffs) staining less intensely for O-GlcNAc29. Interestingly, O-GlcNAc staining on polytene chromosomes colocalizes with Polyhomeotic (PH) protein (which is a component of Polycomb repressive complex 1 (PRC1))25, and genomic profiling of O-GlcNAc in D. melanogaster found enrichment in areas bound by PH and the Pleiohomeotic repressive complex (PHORC)25,26. Moreover, the Polycomb complex PHORC binds to O-GlcNAc lectin columns, suggesting that it is modified by O-GlcNAc26, but the purpose of O-GlcNAc modification or the function of sxc+/− in this complex is not understood. Complete knockout of sxc leads to death, but this can be rescued by the expression of full-length human OGT25. sxc+/− mutants have functional Polycomb complexes that interact with chromatin26, indicating that OGT activity is needed for the proper function of Polycomb complexes, possibly through O-GlcNAcylation of PH. Therefore, the activity and localization of OGT could have a profound effect on chromatin methylation, acetylation and O-GlcNAcylation. In mammals, two different Polycomb complexes exist, PRC1 and PRC2. Which Polycomb complex OGT associates with, or whether OGT associates with both complexes, is unclear. It is possible that OGT overexpression increases trimethylation at H3K27 (Ref. 30), promoting gene repression. Such alterations to epigenetic marks have been characterized in cancer31.
OGT also O-GlcNAcylates the basal transcriptional machinery, including RNA polymerase II and the basal transcription factors, and it probably has a fundamental role at all sites of transcription32–34 (Fig. 1). Other transcriptional regulatory complexes, such as the repressor SIN3A35, the histone acetyltransferase MYST1 (also known as MOF)-containing complex nonspecific lethal (NSL)36, the X-chromosome complex male-specific lethal (MSL)37, and the arginine methyltransferase CARM1 (Refs 38,39) contain OGT and are O-GlcNAcylated. O-GlcNAcylation activates the mixed lineage leukaemia 5 (MLL5) histone methyltransferase complex40. The MLL5 methylates H3K4, which leads to transcriptional activation, but only when OGT is in association with the protein40. Interestingly, several of these complexes also contain the transcriptional activator host cell factor 1 (HCF1), which is heavily modified by O-GlcNAc41,42. O-GlcNAcylation seems to promote the cleavage of the HCF1 precursor, thus promoting HCF1 activity43,44. Together, these data strongly suggest that several chromatin-modifying complexes contain OGT and that the O-GlcNAcylation of subunits is crucial for proper function.
Figure 1. O-GlcNAc modifies the transcriptional machinery.
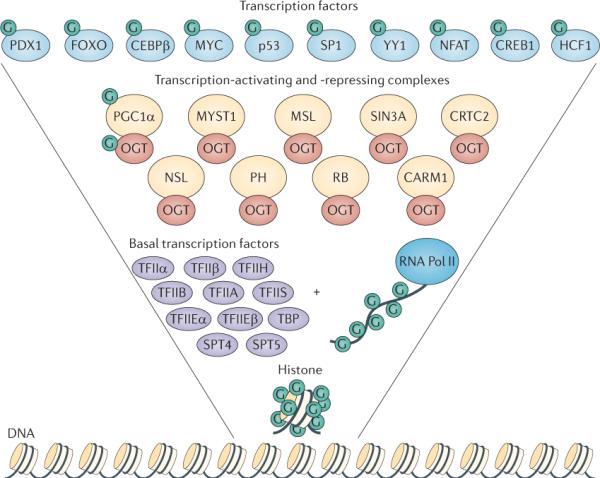
In this inverted pyramid diagram, the transcription-regulating proteins that are modified by β-D-N-acetylglucosamine (GlcNAc; G) are shown. In addition to its catalytic role, O-linked N-acetylglucosamine transferase (OGT; red ovals) interacts with numerous transcription-regulating proteins. Starting from the bottom, histones are dynamically modified by O-GlcNAc; furthermore, the carboxy-terminal domain of RNA polymerase II (RNA Pol II), as well as the basal transcription factors (purple ovals) are also modified by O-GlcNAc. Many of the transcription-activating and transcription-repressing complexes (yellow circles) that contain histone-modifying enzymes, such as histone methyltransferases, histone acetytransferases and histone deacetylases, also interact with OGT, suggesting that OGT is an integral unit in the regulation of the histone code. Finally, many transcription factors (light blue circles) are modified by O-GlcNAc. Together, the collected data from many laboratories have strongly confirmed the regulation of gene transcription by O-GlcNAcylation. CEBPβ, CCAAT/enhancer-binding protein-β; CREB1, cyclicAMP-responsive element-binding protein 1; CRTC2, CREB-regulated transcription coactivator 2; FOXO, forkhead box protein O family; HCF1, host cell factor 1; MSL, male-specific lethal; NFAT, nuclear factor of activated T cells; NSL, nonspecific lethal; PDX1, pancreas/duodenum homeobox protein 1; PGC1α: peroxisome proliferator-activated receptor-γ co-activator 1α; PH, Polyhomeotic protein; TBP, TATA-box-binding protein.
Alterations in OGT expression cause profound effects on chromatin marks during mitosis39. H3K27me3 and H3K9Ac are increased and H3 Ser10 phosphorylation, and H3R17me2 are decreased by OGT overexpression39. OGT overexpression caused mislocalization of the CARM1 methyltransferase at M phase and altered its mitotic phosphorylation, which potentially explains the loss of H3R17me2 during M phase39. Phosphorylation at H3 Ser10 is dependent on aurora kinase B, but OGT overexpression reduces the expression of this kinase42. Methylation at H3K9 increases at the G2/M transition and is required for the proper segregation of the chromosomes45; however, OGT overexpression causes an increase in H3K9Ac suggesting that methylation is considerably decreased at this site. Multiple mechanisms for this switch are possible. OGT might block histone deacetylase (HDAC) binding to H3K9 or might promote histone acetyltransferase (HAT) occupancy. Interestingly, a member of the jumonji family of demethylases (JMJD2C) that targets H3K9 is heavily modified by O-GlcNAc at M phase42, and O-GlcNAcylation of JMJD2C could potentially direct it to chromatin or enhance its activity.
Dynamic regulation of chromatin structure
O-GlcNAcylation of histones is dynamic and rapidly increases after heat shock. O-GlcNAc is found on each of the core histones with sites mapped to H2A Thr101, H2B Ser36 and H4 Ser47 (Ref. 46). Both H2B Ser36 and H4 Ser47 are found on the lateral surface of the nucleosome and contact the DNA either directly (in the case of H2B Ser36) or indirectly (in the case of H4 Ser47)46. H2A Thr101 is in the carboxy-terminal tail domain that is responsible for docking with H3 and stabilizing the histone fold46. All three O-GlcNAc sites are also phosphorylation sites47–49. Phosphorylation at H2B Ser36 enhances the expression of G2/M-regulating genes48, and total histone phosphorylation increases during the early stages of M phase50. This increase in phos-phorylation promotes the compaction of the nucleosome and chromatin segregation51. By contrast, total interphase phosphorylation is correlated with nucleosome relaxation52. Interestingly, total histone O-GlcNAcylation decreases through M phase but steadily increases as the cells enter G1 phase46. Increased histone O-GlcNAcylation induces a more compact nucleosomal structure, as judged by micrococcal nuclease (MNase) chromatin-sensitivity assays46 (Fig. 2). Previous NMR studies show that the O-GlcNAcylation of peptides causes a β-turn structure in the murine oestrogen receptor, suggesting that O-GlcNAc promotes secondary structure motifs53. Much like phosphorylation, O-GlcNAc might alter nucleosome structure by a site-and time-dependent mechanism. Under stress conditions, increased O-GlcNAc can potentially increase nucleosome compaction and protect the DNA from damage; although, in other cases, such as entry in G1 phase, O-GlcNAc might potentially promote nucleosome relaxation by either blocking phosphorylation or promoting acetylation.
Figure 2. Histone O-GlcNAcylation increases after stress.

After a cellular stress event, histone O-GlcNAcylation (the covalent attachment of β-D-N-acetylglucosamine (GlcNAc) sugars to serine or threonine residues of proteins; green dots) increases, and this is correlated with an increase in DNA compaction. Histone O-GlcNAcylation is catalysed by O-GlcNAc transferase (OGT) via the addition of a GlcNAc moiety from UDP-GlcNAc.
Obviously, multiple factors control nucleosome structure, and the crosstalk between different post-translational modifications is incredibly important. Phosphorylation and O-GlcNAcylation might have a competitive relationship on numerous sites at M phase; however, until a histone O-GlcNAc site-specific antibody is made, or quantitative site mapping study by mass spectrometry is carried out, then this competitive relationship will remain correlative.
Overexpression of OGT also reduces total histone acetylation. This indicates that, as for phosphorylation, acetylation may function antagonistically with O-GlcNAcylation. However, after stress that is induced by heat shock, cells showed both an increase in O-GlcNAcylation and an increase in acetylation on H3 and H4 (Ref. 46). What is unclear is whether O-GlcNAcylation of histones under stress conditions promotes an increase in acetylation at genes that are involved in cellular protective responses, or whether OGT expression promotes HAT accumulation at specific genes.
Clearly, histone O-GlcNAcylation alters higher order chromatin structure and probably the ability of histone-modifying proteins to interact with chromatin; however, further investigation is needed to ascertain how O-GlcNAc exerts its effects on chromatin structure and under what conditions the O-GlcNAcylation of chromatin is most relevant. Furthermore, O-GlcNAcylation of histones allows the cell to couple glucose metabolism and cellular energy state to nucleosome structure and gene regulation, an important point given the altered metabolism of cancer cells6. O-GlcNAc, together with other post-translational modifications, can tightly regulate nucleosome structure and function under various cellular conditions and stimuli. However, major unanswered questions remain, as the discovery of O-GlcNAc as part of the histone code was a recent finding. For example, do cancer cells show alterations in the activity of the O-GlcNAc-processing enzymes or the rate of O-GlcNAc cycling, and how might such alterations affect chromatin structure and other histone modifications? Are epigenetic changes in cancer due to altered OGT activity, localization or expression, or do epigenetic changes influence OGT? Undoubtedly, the roles of OGT-mediated chromatin alterations are poorly understood, and a better grasp of this process would potentially facilitate a greater interpretation of gene silencing and activation in cancer.
Competition with other modifications
As discussed above, the modification of histones and DNA-associated proteins is a complex process that involves the interaction between different modifications, which are likely to influence transcription. A preponderance of proteins that are subject to O-GlcNAc modification are transcription factors54, and transcription factors are also regulated by other modifications such as methylation, phosphorylation and acetylation. Alteration in transcription factor activity either by mutation or by gene expression is a well-known mechanism in the progression of cells from a normal state to a cancerous state55. Changes in transcription factor activity can lead to major alterations in gene expression, resulting in increased proliferation, the stimulation of angiogenesis, invasive growth and metastasis55. Almost all O-GlcNAc-modified proteins are also phosphorylated, suggesting interplay between the two modifications6. Indeed, as O-GlcNAcylation and phosphorylation both occur on serine and threonine residues, the two modifications can compete for occupancy at the same site42. However, pharmacologically inhibiting dephosphorylation does not decrease O-GlcNAcylation on all proteins, and inhibition of OGA or overexpression of OGT can produce increases in phosphorylation42,56. These data suggest a complex interplay between the two modifications. However, O-GlcNAcylation and phosphorylation can also be proximal to each other on the protein, and these spatial interactions can alter the ability of proteins to be modified, such as for p53.
More than 50% of all cancers contain mutations in the TP53 gene locus57; thus, p53 is the major `gatekeeper' of genomic stability58. Under conditions of cellular stress, environmental damage or genetic catastrophe, p53 expression and stability increase, followed by the induction of genes that promote cell cycle arrest, apoptosis and autophagy58. Owing to the pivotal role of p53 in maintaining genomic integrity, p53 is tightly regulated by proteolytic degradation59,60 (Fig. 3a). The ubiquitin ligase MDM2 binds to p53, negatively regulating p53 expression by promoting its degradation59. The stability of p53 is also affected by phosphorylation. The amino-terminal domain of the tumour suppressor contains the transactivation domain and several known phosphorylation sites61. Phosphorylation at Ser18 and Ser23 promotes p53 stability and tumour suppression62, and phosphorylation of p53 at Thr155 (which resides in the DNA-binding domain) by the COP9 signalosome promotes p53 degradation63. O-GlcNAcylation of p53 at Ser149 is antagonistic to Thr155 phosphorylation64; thus, O-GlcNAcylation at Ser149 increases p53 stability and activity64. However, as O-GlcNAcylation of Ser149 is induced in conditions of cellular stress, its effect on stability occurs concomitantly with the loss of MDM2 binding64. It is unclear whether O-GlcNAcylation at Ser149 physically blocks the COP9 signalosome from phosphorylating Thr155, whether O-GlcNAcylated p53 no longer preferentially binds to the signalosome or whether stress downregulates the signalosome activity towards p53. Thus, the phosphorylation block by O-GlcNAc needs to be further explored with a special focus on whether reduced levels of Ser149 O-GlcNAcylation have an effect in cancer cells with an intact p53 pathway.
Figure 3. Regulation of transcription factors by O-GlcNAc.

a | The tumour suppressor p53 associates with the protein MDM2, which keeps p53 at low levels in cells by promoting its degradation. Phosphorylation (P) at Thr155 stimulates the rapid degradation of p53, and O-GlcNAcylation (the covalent attachment of β-D-N-acetylglucosamine (GlcNAc) sugars to serine or threonine residues of nuclear and cytoplasmic proteins; G) at Ser149 disrupts MDM2 binding and promotes p53 stability. Mutations abolishing O-GlcNAc on p53 enhances degradation. b | In quiescent cells, the oncoprotein MYC is O-GlcNAcylated at Thr58. Upon growth signals, the O-GlcNAc modification is rapidly removed, and the protein is phosphorylated at Ser62. This site is a priming site for glycogen synthase kinase 3β (GSK3β), which can then phosphorylate Thr58, leading to the rapid degradation of MYC. Numerous solid tumours have mutations at Thr58, which promotes increased MYC stability and altered gene transcription. Inhibition of GSK3β by lithium chloride (LiCl) or mutations at Ser62 increases O-GlcNAcylation at Thr58.
In the case of the oncoprotein MYC, the phosphorylation and O-GlcNAcylation of Thr58 have a clear reciprocal relationship (Fig. 3b). In quiescent cells, Thr58 is O-GlcNAcylated, but very rapidly becomes phosphorylated by GSK3β65 on serum stimulation66,67. Thr58 lies within the transactivation domain, and phosphorylation at this site leads to rapid degradation, resulting in the loss of MYC target gene expression65. However, before GSK3β can phosphorylate Thr58 (Ref. 65), Ser62 needs to be phosphorylated. Phosphorylation of Ser62 is carried out by a variety of kinases and promotes MYC stability68. Mutation of Ser62 to alanine causes a striking increase in Thr58 O-GlcNAcylation66,67. Not surprisingly, the inhibition of GSK3β also increases MYC Thr58 O-GlcNAcylation69. The hierarchical phosphorylation of Ser62 and Thr58 is important in regulating MYC function: Thr58 is mutated in a variety of lymphomas, which leads to more-stable MYC68. Furthermore, mutations at Thr58 disrupt the ability of MYC to promote gene expression of the proapoptotic protein BIM70,71, uncoupling proliferation from apoptosis. O-GlcNAcylation of Thr58 clearly adds another level of complexity to this circuit66,67. However, it is unclear whether Thr58 O-GlcNAcylation or Ser62 phosphorylation alters the modification status of the other site in vivo. Although in vitro studies with synthetic peptides indicate that the presence of O-GlcNAc at Thr58 prevents phosphorylation at both Thr58 and Ser62, it is unclear whether this is true in vivo. Nor is it clear whether the oncogenic effect of the Thr58 mutations is due to the loss of phosphorylation or O-GlcNAcylation or both. Clearly, more detailed studies of this mechanism and the functions of the different forms of MYC are needed, possibly in systems in which the function of each type of modification on Thr58 can be independently evaluated.
As a further example, the C-terminal domain of RNA polymerase II is composed of multiple heptad repeats that can be phosphorylated at Ser2 and Ser5, but the repeat can also be O-GlcNAcylated at Thr4 (Refs 32,34) (Fig. 1). Phosphorylation of the heptad blocks O-GlcNAcylation, and O-GlcNAc blocks phosphorylation. This suggests that, on this protein, these two modifications antagonize each other34.
Regulatory mechanisms become even more complex when O-GlcNAcylation of kinases is considered. In the case of calcium/calmodulin-dependent protein kinase 4 (CAMK4), the enzyme is heavily O-GlcNAcylated when inactive; the O-GlcNAc moiety is removed before activation and an activating phosphorylation event occurs72. Thus, OGT and OGA work together to control the activation of a kinase and the subsequent downstream phosphorylation events. By layering the complexity of signalling with multiple phosphorylation and O-GlcNAcylation events, the cell can then finetune complex regulatory processes, such as responses to nutrients, extracellular stimuli and stress, or can regulate pathways such as apoptosis or the cell cycle73.
Conclusions and future directions
The O-GlcNAc modification is a crucial mechanism for cells to respond to various stimuli. The O-GlcNAc modification allows cells to link nutrient availability and cellular metabolism to the regulation of crucial cellular processes, such as cell cycle regulation, stress response and gene expression. However, many of these processes can become corrupted in cancer. Alteration in O-GlcNAcylation is one mechanism through which cell cycle progression, adaptability to the local environment and changes in gene expression are modulated. Furthermore, changes in O-GlcNAcylation can then disrupt the flow of information from other signalling systems, such as phosphorylation, ubiquitylation and acetylation.
The regulation and targeting of OGT and OGA are crucially important to cells in order to maintain proper cellular function. Elucidating the mechanisms that target these enzymes to their myriad substrates remains a key challenge (BOX 3). Although no human cancers are directly linked to mutations in OGT or OGA, perhaps owing to the lethality of such mutations, alterations in the regulation of these two enzymes have profound effects on cellular functions. Chemotherapeutics targeting the inhibition of OGT or OGA could potentially alter tumour function or could make tumours more susceptible to other chemotherapeutic agents; however, what adverse affects OGT or OGA inhibition might have on normal cells is unknown.
As the field of O-GlcNAc research moves forwards, new tools to tease out the roles of O-GlcNAcylation are needed. Although conditional OGT-knockout mice have been generated, advances in targeted OGT or OGA knockouts in specific tissues are needed. At least in cell culture, the inducible CRE OGT-knockout mouse embryonic cell line will prove useful for many types of proliferative studies17. Recently, an O-GlcNAc sensor protein was generated and showed dynamic O-GlcNAcylation during signalling4. This technology will allow the careful in vivo dissection of the effect of different signalling systems on O-GlcNAc cycling. Additionally, studies using deep sequencing or advanced proteomic techniques such as stable isotope labelling with amino acids in cell culture (SILAC) or isobaric tag for relative and absolute quantitation (iTraq) will provide new insight into the signalling pathways that are regulated by O-GlcNAc. Clearly, as we begin to learn more about the pathways that are altered in cancer and the role of O-GlcNAc in these pathways, the potential exists to develop novel targeted therapeutics to treat this disease.
Box 1 | OGT and OGA: structures and therapeutic targeting.
The crystal structure of human O-linked N-acetylglucosamine transferase (OGT) has recently been solved74. The protein is composed of a tetratricopeptide repeat (TPR) domain in the amino terminus75, and the carboxyl terminus contains a two-part catalytic domain that is bridged by an intermediate motif. The intermediate domain forms a unique structure with a helix of the C-terminal catalytic domain that contains a basic region consisting of ten lysine residues74. Residing within this region is a putative phosphatidylinositol-3-phosphate (PIP3; also known as PtdIns3P)-binding domain76. The TPR domain is separated from the catalytic core by a flexible hinge region that allows the TPR domain to pivot, restricting or allowing access to protein substrates74. The structure also reveals that the peptide-binding domain lies above the UDP-β-D-N-acetylglucosamine (GlcNAc)-binding region, suggesting that the sugar substrate binds first, followed by the peptide74. Interestingly, the apparent Michaelis constant (Km) of OGT for peptide substrates lowers with increasing concentration of the sugar nucleotide, suggesting that small fluctuations in the flux of glucose into UDP-GlcNAc can have wide-ranging effects on peptide–substrate binding77. Hopefully, the crystal structure of OGT will allow for the creation of structure-based, highly selective inhibitors of OGT, which could potentially lead to chemotherapeutics.
Although no crystal structure for eukaryotic O-GlcNAcase exists, its enzymatic mechanism is well established. The N-terminal domain of the enzyme contains the glycoside hydrolyase activity, which falls into the class 84 family of glycosidases78. Crystal structure studies on bacterial homologues79 have helped to elucidate that the hydrolyse activity resides with two aspartic acid residues that function as a general acid catalyst78,80. Interestingly, the C-terminal domain has a putative GCN5-related histone acetyltransferase-like domain; however, whether this domain actually has histone acetyltransferase activity remains controversial81,82. These studies have allowed the synthesis of highly specific inhibitors to OGA83,84. Currently, a relevant question is how O-GlcNAcase inhibition would affect tumour progression and growth.
Box 2 | O-GlcNAc and mitosis.
Overexpression of O-linked N-acetylglucosamine transferase (OGT) in HeLa cells impairs mitotic progression by drastically altering the cyclin-dependent kinase 1 (CDK1) signalling pathway42. Active CDK1 phosphorylates numerous proteins at M phase of the cell cycle, promoting nuclear membrane collapse and spindle formation85. OGT overexpression increases the inhibitory phosphorylation on CDK1 at Thr14 and Tyr15 (Ref. 42). At least two mechanisms are responsible for this: mRNA levels of the dual-specific phosphatase CDC25C are depressed, reducing CDK1 phosphorylation on Thr14 and Tyr15; and the expression levels and activity of protein kinase, membrane-associated tyrosine/threonine 1 (PKMYT1), which also phosphorylates these sites, is increased42 owing to reduced expression of polo-like kinase 1 (PLK1), which phosphorylates and inhibits PKMYT1 (Ref. 42). Importantly, OGT localizes to the same mitotic structures as PLK1 during M phase progression, suggesting the potential for an additional level of regulation between these two proteins42. The OGT is probably altering multiple mitotic pathways to impair mitotic progression.
Not only does OGT slow mitotic progression, but its modest overexpression in cells also promotes aneuploidy. Most solid tumours show signs of aneuploidy, although whether this provides a selective advantage to a cancer cell is controversial86. One potential mechanism for the ability of OGT to promote aneuploidy is its binding to and regulation of aurora kinase B, the loss of which promotes aneuploidy87. OGT overexpression lowers the protein levels of aurora B, as well as its binding partner inner centromere protein (INCENP)42, although there is uncertainty regarding whether this effect is transcriptional or translational or due to increased protein degradation. Aurora kinase B and OGT colocalize to the midbody and precipitate in a complex with protein phosphatase 1 (PP1) and OGA88. PP1 not only dephosphorylates aurora kinase B substrates89, but it also concomitantly targets OGT to these substrates to facilitate their subsequent O-GlcNAcylation (the covalent attachment of β-D-N-acetylglucosamine (GlcNAc) sugars to serine or threonine residues of nuclear and cytoplasmic proteins)35. OGA can remove O-GlcNAc moieties from proteins that can then be immediately phosphorylated by aurora kinase B. How a futile cycle is avoided by this complex at the midbody is unknown. Other accessory proteins or different post-translational modifications could finetune activities towards these substrates.
Box 3 | Substrate specificity of OGT and OGA.
One of the more vexing questions faced in O-β-D-N-acetylglucosamine (O-GlcNAc) research is the question of how one unique O-linked N-acetylglucosamine transferase (OGT) and one unique O-linked N-acetylglucosaminase (OGA) specifically catalyse the cycling of O-GlcNAc on thousands of different proteins and distinct sites within cells. Unlike kinases, the modification of substrates by OGT does not occur at a canonical O-GlcNAcylation consensus sequence; however, the enzyme does tend to prefer a proline-valine before the modified serine or threonine followed by a polar amino acid (see the Database of O-GlcNAcylated Proteins and Sites (dbOGAP) (see Further information))42,74. How does OGT locate the right substrates during a signalling event? Yeast two-hybrid studies have begun to answer this question38. One identified protein was myosin phosphatase targeting subunit 1 (MYPT1), which, when present in a protein phosphatase 1 (PP1) complex, targets PP1 activity to myosin and other proteins38. A novel technique indicated that MYPT1 enabled the O-GlcNAcylation of multiple proteins, suggesting that MYPT1 targets OGT to unique substrates. Similarly, another protein, trafficking kinesin-binding protein 1 (TRAK1; formerly known as OGT-interacting protein 106), targets OGT to RNA polymerase II and GABA receptors90–92. These interactions seem to form transient OGT holoenzyme complexes that are responsive to cellular stimuli. On glucose deprivation, the kinase p38 MAPK interacts with the carboxy-terminal domain of OGT and targets OGT to neurofilament proteins38. One major function of the transcriptional co-activator, peroxisome proliferator-activated receptor-γ co-activator 1α
(PGC1α), is to target OGT to the FOXO transcription factors in liver that has been exposed to nutrient excess20. These data demonstrate that a large number of proteins interact with and target OGT to unique substrates after some sort of cellular stimulus. Many of these proteins are also modified with O-GlcNAc, which might either enhance the interaction and targeting, or disrupt targeting once the OGT is brought to the correct substrate91. As for OGA, even less is known about the specific signals that target this enzyme to its substrates. Interestingly, OGA that is purified from rat spleen cannot be purified to homogeneity but purifies as a multi-protein complex8,93. OGA is also probably targeted to O-GlcNAc-modified proteins by interacting proteins, but this question has not yet been fully addressed.
Acknowledgements
The authors would like to thank K. Sakabe and Q. Zeidan for critical reading of the manuscript. Research is supported by the US National Institutes of Health (R01 DK61671; R01 CA42486; R24 DK084949), the Patrick C. Walsh Prostate Cancer Research Fund, and by N01-HV-00240 to G.W.H. and P20RR024214 to C.S..
Footnotes
Competing interests statement The authors declare competing financial interests. See Web version for details.
References
- 1.Hart GW, Copeland RJ. Glycomics hits the big time. Cell. 2010;143:672–676. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torres CR, Hart GW. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984;259:3308–3317. [PubMed] [Google Scholar]
- 3.Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 4.Carrillo LD, Froemming JA, Mahal LK. Targeted in vivoO-GLcNAc sensors reveal discrete compartment-specific dynamics during signal transduction. J. Biol. Chem. 2010;286:6650–6658. doi: 10.1074/jbc.M110.191627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011;80:825–858. doi: 10.1146/annurev-biochem-060608-102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Slawson C, Copeland RJ, Hart GW. O-GlcNAc signaling: a metabolic link between diabetes and cancer? Trends Biochem. Sci. 2010;35:547–555. doi: 10.1016/j.tibs.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haltiwanger RS, Holt GD, Hart GW. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine:peptide β-N-acetylglucosaminyltransf erase. J. Biol. Chem. 1990;265:2563–2568. [PubMed] [Google Scholar]
- 8.Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-β-D-glucosaminidase from rat spleen cytosol. J. Biol. Chem. 1994;269:19321–19330. [PubMed] [Google Scholar]
- 9.Slawson C, Pidala J, Potter R. Increased N-acetyl-β-glucosaminidase activity in primary breast carcinomas corresponds to a decrease in N-acetylglucosamine containing proteins. Biochim. Biophys. Acta. 2001;1537:147–157. doi: 10.1016/s0925-4439(01)00067-9. [DOI] [PubMed] [Google Scholar]
- 10.Krzeslak A, Pomorski L, Lipinska A. Elevation of nucleocytoplasmic β-N-acetylglucosaminidase (O-GlcNAcase) activity in thyroid cancers. Int. J. Mol. Med. 2010;25:643–648. doi: 10.3892/ijmm_00000387. [DOI] [PubMed] [Google Scholar]
- 11.Gu Y, et al. GlcNAcylation plays an essential role in breast cancer metastasis. Cancer Res. 2010;70:6344–6351. doi: 10.1158/0008-5472.CAN-09-1887. [DOI] [PubMed] [Google Scholar]
- 12.Mi W, et al. O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy. Biochim. Biophys. Acta. 2011;1812:514–519. doi: 10.1016/j.bbadis.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Shi Y, et al. Aberrant O-GlcNAcylation characterizes chronic lymphocytic leukemia. Leukemia. 2010;24:1588–1598. doi: 10.1038/leu.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Slawson C, et al. Perturbations in O-linked β-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J. Biol. Chem. 2005;280:32944–32956. doi: 10.1074/jbc.M503396200. [DOI] [PubMed] [Google Scholar]
- 15.Love DC, et al. Dynamic O-GlcNAc cycling at promoters of Caenorhabditis elegans genes regulating longevity, stress, and immunity. Proc. Natl Acad. Sci. USA. 2010;107:7413–7418. doi: 10.1073/pnas.0911857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whisenhunt TR, et al. Disrupting the enzyme complex regulating O-GlcNAcylation blocks signaling and development. Glycobiology. 2006;16:551–563. doi: 10.1093/glycob/cwj096. [DOI] [PubMed] [Google Scholar]
- 17.Kazemi Z, Chang H, Haserodt S, McKen C, Zachara NE. O-linked β-N-acetylglucosamine (O-GlcNAc) regulates stress-induced heat shock protein expression in a GSK-3β-dependent manner. J. Biol. Chem. 2010;285:39096–39107. doi: 10.1074/jbc.M110.131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caldwell SA, et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. 2010;29:2831–2842. doi: 10.1038/onc.2010.41. [DOI] [PubMed] [Google Scholar]
- 19.Housley MP, et al. O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem. 2008;283:16283–16292. doi: 10.1074/jbc.M802240200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Housley MP, et al. A PGC-1α-O-GlcNAc transferase complex regulates FoxO transcription factor activity in response to glucose. J. Biol. Chem. 2009;284:5148–5157. doi: 10.1074/jbc.M808890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho SR, et al. O-GlcNAcylation enhances FOXO4 transcriptional regulation in response to stress. FEBS Lett. 2010;584:49–54. doi: 10.1016/j.febslet.2009.11.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ho KK, Myatt SS, Lam EW. Many forks in the path: cycling with FoxO. Oncogene. 2008;27:2300–2311. doi: 10.1038/onc.2008.23. [DOI] [PubMed] [Google Scholar]
- 23.Park SY, et al. Snail1 is stabilized by O-GlcNAc modification in hyperglycaemic condition. EMBO J. 2010;29:3787–3796. doi: 10.1038/emboj.2010.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingham PW. A gene that regulates the bithorax complex differentially in larval and adult cells of Drosophila. Cell. 1984;37:815–823. doi: 10.1016/0092-8674(84)90416-1. [DOI] [PubMed] [Google Scholar]
- 25.Sinclair DA, et al. Drosophila O-GlcNAc transferase (OGT) is encoded by the Polycomb group (PcG) gene, super sex combs (sxc) Proc. Natl Acad. Sci. USA. 2009;106:13427–13432. doi: 10.1073/pnas.0904638106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gambetta MC, Oktaba K, Muller J. Essential role of the glycosyltransferase sxc/Ogt in polycomb repression. Science. 2009;325:93–96. doi: 10.1126/science.1169727. [DOI] [PubMed] [Google Scholar]
- 27.Mills AA. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nature Rev. Cancer. 2010;10:669–682. doi: 10.1038/nrc2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah N, Sukumar S. The Hox genes and their roles in oncogenesis. Nature Rev. Cancer. 2010;10:361–371. doi: 10.1038/nrc2826. [DOI] [PubMed] [Google Scholar]
- 29.Kelly WG, Hart GW. Glycosylation of chromosomal proteins: localization of O-linked N-acetylglucosamine in Drosophila chromatin. Cell. 1989;57:243–251. doi: 10.1016/0092-8674(89)90962-8. [DOI] [PubMed] [Google Scholar]
- 30.Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 31.Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev. Cell. 2010;19:698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 32.Kelly WG, Dahmus ME, Hart GW. RNA polymerase II is a glycoprotein. Modification of the COOH-terminal domain by O-GlcNAc. J. Biol. Chem. 1993;268:10416–10424. [PubMed] [Google Scholar]
- 33.Comer FI, Hart GW. O-GlcNAc and the control of gene expression. Biochim. Biophys. Acta. 1999;1473:161–171. doi: 10.1016/s0304-4165(99)00176-2. [DOI] [PubMed] [Google Scholar]
- 34.Comer FI, Hart GW. Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry. 2001;40:7845–7852. doi: 10.1021/bi0027480. [DOI] [PubMed] [Google Scholar]
- 35.Yang X, Zhang F, Kudlow JE. Recruitment of O-GlcNAc transferase to promoters by corepressor mSin3A: coupling protein O-GlcNAcylation to transcriptional repression. Cell. 2002;110:69–80. doi: 10.1016/s0092-8674(02)00810-3. [DOI] [PubMed] [Google Scholar]
- 36.Cai Y, et al. Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the male-specific lethal (MSL) complex. J. Biol. Chem. 2010;285:4268–4272. doi: 10.1074/jbc.C109.087981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendjan S, et al. Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol. Cell. 2006;21:811–823. doi: 10.1016/j.molcel.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 38.Cheung WD, Sakabe K, Housley MP, Dias WB, Hart GW. O-linked β-N-acetylglucosaminyltransfer ase substrate specificity is regulated by myosin phosphatase targeting and other interacting proteins. J. Biol. Chem. 2008;283:33935–33941. doi: 10.1074/jbc.M806199200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sakabe K, Hart GW. O-GlcNAc transferase regulates mitotic chromatin dynamics. J. Biol. Chem. 2010;285:34460–34468. doi: 10.1074/jbc.M110.158170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujiki R, et al. GlcNAcylation of a histone methyltransferase in retinoic-acid-induced granulopoiesis. Nature. 2009;459:455–459. doi: 10.1038/nature07954. [DOI] [PubMed] [Google Scholar]
- 41.Wang Z, Pandey A, Hart GW. Dynamic interplay between O-linked N-acetylglucosaminylation and glycogen synthase kinase-3-dependent phosphorylation. Mol. Cell Proteomics. 2007;6:1365–1379. doi: 10.1074/mcp.M600453-MCP200. [DOI] [PubMed] [Google Scholar]
- 42.Wang Z, et al. Extensive crosstalk between O-GlcNAcylation and phosphorylation regulates cytokinesis. Sci. Signal. 2010;3:ra2. doi: 10.1126/scisignal.2000526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Capotosti F, et al. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell. 2011;144:376–388. doi: 10.1016/j.cell.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 44.Daou S, et al. Crosstalk between O-GlcNAcylation and proteolytic cleavage regulates the host cell factor-1 maturation pathway. Proc. Natl Acad. Sci. USA. 2011;108:2747–2752. doi: 10.1073/pnas.1013822108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heit R, Rattner JB, Chan GK, Hendzel MJ. G2 histone methylation is required for the proper segregation of chromosomes. J. Cell Sci. 2009;122:2957–2968. doi: 10.1242/jcs.045351. [DOI] [PubMed] [Google Scholar]
- 46.Sakabe K, Wang Z, Hart GW. β-N-acetylglucosamine (O-GlcNAc) is part of the histone code. Proc. Natl Acad. Sci. USA. 2010;107:19915–19920. doi: 10.1073/pnas.1009023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maile T, Kwoczynski S, Katzenberger RJ, Wassarman DA, Sauer F. TAF1 activates transcription by phosphorylation of serine 33 in histone H2B. Science. 2004;304:1010–1014. doi: 10.1126/science.1095001. [DOI] [PubMed] [Google Scholar]
- 48.Olson LE, et al. Homeodomain-mediated β-catenin-dependent switching events dictate cell-lineage determination. Cell. 2006;125:593–605. doi: 10.1016/j.cell.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 49.Mayya V, et al. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals system-wide modulation of protein-protein interactions. Sci. Signal. 2009;2:ra46. doi: 10.1126/scisignal.2000007. [DOI] [PubMed] [Google Scholar]
- 50.Perez-Cadahia B, Drobic B, Davie JR. H3 phosphorylation: dual role in mitosis and interphase. Biochem. Cell Biol. 2009;87:695–709. doi: 10.1139/O09-053. [DOI] [PubMed] [Google Scholar]
- 51.Wei Y, Yu L, Bowen J, Gorovsky MA, Allis CD. Phosphorylation of histone H3 is required for proper chromosome condensation and segregation. Cell. 1999;97:99–109. doi: 10.1016/s0092-8674(00)80718-7. [DOI] [PubMed] [Google Scholar]
- 52.Mahadevan LC, Willis AC, Barratt MJ. Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell. 1991;65:775–783. doi: 10.1016/0092-8674(91)90385-c. [DOI] [PubMed] [Google Scholar]
- 53.Chen YX, et al. Alternative O-GlcNAcylation/O-phosphorylation of Ser16 induce different conformational disturbances to the N terminus of murine estrogen receptor β. Chem. Biol. 2006;13:937–944. doi: 10.1016/j.chembiol.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 54.Ozcan S, Andrali SS, Cantrell JE. Modulation of transcription factor function by O-GlcNAc modification. Biochim. Biophys. Acta. 2010;1799:353–364. doi: 10.1016/j.bbagrm.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 56.Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl Acad. Sci. USA. 2008;105:13793–13798. doi: 10.1073/pnas.0806216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hollstein M, et al. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22:3551–3555. [PMC free article] [PubMed] [Google Scholar]
- 58.Brady CA, Attardi LD. p53 at a glance. J. Cell Sci. 2010;123:2527–2532. doi: 10.1242/jcs.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jones SN, Roe AE, Donehower LA, Bradley A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature. 1995;378:206–208. doi: 10.1038/378206a0. [DOI] [PubMed] [Google Scholar]
- 60.Jones NC, et al. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nature Med. 2008;14:125–133. doi: 10.1038/nm1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 2010;16:528–536. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chao C, Herr D, Chun J, Xu Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 2006;25:2615–2622. doi: 10.1038/sj.emboj.7601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bech-Otschir D, et al. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 2001;20:1630–1639. doi: 10.1093/emboj/20.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang WH, et al. Modification of p53 with O-linked N-acetylglucosamine regulates p53 activity and stability. Nature Cell Biol. 2006;8:1074–1083. doi: 10.1038/ncb1470. [DOI] [PubMed] [Google Scholar]
- 65.Gregory MA, Qi Y, Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol. Chem. 2003;278:51606–51612. doi: 10.1074/jbc.M310722200. [DOI] [PubMed] [Google Scholar]
- 66.Chou TY, Dang CV, Hart GW. Glycosylation of the c-Myc transactivation domain. Proc. Natl Acad. Sci. USA. 1995;92:4417–4421. doi: 10.1073/pnas.92.10.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chou TY, Hart GW, Dang CV. c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. J. Biol. Chem. 1995;270:18961–18965. doi: 10.1074/jbc.270.32.18961. [DOI] [PubMed] [Google Scholar]
- 68.Vervoorts J, Luscher-Firzlaff J, Luscher B. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 2006;281:34725–34729. doi: 10.1074/jbc.R600017200. [DOI] [PubMed] [Google Scholar]
- 69.Kamemura K, Hayes BK, Comer FI, Hart GW. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: alternative glycosylation/phosphorylation of THR-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens. J. Biol. Chem. 2002;277:19229–19235. doi: 10.1074/jbc.M201729200. [DOI] [PubMed] [Google Scholar]
- 70.Egle A, Harris AW, Bouillet P, Cory S. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc. Natl Acad. Sci. USA. 2004;101:6164–6169. doi: 10.1073/pnas.0401471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hemann MT, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436:807–811. doi: 10.1038/nature03845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dias WB, Cheung WD, Wang Z, Hart GW. Regulation of calcium/calmodulin-dependent kinase IV by O-GlcNAc modification. J. Biol. Chem. 2009;284:21327–21337. doi: 10.1074/jbc.M109.007310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zeidan Q, Hart GW. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J. Cell Sci. 2010;123:13–22. doi: 10.1242/jcs.053678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lazarus MB, Nam Y, Jiang J, Sliz P, Walker S. Structure of human O-GlcNAc transferase and its complex with a peptide substrate. Nature. 2011;469:564–567. doi: 10.1038/nature09638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J. Biol. Chem. 1997;272:9308–9315. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- 76.Yang X, et al. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature. 2008;451:964–969. doi: 10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- 77.Kreppel LK, Hart GW. Regulation of a cytosolic and nuclear O-GlcNAc transferase. Role of the tetratricopeptide repeats. J. Biol. Chem. 1999;274:32015–32022. doi: 10.1074/jbc.274.45.32015. [DOI] [PubMed] [Google Scholar]
- 78.Cetinbas N, Macauley MS, Stubbs KA, Drapala R, Vocadlo DJ. Identification of Asp174 and Asp175 as the key catalytic residues of human O-GlcNAcase by functional analysis of site-directed mutants. Biochemistry. 2006;45:3835–3844. doi: 10.1021/bi052370b. [DOI] [PubMed] [Google Scholar]
- 79.Rao FV, et al. Structural insights into the mechanism and inhibition of eukaryotic O-GlcNAc hydrolysis. EMBO J. 2006;25:1569–1578. doi: 10.1038/sj.emboj.7601026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.He Y, Macauley MS, Stubbs KA, Vocadlo DJ, Davies GJ. Visualizing the reaction coordinate of an O-GlcNAc hydrolase. J. Am. Chem. Soc. 2010;132:1807–1809. doi: 10.1021/ja9086769. [DOI] [PubMed] [Google Scholar]
- 81.Toleman C, Paterson AJ, Whisenhunt TR, Kudlow JE. Characterization of the histone acetyltransferase (HAT) domain of a bifunctional protein with activable O-GlcNAcase and HAT activities. J. Biol. Chem. 2004;279:53665–53673. doi: 10.1074/jbc.M410406200. [DOI] [PubMed] [Google Scholar]
- 82.Butkinaree C, et al. Characterization of β-N-acetylglucosaminidase cleavage by caspase-3 during apoptosis. J. Biol. Chem. 2008;283:23557–23566. doi: 10.1074/jbc.M804116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yuzwa SA, et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nature Chem. Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- 84.Dorfmueller HC, van Aalten DM. Screening-based discovery of drug-like O-GlcNAcase inhibitor scaffolds. FEBS Lett. 2010;584:694–700. doi: 10.1016/j.febslet.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dephoure N, et al. A quantitative atlas of mitotic phosphorylation. Proc. Natl Acad. Sci. USA. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 87.Adams RR, Maiato H, Earnshaw WC, Carmena M. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J. Cell Biol. 2001;153:865–880. doi: 10.1083/jcb.153.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wells L, Kreppel LK, Comer FI, Wadzinski BE, Hart GW. O-GlcNAc transferase is in a functional complex with protein phosphatase 1 catalytic subunits. J. Biol. Chem. 2004;279:38466–38470. doi: 10.1074/jbc.M406481200. [DOI] [PubMed] [Google Scholar]
- 89.Sugiyama K, et al. Aurora-B associated protein phosphatases as negative regulators of kinase activation. Oncogene. 2002;21:3103–3111. doi: 10.1038/sj.onc.1205432. [DOI] [PubMed] [Google Scholar]
- 90.Iyer SP, Akimoto Y, Hart GW. Identification and cloning of a novel family of coiled-coil domain proteins that interact with O-GlcNAc transferase. J. Biol. Chem. 2003;278:5399–5409. doi: 10.1074/jbc.M209384200. [DOI] [PubMed] [Google Scholar]
- 91.Iyer SP, Hart GW. Roles of the tetratricopeptide repeat domain in O-GlcNAc transferase targeting and protein substrate specificity. J. Biol. Chem. 2003;278:24608–24616. doi: 10.1074/jbc.M300036200. [DOI] [PubMed] [Google Scholar]
- 92.Beck M, et al. Identification, molecular cloning, and characterization of a novel GABAA receptor-associated protein, GRIF-1. J. Biol. Chem. 2002;277:30079–30090. doi: 10.1074/jbc.M200438200. [DOI] [PubMed] [Google Scholar]
- 93.Wells L, et al. Dynamic O-glycosylation of nuclear and cytosolic proteins: further characterization of the nucleocytoplasmic β-N-acetylglucosaminidase, O-GlcNAcase. J. Biol. Chem. 2002;277:1755–1761. doi: 10.1074/jbc.m109656200. [DOI] [PubMed] [Google Scholar]