

SUMMARY
Altered function of Cdk5 kinase is associated with many forms of neurodegenerative disease in humans. We show here that inactivating the Drosophila Cdk5 ortholog, by mutation of its activating subunit, p35, causes adult-onset neurodegeneration in the fly. In the mutants, a vacuolar neuropathology is observed in a specific structure of the central brain, the ‘mushroom body’, which is the seat of olfactory learning and memory. Analysis of cellular phenotypes in the mutant brains reveals some phenotypes that resemble natural aging in control flies, including an increase in apoptotic and necrotic cell death, axonal fragmentation, and accumulation of autophagosomes packed with crystalline-like depositions. Other phenotypes are unique to the mutants, notably age-dependent swellings of the proximal axon of mushroom body neurons. Many of these phenotypes are also characteristic of mammalian neurodegenerative disease, suggesting a close relationship between the mechanisms of Cdk5-associated neurodegeneration in fly and human. Together, these results identify the cellular processes that are unleashed in the absence of Cdk5 to initiate the neurodegenerative program, and they provide a model that can be used to determine what part each process plays in the progression to ultimate degeneration.
INTRODUCTION
Progression of neurodegenerative diseases (NDDs), including Alzheimer’s disease (AD), leads to brain atrophy with neuronal loss. These diseases are associated with a complex array of cellular and molecular pathologies. Pathological phenotypes typically include some combination of accumulation of intracellular depositions and of extracellular amyloid plaques, defects in axonal transport, synapse and mitochondrial dysfunction, and more (Muller et al., 2010; Palop and Mucke, 2010; Wong and Cuervo, 2010). One of the key issues in neurodegenerative disease is to understand the relationships among this array of phenotypes: which ones initiate the disease and which are late manifestations of neuronal dysfunction? One confounding factor in understanding neurodegenerative disease is that many of its features mimic the changes observed in normal aging, making it challenging to isolate the molecular mechanisms that are specific for disease.
One protein that is thought to be intimately involved in the development and progression of mammalian neurodegeneration is the cyclin-dependent protein kinase Cdk5 (for a review, see Ip and Tsai, 2008). Cdk5 is one of the major kinases that hyperphosphorylates tau to create the forms present in the neurofibrillary tangles that are characteristic of AD and other neurodegenerative diseases, and Cdk5 hyperphosphorylates neurofilament to produce forms that are mislocalized to the cell soma in motor neuron disease (Lee and Tsai, 2003; Noble et al., 2003). Moreover, a mislocalized and deregulated form of Cdk5 is found associated with degenerating tissue in AD brains (Patrick et al., 1999), and the protein has been associated with many cellular pathologies that are linked to degeneration, including altered autophagy and mitochondrial disruption (Meuer et al., 2007; Qu et al., 2007; Furuya et al., 2010; Wong et al., 2011). Both increased and decreased Cdk5 activity causes cell death in vitro and in in vivo models (Jessberger et al., 2009; Zhang et al., 2010), and both are associated with neurodegeneration in animal models (Ohshima et al., 1996; Patrick et al., 1999; Takahashi et al., 2010).
Activity of Cdk5 kinase is absolutely dependent on its association with one of two related regulatory subunits, p35 or p39 (Ko et al., 2001). In mammals, the Cdk5 catalytic subunit is expressed ubiquitously but the p35 and p39 regulatory subunits are largely neural-specific, accounting for the restriction of Cdk5 activity largely to the nervous system (Zheng et al., 1998). In addition to its role in neuronal maintenance, Cdk5 is essential for proper radial migration of neurons during the development of the mammalian cortex (Ohshima et al., 1996), and also has effects on axon growth (Connell-Crowley et al., 2000) and guidance (Nikolic et al., 1996), and on synaptic function (Jessberger et al., 2009). The widespread functions of Cdk5 in nervous system development have confounded efforts to understand the role of mammalian Cdk5 in neurodegeneration in vivo. Moreover, we do not know which of the neuronal targets of Cdk5 are altered during degeneration in the mouse Cdk5-knockout model because this was not investigated (Takahashi et al., 2010).
To overcome the challenges of disentangling the many functions of Cdk5, we set out to investigate its function in Drosophila. The fly has only a single p35 family member, and no p39 ortholog, simplifying genetic analysis. Moreover, development of the fly brain does not involve widespread cell migration so brain structure is not grossly disturbed in mutants of p35 (Connell-Crowley et al., 2007) or Cdk5 (Kissler et al., 2009). Flies bearing null mutations of either gene are viable and fertile, and the phenotypes of the two mutants seem to be identical. Many studies have shown that Drosophila offers a valuable model for investigating the basic cellular mechanisms of neurodegenerative disease, reflecting the widespread phylogenetic conservation of fundamental molecular mechanisms between vertebrates and invertebrates. In order to assay the wild-type function of Cdk5-p35 kinase, we therefore characterized the neural phenotypes resulting from a p35 null mutation in Drosophila.
We have shown previously that p35 null mutant flies are grossly normal behaviorally at adult eclosion, but rapidly develop age-dependent loss of motor function, progressing to rigidity, and then death, with a lifespan of about half that of the wild type (Connell-Crowley et al., 2007). We now show that aging p35 mutant flies suffer adult-onset neurodegeneration, with overt tissue disruption in a specific structure of the central brain, the ‘mushroom bodies’ (MB), which are the seat of olfactory learning and memory. These flies further display a syndrome of phenotypes that are reminiscent of defects that occur in mammalian neurodegenerative disease, including axonal fragmentation, swellings of the proximal axon, accumulation of organelles characteristic to the lysosomal-autophagosomal degradation pathway, crystalline-appearing intracellular depositions and neuronal cell death or fragility. Some of these phenotypes are consistent with the idea that there is an acceleration of the natural aging process in p35 mutants, whereas others seem to be specific to Cdk5-associated neurodegeneration per se. These data therefore distinguish several distinct cellular mechanisms that together contribute to pathological neurodegeneration in the absence of Cdk5 activity.
RESULTS
Vacuolar pathology in the MBs of aging p35 mutant brains
The progressive deterioration of adult motor function and shortened lifespan observed in p35 mutant flies (Connell-Crowley et al., 2007) prompted us to examine the brain for evidence of adult neurodegeneration. In Drosophila, the tissue loss of neurodegeneration is typically detected as accumulation of degenerative vacuoles, or ‘holes’, in the brain, as assayed in histological sections (Wittmann et al., 2001; Muhlig-Versen et al., 2005). We therefore examined integrity of brain tissue in sections derived from wild-type or mutant animals at different ages. Because p35 mutants have a substantially reduced lifespan, we designed our experiment to compare wild-type and mutant animals of the same absolute chronological age and also animals at equivalent positions in their mortality curves, hypothesizing that this would more accurately reflect the effective ‘physiological age’ of the animals (Fig. 1A). Hereafter, we refer to the relative aging periods as follows: young (3–5 days after adult eclosion), middle-aged (at ∼50% mortality of the population) and old (∼80% population mortality).
Fig. 1.

Neurodegeneration in p35null central brain. (A) Absolute chronological and relative physiological aging process. To account for their shortened lifespan, p35 mutants (p35−; point 1) were compared with age-matched wild-type control w− flies (point 2; black arrow for chronologically matched points) and also with control flies that had reached a similar position on their mortality curve (point 3; white arrow for physiologically matched points). (B,C) Young control and p35 mutant brains develop normally: C, cortex, Kenyon cells somatic region; ca, neuropil/calyx. (D,E) Vacuoles appeared randomly in old control brains but were accumulated in the MB area in old p35 mutants (red arrows). Vacuoles were present both in somatic and neuropil domains. Five brains for each genotype obtained in two independent experiments were sequentially sectioned. Thick (1 μm) plastic sections were stained with Toluidine Blue. Scale bar: 20 μm. (F–I) Actin cytoskeleton was deformed or deteriorated (red arrows) in old p35− brains. Identical regions in the MB at the level covering Kenyon cell bodies, calyx (white dashed line), primary neurites and beginning of peduncle (blue dashed line) are shown in merged confocal images of whole-mount brain preparation. Optic slice: 1.6 μm. Scale bar: 20 μm.
In young flies, wild-type and p35 mutant brains were indistinguishable at the histological level. As wild-type flies aged (60 days after eclosion, corresponding to ∼80% mortality of the population), examination of the brains of surviving flies revealed a low frequency of vacuolar lesions; when present, these lesions were distributed homogeneously throughout the brain (Fig. 1B,D). Examination of p35 mutant flies at a similar level of population mortality (45 days after eclosion) revealed a similar pattern of homogeneously distributed lesions. In addition, however, the mutant flies showed a selective accumulation of vacuolar lesions in the MB of the central brain (n=6) (Fig. 1C,E). This brain region is the seat of olfactory learning and memory in the fly. Most of these lesions were in the MB neuropil, particularly the calyx (i.e. dendrites), although some were found in the cell body region (cortex) of the structure. Accumulation of lesions in the MB was not observed in wild type (n=6), nor in homozygous mutant animals that carried a rescuing p35+ genomic transgene (n=3). Thus, histological analysis of the brains of aging p35 mutant flies presented evidence of general degeneration throughout the central brain and selective, age-dependent tissue disruption in a specific structure, the MB.
To further assess the molecular and cellular nature of the vacuolar lesions in p35 mutants, we isolated mutant and wild-type brains and performed whole-mount labeling with fluorescently-tagged phalloidin, to reveal actin structure, and with the pan-neuronal cell surface marker anti-HRP, to reveal general neuronal morphology (Fig. 1F–I). The whole-mount procedure is gentler than histological sectioning because it does not require dehydration and paraffin or plastic embedding. By this method, rather than overt histological vacuoles, we observed regions of reduced tissue density, as reflected by locally reduced F-actin labeling, and local tissue disorganization. As in the sectioned material, it was most apparent in the basal calyx of the MB in p35 mutants, and was observed only in aged, mutant animals. We infer that the ‘holes’ seen in sectioned material probably evolve from these regions of compromised tissue integrity during the processing of material for sectioning. Nonetheless, immunofluorescent assay confirms the result from histological sectioning that absence of Cdk5-p35 activity causes central brain degeneration, particularly affecting the central brain region responsible for olfactory learning and memory.
Aging was accompanied by a progressive increase in cell death in both wild type and p35 mutants
One simple hypothesis to explain the tissue loss in p35 mutants would be an increase in cell death. We therefore examined apoptotic cell death by TUNEL staining in whole-mount wild-type and mutant brains. In both controls and p35 mutants, we found an age-dependent progression in the frequency of apoptotic cell death (Fig. 2A,B). A low, and approximately equal, number of TUNEL-positive cells was observed in the brains of young animals of both genotypes, and included both neurons and glia (as assessed by molecular markers) as well as a small proportion of cells whose identity could not be ascertained. In controls the rate of neuronal cell death increased twofold by the middle of the lifespan and threefold in old adults over young animals (Fig. 2C). Glial cell apoptosis increased by 50% over the initial level in old controls (Fig. 2D). The portion of non-identifiable apoptotic cells also increased progressively, showing dynamics similar to neurons (not shown). TUNEL-positive cells were observed in all compartments of the central brain and did not reveal any compartment-specific accumulation.
Fig. 2.

Age-dependent progression of apoptotic cell death. Representative examples show TUNEL-positive nuclei in neurons (Elav positive; A–A″) and glia (Repo positive; B–B″) in p35 mutant brains. Note the weaker Elav signal in the apoptotic neuron in comparison with surrounding cells (white arrow in A–A″) and the weaker Repo signal in dying glial cell (white arrowhead in B–B″). Single confocal plan, optic slice: 1.6 μm. Scale bar: 10 μm. (C,D) The absolute number of TUNEL-positive cells per hemisphere is presented on the y axis (n≥6; mean ± s.e.m.). (C) Apoptosis in neurons. The pan-neuronal marker Elav was used to identify neurons (n≥6; mean ± s.e.m.) in three independent experiments. c, control. (D) Apoptosis in glia. The pan-glial marker Repo was used to identify glia (n≥6; mean ± s.e.m.). Time points under the chart show absolute age in days and refer to the position on the mortality curve; results are grouped according to the chronological points. For instance: 45-day-old p35 mutant flies reached 80% of mortality in population and were analyzed simultaneously with 45-day-old wild-type controls that reached only 20% in mortality.
In p35 mutants, apoptosis also increased progressively but to a similar or even lesser extent than for wild-type animals of the same chronological age. Moreover, the dynamics of apoptosis progression were different relative to position in the lifespan. In contrast to controls, at the middle-aged point in the mutants (∼50% population mortality) we did not observe a significant difference in comparison with young adults. In old p35 mutants (80% of mortality), apoptosis was increased in all three groups of cells to close to the level of the old controls. Also similar to controls, the increase in neuronal cell death was higher than that in glia. There was no point in the lifespan at which the total number of TUNEL-positive neurons or the number of TUNEL-positive glia was higher in mutants as compared with the physiologically matched controls. Moreover, we did not observe any preference for development of TUNEL-positive cells in the MB. We concluded that absence of Cdk5 activity in p35 null mutants neither prevented apoptosis nor accelerated it over control level in the central brain. Thus, the increase in apoptotic cell death does not seem to account for the selective degeneration of the MBs in p35 mutants.
The failure to detect enhanced apoptosis in the central brain in p35 mutants led us to investigate the possibility of an increase in necrosis as an alternative type of cell death (Ullman et al., 2008; Vandenabeele et al., 2010). Whole-brain staining for necrosis in vivo was adapted from cell culture protocols. Briefly, brains of wild-type or p35 mutant flies of different ages were explanted and cultured briefly in propidium iodide (PI), and the fraction of necrotic cells in the MB cortex was quantified based on loss of PI exclusion and nuclear engorgement (see Methods for details) (Fig. 3A,B). We found that necrosis increased progressively with aging in the central brain of controls, reaching a maximum level at the oldest time point. In p35 mutant brains, necrosis in young animals was at the same low level as in controls but reached a maximum in middle-aged brains and stayed at this level in old brains (Fig. 3C). At each time point, however, the amount of necrosis in p35 mutants correlated closely with the amount of necrosis observed in wild-type flies from a population that had been aged to a similar percent mortality. We interpret this to mean that the level of necrosis that we detected in the mutant tracked with the ‘physiological age’ of the flies, as defined by percent mortality of the population. Interestingly, in p35 mutants the increase and plateau in necrotic cell death happened before the increase of apoptosis, whereas, in controls, the dynamics of apoptotic and necrotic cell death were closely correlated. We conclude that, as in wild type, cell death increased in p35 mutants upon aging and, in particular, that the p35 null mutation predisposed central brain cells to undergo necrosis.
Fig. 3.

Progressive increase in necrotic cell death. Necrotic cells were identified as propidium iodide (PI)- and Hoechst (HO)-positive nuclei. (A) Z-stack of confocal images through the MB area near the top of the brain (12 μm). Small trachea tubes surround the calyx (ca) of the MB and stain positive for PI (yellow arrow). Note the significant number of PI-positive cells due to physical damage: this portion of the brain was excluded from analysis. (B–B″) Example of a more-internal 5-μm-thick area included in analysis (outer white dotted line). PI-positive nuclei also exhibit bright HO signal and often have a larger diameter (hollow white arrow). Many cells have very low HO signal (white arrowheads). Necrotic cells were often clustered in both controls and mutants (dashed rectangles). Abundance and position of PI-positive cells implied that the majority of necrotic cells were neurons. Scale bar: 20 μm. (C) Increase in the percentage of necrotic cells with age; comparison of PI- and HO-positive versus HO-positive cells (n≥6; mean ± s.e.m.). Age-dependent time points are described in the legend to Fig. 1A and Fig. 2C,D. c, control.
Accelerated and enhanced accumulation of lysosomal and/or autophagosomal organelles in aged p35 mutants
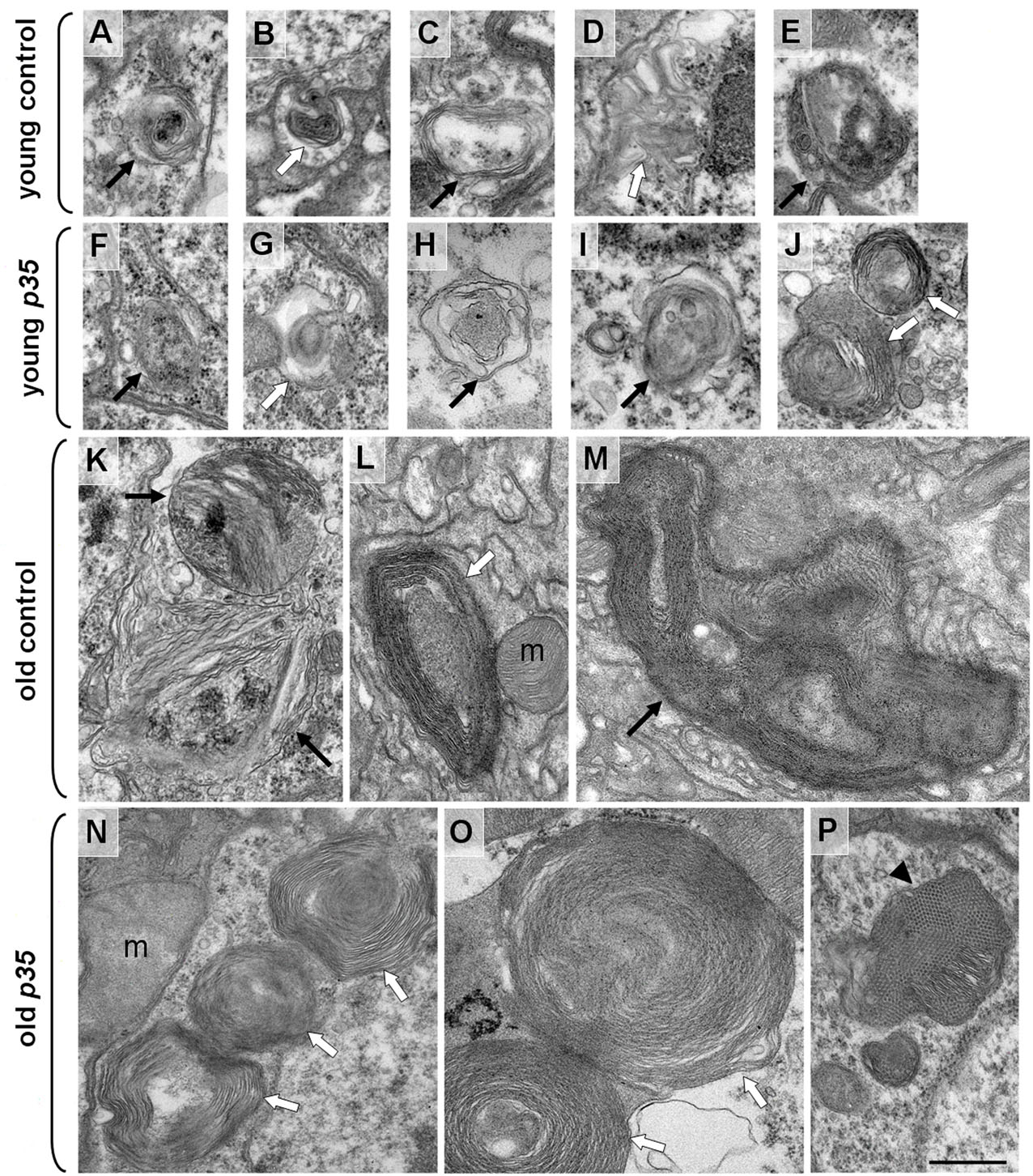
To try to identify cellular and molecular changes that might underlie the MB defects in p35 mutants, we extended our analysis to the ultrastructural level using electron microscopy (EM). We found that flies aged to 80% population mortality, both p35 mutants (45-days old) and wild-type controls (60-days old), accumulated organelles that are characteristic of the lysosomal-autophagosomal degradation pathway, including autophagosomes (APGs), multilamellar bodies (MLBs) and autophagolysosomes (APGLs) (Fig. 4). These were observed in young flies of either genotype at much lower frequencies (n∼100 cells analyzed). On average, MLBs and APGs were smaller and less mature then organelles in old flies of both genotypes (supplementary material Fig. S1). Organelles of the lysosomal-autophagosomal degradation pathway were detected in the soma as well as neurites in old neurons, and these structures were also detected in glia (supplementary material Fig. S2). We noted that cells containing two or more autophagosomal organelles were often clustered (Fig. 4D). Accumulation of large MLBs and APGs is consistent with previously published aging phenotypes in various cell types of 85- to 100-day-old flies (Miquel et al., 1974) and in a fly model of spinobulbar muscular atrophy (Pandey et al., 2007). Moreover, old p35 mutant neurons accumulated significantly more organelles than their physiologically matched controls (Fig. 4G).
Fig. 4.

Accumulation of autophagosomal organelles in old p35 mutant brains. Electron micrographs of MB neurons from control wild-type and p35 mutant brains at different time points. (A,B) Representative images of a middle-aged wild-type neuron (A) and young p35 mutant neuron (B). Boundaries of individual cells are highlighted by colored dotted lines. N, nucleus. (C,D) Age-dependent accumulation of autophagosomes (arrow) in both controls (C) and p35 mutants (D). Note the clusters of adjacent cells containing multiple ATG organelles. C′–F′ are high-magnification images of organelles highlighted in C–F, respectively. (E,F) Characteristic appearance of age-dependent multi-molecular depositions (arrowhead) in controls (E) and p35 mutants (F). Depositions with hexagonal symmetry were often located in close proximity to multilamellar structures. Scale bar: 500 nm. (G) Increase in the average number of autophagic organelles (AO) per neuron in old p35 mutants. Because APGs containing multilamellar structures represent the vast majority of all autophagic organelles that we observed in this project, we combined data for APGs, MLBs and APGLs. A minimum of 20 neurons per brain were analyzed. Values are mean ± s.e.m. (n≥3 brains). c, control.
We also observed accumulation of hexagonally-packed ‘crystalline-like’ depositions of electron-dense material in glia and neuronal soma in the brains of both aged wild-type and p35 mutant flies (Fig. 4E,F; supplementary material Fig. S3). The single deposition unit was 15–17 nm in diameter and had a cylindrical appearance. These depositions were observed in the cytoplasm as well as enclosed in single or double membranes, and sometimes those depositions were observed in late APGs together with other cytoplasmic and lamellar materials (Fig. 4; supplementary material Fig. S3). Such structures were observed at similar frequencies in old mutant and control neurons, implying that the recognition of depositions and their engulfment into APGs were not blocked by the p35 mutation. Because, by contrast, the overall frequency of autophagosomal organelles was notably increased in the mutant, it seems probable that the autophagic process in p35 mutants was disrupted at some later stage, although testing this will require further study. Interestingly, intracellular depositions of similar size and appearance are observed in young Drosophila neurons when autophagy is suppressed (Simonsen et al., 2008). Accumulation of aggregated depositions and APGs are another common feature noted in many neurodegenerative diseases, such as Parkinson’s disease and AD (Zhu et al., 2003; Nixon et al., 2005; Bandyopadhyay and Cuervo, 2007; Shacka et al., 2008), as well as in animal models, including a Drosophila model of spinobulbar muscular atrophy (Pandey et al., 2007).
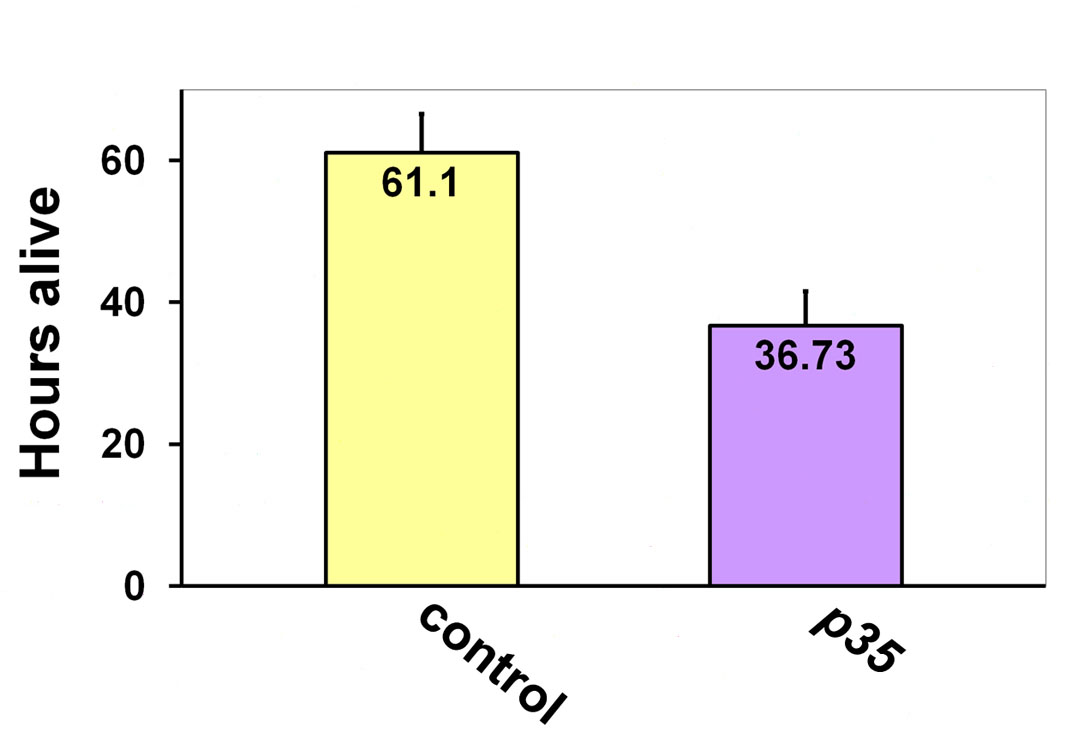
To further assess involvement of Drosophila Cdk5 in autophagy regulation, we performed a starvation test, because reduced survival under starvation stress is a common indicator of compromised autophagy (Simonsen et al., 2008; Chang and Neufeld, 2010) (supplementary material Fig. S4). The median survival time of p35 null mutants was reduced by 37% upon starvation. These data are also consistent with Drosophila Cdk5 potentially having a role in the autophagic process.
Altogether, ultrastructural analysis showed that deletion of the p35 gene caused multiple intracellular changes that resembled the accelerated and enhanced appearance of cellular markers of brain aging, including alterations and perhaps impairment of the autophagy process.
Morphological phenotype of aging p35 mutant and wild-type MB projection neurons
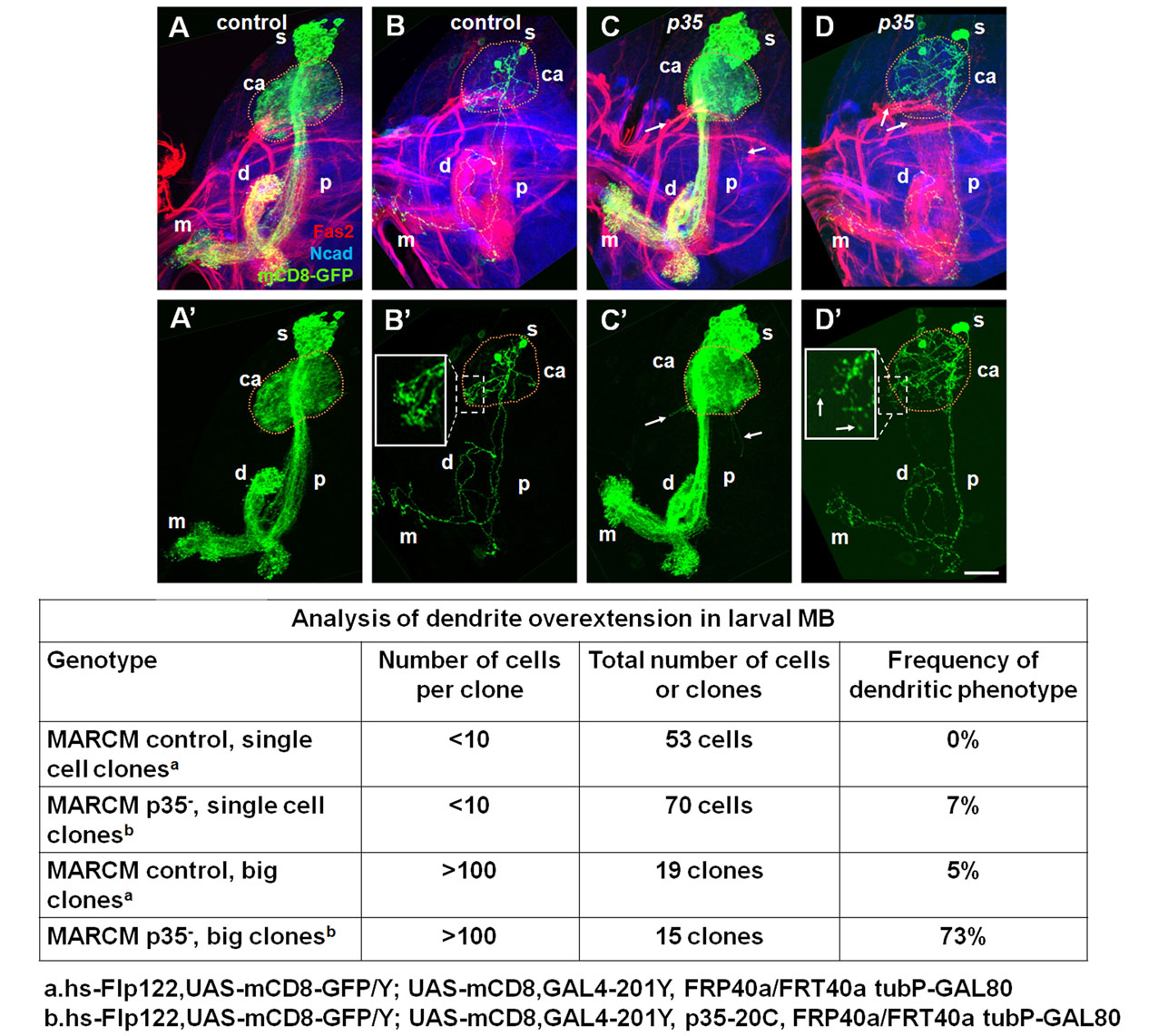
To gain further insight into the p35 phenotype in MB neurons, we examined the morphology of these neurons in detail (supplementary material Figs S5, S6). Visualizing single wild-type or mutant MB neurons in young adults by MARCM labeling with mCD8-GFP (Lee and Luo, 1999) revealed that the vast majority of mutant cells (93%: n=71) initially developed with a wild-type morphology (Lee, T. et al., 1999). The dendritic arbor branched appropriately into the calyx, and the axon branches projected in the typical lobar arrangement, both in small mutant clones and in wholly mutant animals. A morphological defect was observed in a small fraction of neurons (7%), with dendrites projecting out beyond the boundaries of the MB along a Fasciclin-2-positive nerve that normally enters the calyx. These ‘overshooting’ dendrites also lacked a ‘claw-like’ termination that is found at the tips of wild-type dendrites. Other aspects of overall MB neuron morphology seemed normal in the mutant, and the presence of a small number of mutant neurons did not affect the lifespan of the animals (supplementary material Fig. S6).
As flies aged, we began to detect fragmentation of the axons and dendrites of MB projection neurons both in wild-type and p35 mutant neurons using MARCM mosaics (Fig. 5). Age-related morphological changes in neurons of both genotypes were very similar and largely resembled changes that occur during developmental pruning of γ-neurons at the pupal stage (Fig. 5). These included fragmentation of dendrites and distal axonal lobes, clearance of degenerating neurites and protection of the peduncle from disassembly (Lee et al., 2000). In contrast to developmental pruning, however, in age-related degeneration, we observed breaks in the proximal neurite that occurred when the peduncle was intact and the remains of distal neurites were present. We conclude that a proportion of p35 mutant MB neurons, like wild type, undergo age-dependent fragmentation and loss.
Fig. 5.

Age-related neuronal degeneration. MARCM clones were analyzed in old flies above 80% of mortality of population. Images are Z-stack projections of sequential confocal slices covering the whole MB area. Brain midline is on the left, dorsal side is up. (A,A′) The first sign of degeneration in controls is when the axonal medial (m) lobe of γ-neuron appears thin and partially fragmented (arrowhead). (B,B′) Control γ-neuron with breaks in the proximal neurite (white dashed arrow) and completely missing axonal lobes (white dotted line). Note the end of the existing axon correlated with distal peduncle border (white hollow arrow). (C,C′) p35 mutant MB neuron has multiple breaks in the proximal neurite (dashed arrow), and the dorsal and medial axonal lobes (arrows). Note that the peduncle part of the proximal axon exhibits a reduced amount of fragmentation. Present parts of the proximal neurite (pn), peduncle (p), dorsal (d) and medial (m) axonal lobes are highlighted with a single dashed line. Missing links of neurites are highlighted with fine white dotted line. Other non-MB neuronal types labeled with GFP are marked with a star. Scale bar: 30 μm.
Gross swelling of the proximal axon in MB neurons of aged p35 mutants
p35 mutant MB projection neurons often displayed one unique morphological defect in aged adults: gross swelling of the proximal region of the axon (Fig. 6). These swellings could be substantially larger than the diameter of the wild-type axon, and sometimes contained large, membrane-containing organelles. Swellings were observed in ∼17% of mutant neurons (n=71) and were found in all three classes of MB projection neurons (α, β and γ), but were never observed in wild-type neurons at any age. Axonal swellings and transport organelle accumulations are commonly observed in mammalian models of AD (Stokin et al., 2008), and ‘traffic jams’ have been reported in Drosophila axons in the context of mutations that disrupt axonal transport (Gunawardena and Goldstein, 2001). We concluded that deletion of the p35 gene did not significantly affect the morphology of neurons in young adults but caused a characteristic axonal defect with aging.
Fig. 6.
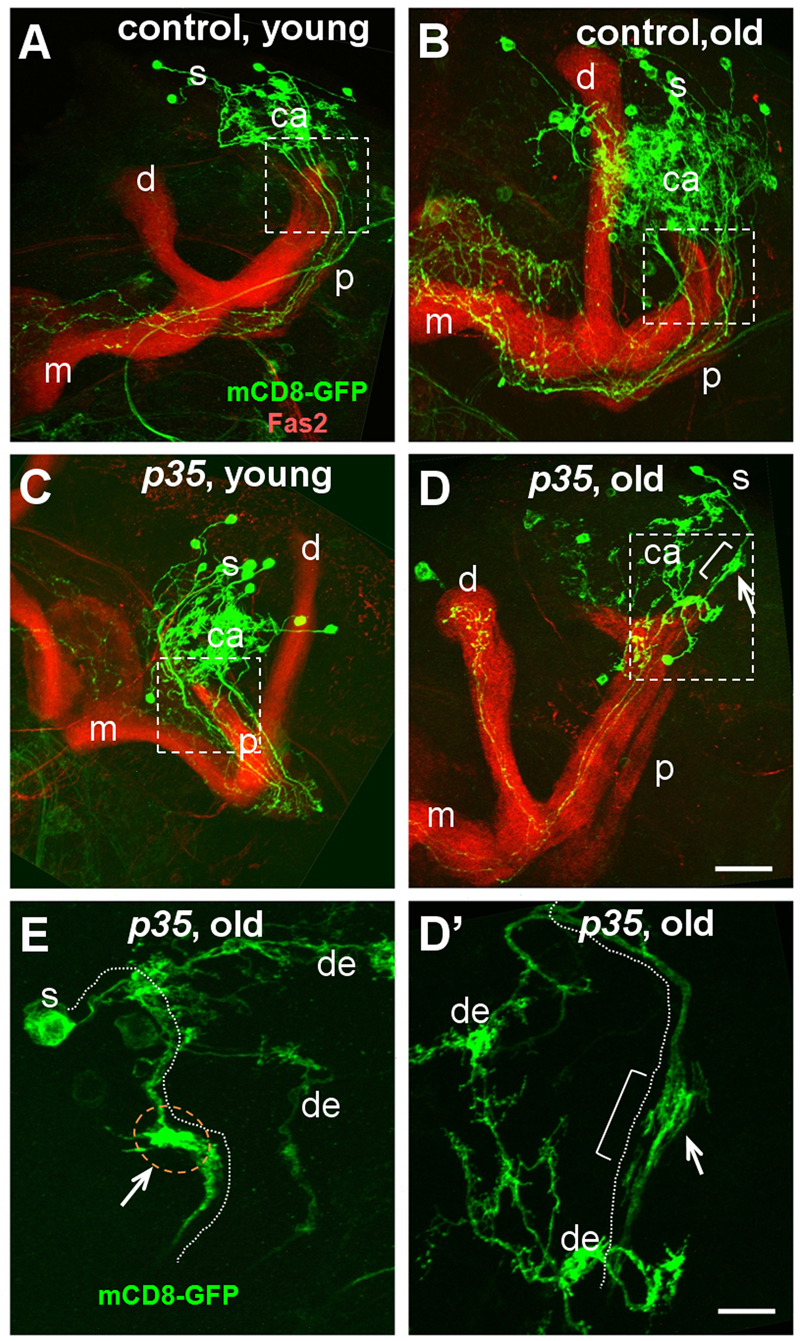
Age-dependent changes in morphology of p35 mutant neurons. MARCM clones were analyzed in young (5-day old) and old flies above 80% of mortality in population. Images are Z-stack projections of the whole MB area. For orientation and labeling details, see legend to Fig. 5. Young (A) and old (B) controls and young p35 mutants (C) have normal morphology. Old p35 mutants (D) develop swelling (white arrow and bracket) in the area of AIS compartment (white rectangle). Scale bar: 30 μm. (D′,E) Higher magnification showing details of swellings. (D′) Boxed area in D; note bright GFP clog. (E) Example of axonal swelling from another brain. Position of the axon is highlighted by a white dotted line; dendrites are marked as ‘de’. Scale bar: 10 μm.
DISCUSSION
We have shown here that mutation of the gene encoding the Cdk5-activating subunit p35 causes adult-onset neurodegeneration of the MBs of the Drosophila central brain. As the mutant flies age, they develop domains of reduced tissue density and integrity in the MB that manifest as a vacuolar pathology in histological sections, and they display gross swelling of the proximal axon in a significant fraction of MB projection neurons. In addition to these mutant-specific phenotypes, we also observe acceleration of a variety of phenotypes that are characteristic of normal aging, including axonal fragmentation, accumulation of lysosomal and/or autophagosomal organelles, development of ‘crystalline’ depositions of endogenous cellular materials, susceptibility to necrosis, and a low level of generalized neuronal degeneration throughout the brain. All of these Drosophila p35 phenotypes are reminiscent of the phenotypes observed in Cdk5-associated neurodegenerative diseases in humans (Nixon et al., 2005; Pimplikar et al., 2010), suggesting that the fly might offer insights into the molecular mechanism of human disease.
Given the extensive literature linking Cdk5 to neuronal apoptosis in mammals (Shelton and Johnson, 2004; Zhang et al., 2010), it was surprising that we did not detect an increase in apoptosis in the brain during aging of p35 mutants. Indeed, the level of apoptosis was slightly decreased in middle-aged mutants in comparison with controls. Formally, we cannot rule out the possibility of an increase in the rate of phagocytic clearance that exactly balances an undetected increase in apoptosis, but this seems unlikely. One possibility is that a different form of cell death, such as necrosis, which was previously reported as Cdk5-dependent under acute stress (Maccioni et al., 2001), and/or autophagy (Denton et al., 2009), is primarily responsible for cell loss in the old p35 brain. Alternatively, it is possible that tissue loss in the degenerating fly brain arises from disassembly of neurites rather than neuronal death (Ayaz et al., 2008), and that the behavioral deficits in Cdk5-associated neurodegeneration (Connell-Crowley et al., 2007) arise largely from neuronal disconnection and improper circuit function, and not from cell death.
We did observe an increase in susceptibility to necrosis in MB neurons as mutant flies aged. Because the necrosis assay requires dissection and transient culturing of the brain, and thus places the tissue under enormous stress, we cannot distinguish between cells that were already necrotic prior to dissection and those that initiate the necrotic process during the procedure. We therefore interpret the necrosis assay as quantifying the fragility of the cells, and their susceptibility to necrosis, rather than as a direct measure of the number of necrotic cells present in vivo. It is striking that, in wild-type flies, increases in apoptosis and necrosis with aging are temporally well correlated, whereas, in p35 nulls, the age-related rise in necrosis precedes that in apoptosis: necrosis tracks with or ahead of the mortality curve, whereas apoptosis essentially follows the chronological age of the flies as it does in wild type. This relative acceleration of the increase in necrosis lends further weight to the notion that stress-related processes, rather than apoptosis, are apt to be linked to Cdk5-associated mortality.
Ultrastructural analysis of the p35 mutant revealed aggregated depositions of endogenous material, as well as enhanced accumulation of APGs. We cannot distinguish whether the accumulation of autophagic organelles reflects a decrease in the efficacy of turnover of materials by the autophagosomal pathway, or whether the pathway is being overwhelmed by an increase in the bulk of material that enters it. We note, however, that the observation of a similar frequency of both free and engulfed ‘crystalline’ depositions in mutants and controls hints that induction of the autophagic process is not seriously disturbed in p35 mutants. Either way, it is possible that the accumulation of autophagic organelles could contribute to cell death. Incomplete or compromised autophagocytosis can stimulate necrosis in other systems (Samara et al., 2008), particularly in contexts in which the apoptotic pathway is not accessible (Ullman et al., 2008). Other models are possible, however, and additional experiments will be required to investigate this issue. Moreover, increased apoptosis, necrosis and accumulation of autophagic organelles have been observed in AD brains (Velez-Pardo et al., 2001; Boland et al., 2008; Vandenabeele et al., 2010), in which Cdk5 activity is thought to be significantly increased (Lee, K. Y. et al., 1999; Patrick et al., 1999). So, it could be that Cdk5 acts at multiple steps, and in multiple ways, in the initiation and progression of autophagy (Furuya et al., 2010; Wong et al., 2011).
It is interesting to compare the Drosophila p35 null phenotypes with the neurodegeneration observed in a conditional knockout of mouse Cdk5. Our observation of enhanced propensity for necrosis in neurons lacking Cdk5 activity could plausibly account for the induction of the massive inflammatory response that was reported in the mouse knockout. In those experiments, however, a major causative role in degeneration was ascribed to the miswiring of neurons due to cell migration defects (Ohshima et al., 1996; Takahashi et al., 2010). In the fly brain, by contrast, neuronal migration during development is minimal, and neuronal morphology is essentially wild type at the single-cell level in p35 null flies (Fig. 6; supplementary material Fig. S4). This shows that age-related neurodegeneration occurs in the Drosophila model even in the absence of overt defects in neuronal morphogenesis.
Our most striking finding was cell-autonomous swelling of the proximal axon of MB projection neurons in aged p35 mutants. Along with tissue deterioration per se, this was the only phenotype that was specific to the mutant animals, and never observed in normal aging. Axonal swellings are a common feature of many neurodegenerative diseases (Nixon et al., 2005), and are often thought to arise from defects in axon transport (Stokin et al., 2008). Consistent with this, in addition to its prominent activity phosphorylating the microtubule-associated protein tau, Cdk5 is known to regulate microtubule-dependent motors, particularly cytoplasmic dynein (Niethammer et al., 2000), and proteins involved in the development and maintenance of microtubule organization (Xie et al., 2003). It is also remarkable that the axonal swellings in the mutant were not distributed uniformly throughout the axon. Rather, they were concentrated in a proximal portion of the MB axon that we have elsewhere identified as the axon initial segment (AIS) (Trunova et al., 2011). This is a highly specialized portion of the axon where action potentials initiate, and we have shown that the structure and maintenance of the AIS are exquisitely sensitive to Cdk5 activity. Disrupting the structure of this portion of the axon would be expected to have profound effects on the physiological properties of the neuron and its subcellular organization, and perhaps, therefore, contribute to instability of the structure of the neuron (Trunova et al., 2011).
Several of the phenotypes that we characterized in p35 mutants, such as sensitivity to necrosis, axonal fragmentation and accumulation of APGs, are also observed in normal aging. What, then, is their relationship to the localized neurodegeneration we observe in the MB? Presumably, the age-related pathology we observe throughout the brain cannot be wholly identical to the Cdk5-associated processes occurring in the MB, because the large-scale tissue disruption and accumulated holes we detect in the MB of the mutant do not occur in the wild type at any age. It might be, however, that these age-related processes predispose or sensitize the neuron to other insults that are linked more directly to the structural disruption of the cell, or else perhaps they accelerate the process of neuronal disassembly. Either way, we imagine that altered Cdk5 activity must produce another, more-specific, signal, perhaps one related to the MB axonal swellings, that cooperates with accelerated aging to produce frank, tissue-level degeneration. Whether such a dual role of Cdk5 occurs in humans and plays an analogous part in the initiation and progression of human neurodegenerative disease will be an important subject for future investigation.
In summary, the experiments reported here allow us to draw three crucial conclusions. First, Drosophila lacking p35 undergo adult-onset neurodegeneration, particularly of the MB in the central brain. Second, the degeneration we observe has many cellular and molecular features that resemble aspects of mammalian neurodegenerative disease. Third, those degeneration-associated processes can be separated into a subset that resemble an accelerated form of the natural aging process, and others that seem to be specific to degeneration per se. Future experiments will focus on identifying the direct molecular targets of Cdk5-p35 kinase that link it to each of the broad range of downstream processes that together make up neurodegeneration.
METHODS
Drosophila techniques
To compare lifespan among different genotypes, mutations were introduced into the same genetic background by consecutive crossing with the same balancing stocks. Wild-type control w− flies were used to saturate balancing stocks with X, 3rd and 4th chromosomes. To generate heteroallelic flies, mutant w−;p3520c females were crossed to w−;DfC2/Cy-GFP males. For genomic rescue, w−;p3520c females were crossed to w−;p3520c;P[w+;Tn p35+]R157 males. All flies were raised on standard media at 25°C and kept at low-to-medium density. Unless specified, w− flies were used as the wild-type controls. For all aging experiments, 3-day-old healthy flies were set up as ten females and 14 males per vial and transferred to fresh food every 3 days, but assays were performed only with males. Each experiment started with 120–140 males.
MARCM genotypes
hs-Flp122,UAS-mCD8-GFP; UAS-mCD8,GAL4-201Y, FRT40A/FRT40a tubP-GAL80 crossed to w; FRT40A males (Lee and Luo, 1999); hs-Flp122,UAS-mCD8-GFP/Y; UAS-mCD8,GAL4-201Y, p3520c, FRT40A/FRT40a tubP-GAL80 crossed to w; p3520c,FRT40A males. MARCM clones were generated by heat shocking first instar larvae at 37°C for 5 minutes in a water bath.
Starvation experiment
We chose strict starvation conditions (Simonsen et al., 2007) to test survivorship of p3520c/DfC2 mutant and w− control flies on wet starvation condition/no calories diet. Flies were allowed to develop at low density conditions on regular food and young (3-to 5-days old) males were subjected to starvation. 14 males per tube were placed on small, equally sized paper pads that had been wet with drinking water. Every 12 hours, water was replaced and survivorship was analyzed.
Transmission electron microscopy
Fly brains were prepared following standard protocols (Muhlig-Versen et al., 2005; Pandey et al., 2007). Before Epon polymerization, heads were oriented individually with the flat, dorsoposterior side facing down. To simplify morphological analysis, blocks were pre-cut and re-oriented to bring the dorsal side of the head (MB area) closest to the cutting blade. For histochemical analysis, 1-μm sections were collected sequentially every 15–20 μm. For ultrastructural analysis, brains were sectioned until the area of the MB was reached (50–75 μm) and 0.1-μm ultrathin sections were collected. The MB was identified based on characteristic cortex and neuropil morphology. All neural cells from EM grid sections positioned in the MB cortex were included in the analysis. 30–35 neurons per brain from three to four heads per condition were analyzed for the presence of macromolecular depositions and lysosomal-autophagosomal organelles. A cell was included in the analysis if it contained a well-defined nucleus. Sections were examined with a SEOL 200CX electron microscope at 120 kV and analyzed with AmtV601 software.
Autophagic organelles were characterized following published classification (Nixon, 2007; Boland et al., 2008; Lee et al., 2010). Additionally, multivesicular bodies also containing cytoplasmic materials were classified as ATGs; single- and double-membrane-limited depositions were classified as MLBs.
Immunochemistry and confocal microscopy
Immunostaining of whole-mount adult brains was described elsewhere (Lee, T. et al., 1999; Trunova et al., 2011). Primary antibodies used were: mouse anti-Fas2 and anti-Repo, rat anti-N-Cad and anti-Elav from the Developmental Studies Hybridoma Bank, and chicken anti-GFP (Aves). Secondary antibodies used were: goat anti-mouse and anti-rat conjugated with A488, A568, A633 fluorochromes (Invitrogen), and anti-chicken FITC (Aves). Filamentous actin was detected with Phalloidin-A658 and -A635 (Invitrogen). Samples were imaged on a Zeiss LSM 510 confocal microscope; scoring of cell death markers was performed with 3D application of AimImageBrowser (Zeiss).
TRANSLATIONAL IMPACT.
Clinical issue
In humans, the protein kinase Cdk5 plays a central role in the progression of many forms of neurodegenerative disease, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis and others, and this is reflected in mouse models of Cdk5 gain- and loss-of-function. Cdk5 has been linked molecularly to a wide array of cellular processes, many of which could plausibly be involved in the process of neurodegeneration. However, it has been extremely difficult to discriminate the relative roles of different pathological processes to the eventual outcome of frank degeneration in complex mammalian models. Indeed, it even remains unclear whether Cdk5 has a protective or damaging effect at individual steps of disease onset and progression.
Results
The results of this paper show that inactivating Cdk5 in Drosophila, by mutation of the gene encoding its obligate activating subunit, p35, causes an adult-onset neurodegenerative syndrome that bears striking similarity to features of mammalian neurodegenerative disease. These include reduced lifespan with progressive motor decline, selective atrophy of a brain region dedicated to learning and memory, increased neuronal propensity to necrosis, accumulation of autophagosomal organelles and aggregated cellular material, axonal swellings, and axonal and dendritic fragmentation. Some of these phenotypes resemble an accelerated version of the normal aging process, whereas others are specific for degeneration per se. Remarkably, enhanced neuronal apoptosis was not among the phenotypes observed.
Implications and future directions
These data suggest that analysis of neurodegeneration in Drosophila lacking Cdk5 activity will provide a valuable model for investigating the molecular and cellular events underlying mammalian neurodegenerative disease. Given the simplicity of the fly brain, and the possibility to both manipulate it genetically in vivo and interrogate its structure and function at cellular and subcellular resolution, this model offers unique opportunities to separate the contributions of the many cellular processes that are correlated with neurodegeneration. For example, the finding that loss of Cdk5 causes frank neurodegeneration with tissue atrophy even in the absence of any detectable effect on apoptosis raises the question of whether treatments aimed at modulating the apoptotic process will be effective in modifying disease course. Moreover, the data indicate that it might be important for drug discovery strategies to maintain Cdk5 activity at wild-type levels, rather than activating or inactivating it excessively. Finally, the observation that only selected degeneration-associated Cdk5-dependent processes mimic normal aging suggests that it will be possible to use this model to determine the contribution of aging to the overall process of neurodegeneration in this system.
Apoptosis staining
TUNEL staining was performed following previously described protocol (Denton et al., 2009) with modifications. Cell identity was determined by colocalization with transcription factor Repo for glia or Elav for neurons. In the case of strong TUNEL signal (diameter ≥5 μm) we often were not able to detect cell-type-specific signal and classified those cells as not identifiable. Signal from cells underlying the stomach canal were not included. Quantification was done manually using Zeiss software for 3D imaging; only well distinguishable individual TUNEL-positive cells (≥2 μm) were analyzed; one central brain hemisphere from each brain was included; samples were collected from three independent aging crosses; six to eight heads per condition were analyzed.
Necrosis staining
Whole-brain staining for necrosis was adapted from cell culture protocols (http://cyto.mednet.ucla.edu/protocols/simultta.htm). Briefly, brains were quickly dissected in standard saline for neurophysiology (Jan and Jan, 1976) at room temperature, transferred to small Petri dishes (1 ml) and incubated with Hoechst (HO; Invitrogen) for 15 minutes (1 μm/ml in saline for neurophysiology). Brains were then transferred to 4°C cold media with PI solution (Invitrogen; 1 μm/ml in saline for neurophysiology) and incubated for 5 minutes; after that PI was immediately washed out and samples were prepared for confocal imaging. After PI staining, and until imaging, brains and slides were kept on ice. Preparation time altogether was less than 30 minutes. The area of MB cortex analyzed was to the depth of 15–20 μm inside the tissue to monitor PI and HO penetration. Stacks of five 1-μm-thick optic slices located 10- to 15-μm under the brain surface were chosen for analysis.
We focused our analysis only on the MB cortex owing to a high number of PI-positive cells per brain produced by this method. MB cortex was chosen because it is easily identifiable based on trachea branching pattern that leaves the dorsal side of MB area clear of big tracheal branches. In general, PI-positive nuclei were randomly distributed through the brain and we did not observe any increase in MB area. Scoring was performed manually using AimImageBrowser. One optic field covering ≥100 cells per central brain hemisphere from each brain was included; samples were collected from two to three independent aging crosses. Five to seven heads per condition were analyzed.
Supplementary Material
Acknowledgments
We thank all the members of our lab for their advice and assistance with these experiments, particularly Ranjini Prithviraj for much help and many useful discussions, and Irina Kuzina and Jamie Wagner for outstanding technical assistance. We also thank Chi-Hon Lee for much useful advice, Stephen Wincovitch and the NHGRI Imaging Facility for assistance with fluorescence microscopy, and Susan Cheng, Rita Azzam and Virginia Croker of the NINDS Electron Microscopy Core Facility, as well as Tai Min and Karen Chang, for their invaluable assistance with the EM analyses. For critical reading of the manuscript, we thank Craig Blackstone, Zu-Hang Sheng, Andrew Singleton and Richard Youle. We thank Liqun Luo, Chi-hon Lee and Eric Baehrecke for sharing protocols and reagents.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
FUNDING
This work was supported by the Basic Neuroscience Program of the Intramural Research Program, NINDS, National Institutes of Health (NIH) [Z01 NS003106].
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.008847/-/DC1
REFERENCES
- Ayaz D., Leyssen M., Koch M., Yan J., Srahna M., Sheeba V., Fogle K. J., Holmes T. C., Hassan B. A. (2008). Axonal injury and regeneration in the adult brain of Drosophila. J. Neurosci. 28, 6010–6021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay U., Cuervo A. M. (2007). Chaperone-mediated autophagy in aging and neurodegeneration: lessons from alpha-synuclein. Exp. Gerontol. 42, 120–128 [DOI] [PubMed] [Google Scholar]
- Boland B., Kumar A., Lee S., Platt F. M., Wegiel J., Yu W. H., Nixon R. A. (2008). Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 28, 6926–6937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. Y., Neufeld T. P. (2010). Autophagy takes flight in Drosophila. FEBS Lett. 584, 1342–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell-Crowley L., Le Gall M., Vo D. J., Giniger E. (2000). The cyclin-dependent kinase Cdk5 controls multiple aspects of axon patterning in vivo. Curr. Biol. 10, 599–602 [DOI] [PubMed] [Google Scholar]
- Connell-Crowley L., Vo D., Luke L., Giniger E. (2007). Drosophila lacking the Cdk5 activator, p35, display defective axon guidance, age-dependent behavioral deficits and reduced lifespan. Mech. Dev. 124, 341–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D., Shravage B., Simin R., Mills K., Berry D. L., Baehrecke E. H., Kumar S. (2009). Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr. Biol. 19, 1741–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya T., Kim M., Lipinski M., Li J., Kim D., Lu T., Shen Y., Rameh L., Yankner B., Tsai L. H., et al. (2010). Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol. Cell 38, 500–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S., Goldstein L. S. (2001). Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 32, 389–401 [DOI] [PubMed] [Google Scholar]
- Ip N. Y., Tsai L. H. (2008). Cyclin Dependent Kinase 5 (Cdk5). New York, NY: Springer Science & Business Media [Google Scholar]
- Jan L. Y., Jan Y. N. (1976). Properties of the larval neuromuscular junction in Drosophila melanogaster. J. Physiol. 262, 189–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S., Gage F. H., Eisch A. J., Lagace D. C. (2009). Making a neuron: Cdk5 in embryonic and adult neurogenesis. Trends Neurosci. 32, 575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissler A. E., Pettersson N., Frolich A., Sigrist S. J., Suter B. (2009). Drosophila cdk5 is needed for locomotive behavior and NMJ elaboration, but seems dispensable for synaptic transmission. Dev. Neurobiol. 69, 365–377 [DOI] [PubMed] [Google Scholar]
- Ko J., Humbert S., Bronson R. T., Takahashi S., Kulkarni A. B., Li E., Tsai L. H. (2001). p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J. Neurosci. 21, 6758–6771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J. Y., Koga H., Kawaguchi Y., Tang W., Wong E., Gao Y. S., Pandey U. B., Kaushik S., Tresse E., Lu J., et al. (2010). HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. Y., Clark A. W., Rosales J. L., Chapman K., Fung T., Johnston R. N. (1999). Elevated neuronal Cdc2-like kinase activity in the Alzheimer disease brain. Neurosci. Res. 34, 21–29 [DOI] [PubMed] [Google Scholar]
- Lee M. S., Tsai L. H. (2003). Cdk5: one of the links between senile plaques and neurofibrillary tangles? J. Alzheimers Dis. 5, 127–137 [DOI] [PubMed] [Google Scholar]
- Lee T., Luo L. (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461 [DOI] [PubMed] [Google Scholar]
- Lee T., Lee A., Luo L. (1999). Development of the Drosophila mushroom bodies: sequential generation of three distinct types of neurons from a neuroblast. Development 126, 4065–4076 [DOI] [PubMed] [Google Scholar]
- Lee T., Marticke S., Sung C., Robinow S., Luo L. (2000). Cell-autonomous requirement of the USP/EcR-B ecdysone receptor for mushroom body neuronal remodeling in Drosophila. Neuron 28, 807–818 [DOI] [PubMed] [Google Scholar]
- Maccioni R. B., Munoz J. P., Barbeito L. (2001). The molecular bases of Alzheimer’s disease and other neurodegenerative disorders. Arch. Med. Res. 32, 367–381 [DOI] [PubMed] [Google Scholar]
- Meuer K., Suppanz I. E., Lingor P., Planchamp V., Goricke B., Fichtner L., Braus G. H., Dietz G. P., Jakobs S., Bahr M., et al. (2007). Cyclin-dependent kinase 5 is an upstream regulator of mitochondrial fission during neuronal apoptosis. Cell Death Differ. 14, 651–661 [DOI] [PubMed] [Google Scholar]
- Miquel J., Tappel A. L., Dillard C. J., Herman M. M., Bensch K. G. (1974). Fluorescent products and lysosomal components in aging Drosophila melanogaster. J. Gerontol. 29, 622–637 [DOI] [PubMed] [Google Scholar]
- Muhlig-Versen M., da Cruz A. B., Tschape J. A., Moser M., Buttner R., Athenstaedt K., Glynn P., Kretzschmar D. (2005). Loss of Swiss cheese/neuropathy target esterase activity causes disruption of phosphatidylcholine homeostasis and neuronal and glial death in adult Drosophila. J. Neurosci. 25, 2865–2873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W. E., Eckert A., Kurz C., Eckert G. P., Leuner K. (2010). Mitochondrial dysfunction: common final pathway in brain aging and Alzheimer’s disease-therapeutic aspects. Mol. Neurobiol. 41, 159–171 [DOI] [PubMed] [Google Scholar]
- Niethammer M., Smith D. S., Ayala R., Peng J., Ko J., Lee M. S., Morabito M., Tsai L. H. (2000). NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron 28, 697–711 [DOI] [PubMed] [Google Scholar]
- Nikolic M., Dudek H., Kwon Y. T., Ramos Y. F., Tsai L. H. (1996). The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 10, 816–825 [DOI] [PubMed] [Google Scholar]
- Nixon R. A. (2007). Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 120, 4081–4091 [DOI] [PubMed] [Google Scholar]
- Nixon R. A., Wegiel J., Kumar A., Yu W. H., Peterhoff C., Cataldo A., Cuervo A. M. (2005). Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 64, 113–122 [DOI] [PubMed] [Google Scholar]
- Noble W., Olm V., Takata K., Casey E., Mary O., Meyerson J., Gaynor K., LaFrancois J., Wang L., Kondo T., et al. (2003). Cdk5 is a key factor in tau aggregation and tangle formation in vivo. Neuron 38, 555–565 [DOI] [PubMed] [Google Scholar]
- Ohshima T., Ward J. M., Huh C. G., Longenecker G., Veeranna Pant H. C., Brady R. O., Martin L. J., Kulkarni A. B. (1996). Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc. Natl. Acad. Sci. USA 93, 11173–11178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop J. J., Mucke L. (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci 13, 812–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey U. B., Nie Z., Batlevi Y., McCray B. A., Ritson G. P., Nedelsky N. B., Schwartz S. L., DiProspero N. A., Knight M. A., Schuldiner O., et al. (2007). HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447, 859–863 [DOI] [PubMed] [Google Scholar]
- Patrick G. N., Zukerberg L., Nikolic M., de la Monte S., Dikkes P., Tsai L. H. (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622 [DOI] [PubMed] [Google Scholar]
- Pimplikar S. W., Nixon R. A., Robakis N. K., Shen J., Tsai L. H. (2010). Amyloid-independent mechanisms in Alzheimer’s disease pathogenesis. J. Neurosci. 30, 14946–14954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu D., Rashidian J., Mount M. P., Aleyasin H., Parsanejad M., Lira A., Haque E., Zhang Y., Callaghan S., Daigle M., et al. (2007). Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron 55, 37–52 [DOI] [PubMed] [Google Scholar]
- Samara C., Syntichaki P., Tavernarakis N. (2008). Autophagy is required for necrotic cell death in Caenorhabditis elegans. Cell Death Differ. 15, 105–112 [DOI] [PubMed] [Google Scholar]
- Shacka J. J., Roth K. A., Zhang J. (2008). The autophagy-lysosomal degradation pathway: role in neurodegenerative disease and therapy. Front. Biosci. 13, 718–736 [DOI] [PubMed] [Google Scholar]
- Shelton S. B., Johnson G. V. (2004). Cyclin-dependent kinase-5 in neurodegeneration. J. Neurochem. 88, 1313–1326 [DOI] [PubMed] [Google Scholar]
- Simonsen A., Cumming R. C., Finley K. D. (2007). Linking lysosomal trafficking defects with changes in aging and stress response in Drosophila. Autophagy 3, 499–501 [DOI] [PubMed] [Google Scholar]
- Simonsen A., Cumming R. C., Brech A., Isakson P., Schubert D. R., Finley K. D. (2008). Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184 [DOI] [PubMed] [Google Scholar]
- Stokin G. B., Almenar-Queralt A., Gunawardena S., Rodrigues E. M., Falzone T., Kim J., Lillo C., Mount S. L., Roberts E. A., McGowan E., et al. (2008). Amyloid precursor protein-induced axonopathies are independent of amyloid-beta peptides. Hum. Mol. Genet. 17, 3474–3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S., Ohshima T., Hirasawa M., Pareek T. K., Bugge T. H., Morozov A., Fujieda K., Brady R. O., Kulkarni A. B. (2010). Conditional deletion of neuronal cyclin-dependent kinase 5 in developing forebrain results in microglial activation and neurodegeneration. Am. J. Pathol. 176, 320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trunova S., Baek B., Giniger E. (2011). Cdk5 regulates the size of an axon initial segment-like compartment in mushroom body neurons of the Drosophila central brain. J. Neurosci. 31, 10451–10462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman E., Fan Y., Stawowczyk M., Chen H. M., Yue Z., Zong W. X. (2008). Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ. 15, 422–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 [DOI] [PubMed] [Google Scholar]
- Velez-Pardo C., Arroyave S. T., Lopera F., Castano A. D., Jimenez Del, Rio M. (2001). Ultrastructure evidence of necrotic neural cell death in familial Alzheimer’s disease brains bearing presenilin-1 E280A mutation. J. Alzheimers Dis. 3, 409–415 [DOI] [PubMed] [Google Scholar]
- Wittmann C. W., Wszolek M. F., Shulman J. M., Salvaterra P. M., Lewis J., Hutton M., Feany M. B. (2001). Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714 [DOI] [PubMed] [Google Scholar]
- Wong A. S., Lee R. H., Cheung A. Y., Yeung P. K., Chung S. K., Cheung Z. H., Ip N. Y. (2011). Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson’s disease. Nat. Cell Biol. 13, 734. [DOI] [PubMed] [Google Scholar]
- Wong E., Cuervo A. M. (2010). Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 13, 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z., Sanada K., Samuels B. A., Shih H., Tsai L. H. (2003). Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell 114, 469–482 [DOI] [PubMed] [Google Scholar]
- Zhang J., Li H., Yabut O., Fitzpatrick H., D’Arcangelo G., Herrup K. (2010). Cdk5 suppresses the neuronal cell cycle by disrupting the E2F1-DP1 complex. J. Neurosci. 30, 5219–5228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M., Leung C. L., Liem R. K. (1998). Region-specific expression of cyclin-dependent kinase 5 (cdk5) and its activators, p35 and p39, in the developing and adult rat central nervous system. J. Neurobiol. 35, 141–159 [DOI] [PubMed] [Google Scholar]
- Zhu J. H., Guo F., Shelburne J., Watkins S., Chu C. T. (2003). Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol. 13, 473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}