

SUMMARY
Long QT syndrome (LQTS) is caused by functional alterations in cardiac ion channels and is associated with prolonged cardiac repolarization time and increased risk of ventricular arrhythmias. Inherited type 2 LQTS (LQT2) and drug-induced LQTS both result from altered function of the hERG channel. We investigated whether the electrophysiological characteristics of LQT2 can be recapitulated in vitro using induced pluripotent stem cell (iPSC) technology. Spontaneously beating cardiomyocytes were differentiated from two iPSC lines derived from an individual with LQT2 carrying the R176W mutation in the KCNH2 (HERG) gene. The individual had been asymptomatic except for occasional palpitations, but his sister and father had died suddenly at an early age. Electrophysiological properties of LQT2-specific cardiomyocytes were studied using microelectrode array and patch-clamp, and were compared with those of cardiomyocytes derived from control cells. The action potential duration of LQT2-specific cardiomyocytes was significantly longer than that of control cardiomyocytes, and the rapid delayed potassium channel (IKr) density of the LQT2 cardiomyocytes was significantly reduced. Additionally, LQT2-derived cardiac cells were more sensitive than controls to potentially arrhythmogenic drugs, including sotalol, and demonstrated arrhythmogenic electrical activity. Consistent with clinical observations, the LQT2 cardiomyocytes demonstrated a more pronounced inverse correlation between the beating rate and repolarization time compared with control cells. Prolonged action potential is present in LQT2-specific cardiomyocytes derived from a mutation carrier and arrhythmias can be triggered by a commonly used drug. Thus, the iPSC-derived, disease-specific cardiomyocytes could serve as an important platform to study pathophysiological mechanisms and drug sensitivity in LQT2.
INTRODUCTION
Signal propagation between cardiomyocytes is a very tightly regulated system. Mutations in ion channels involved in this system can cause electrical alterations that, in certain circumstances, such as during exercise or emotional stress, could trigger arrhythmias. Sudden death in a young healthy person can be the first devastating presentation of an underlying genetic disease. Long QT syndrome (LQTS) can either be genetic or acquired (e.g. drug-induced) in nature and is due to defective functioning of cardiac ion channels. LQTS is characterized by a prolonged cardiac repolarization phase resulting in a long QT interval in the surface electrocardiogram (ECG). The clinical symptoms of LQTS include palpitations, syncope, seizures and even sudden cardiac death. A special type of polymorphic ventricular tachycardia [torsade de pointes (TdP)] is associated with LQTS. Intriguingly, many mutation carriers are without any symptoms. A total of 12 congenital LQTS subtypes are presently known (Hedley et al., 2009). Two of these subtypes account for more than 90% of all genetically identified LQTS cases and both are due to defective functioning of potassium channels. LQTS type 1 (LQT1) is the most common subtype, resulting from mutations in the KCNQ1 gene, which encodes the α-subunit of the slow component of the delayed rectifier potassium current (IKs) channel (Chiang and Roden, 2000). Individuals with LQT1 typically have symptoms during exercise (Schwartz, 2001; Roden, 2008). LQTS type 2 (LQT2) is due to defective functioning of the α-subunit of the rapid delayed potassium channel (IKr), encoded by the KCNH2 [also known as human ether-a-go-go-related gene (HERG)] gene (Curran et al., 1995). Typically, individuals with LQT2 have clinical symptoms after abrupt auditory stimuli, often during sleep when the heart rate is slow (Schwartz, 2001; Roden, 2008). The acquired form of LQTS is also due to altered functioning of the same KCNH2 ion channel.
Induced pluripotent stem cell (iPSC) technology (Takahashi et al., 2007; Yu et al., 2007) has revolutionized research on genetic diseases. iPSCs can be generated from somatic cells of any individual and these pluripotent cells can be differentiated into the desired cell type. Accordingly, it is possible to create genotype-specific cell models with a correct functional intracellular environment. However, a major challenge in the iPSC approach is to reproduce the phenotype of the disease or the individual in iPSC-derived cells. An appropriate disease phenotype has been reproduced with iPSC technology from individuals with LQT1 (Moretti et al., 2010), LQT2 (Itzhaki et al., 2011; Matsa et al., 2011) and with Timothy syndrome (Yazawa et al., 2011). Considering non-cardiac disorders, a disease phenotype or pathogenesis in iPSC-derived models has been demonstrated only for a few neurological diseases (Lee and Studer, 2010) and for the LEOPARD syndrome (Carvajal-Vergara et al., 2010).
The penetrance of the clinical symptoms of LQTS is low and there is considerable variation in phenotypic expression even within families carrying the same mutation (Priori et al., 1999). In addition, it has been proposed that the population prevalence of milder LQTS mutations might be high, suggesting that the prevalence of latent or concealed LQTS, i.e. relatively asymptomatic individuals, is higher than currently anticipated (Marjamaa et al., 2009). For these reasons, LQTS is clinically very challenging. The previous LQT2 iPSC reports used individuals with severe symptoms and the severity of their symptoms was translated to the cardiomyocytes derived from the patient-specific iPSCs. However, a cell model for asymptomatic LQT2 mutation carriers would be valuable to help with clinical decisions about medical treatments and lifestyle restrictions for relatively asymptomatic patients.
A more thorough understanding of the molecular mechanisms underlying LQTS would be very helpful for the pharmaceutical industry. Drug-induced forms of LQTS often arise as a result of inhibition of the hERG channel gating, and is thus analogous to LQT2 (Hancox et al., 2008). These adverse cardiac effects have led to labeling restrictions on both cardiac and non-cardiac drugs as well as to withdrawal from the market (Roden, 2004). Currently, preclinical testing of new chemical entities (NCEs) for proarrhythmic potential relies on animal experiments and ectopic expression of individual ion channels in non-cardiac cells (Pollard et al., 2008). However, current models lack the relevant human physiological environment that might regulate or modify cellular responses (Pollard et al., 2008). Thus, some NCEs could be unnecessarily discarded in the preclinical phase, and others already in clinical use might in fact elicit adverse cardiac side effects. Functional cardiomyocytes derived from both symptomatic and, possibly even more importantly, asymptomatic LQTS individuals would add to and complement presently used models. These cell models would provide the relevant cellular milieu to study genetic and non-genetic interactions influencing the phenotype.
In the present study, we developed an in vitro cell model of LQT2. In contrast to the previous reports (Itzhaki et al., 2011; Matsa et al., 2011), we aimed at generating a model from cells of an individual with LQT2 without severe symptoms. To that end, iPSC lines were derived from a patient’s fibroblasts carrying a mutation for LQT2. Although there is a family history of overt LQTS, this individual was asymptomatic except for occasional palpitations and his 12-lead ECG exhibited a heart-rate-corrected QT time (QTc) of 437 ms. This model for LQT2 provides an important platform to study the pathophysiology of LQT2 and to evaluate adverse cardiac effects of drugs with the potential to prolong the QT interval.
RESULTS
Patient characteristics
A skin biopsy was obtained from a 61-year-old man with a missense mutation in KCNH2 causing an arginine-to-tryptophan substitution at position 176 (R176W, hERG-FinB; Fig. 1A). Although there is a family history of overt LQTS, this individual was asymptomatic except for occasional palpitations. His 12-lead ECG exhibited a QTc of 437 ms (Fig. 1B). His sister was diagnosed with LQTS having a QT(U)c interval of 550 ms (Fig. 1C), presence of palpitations and sudden death at the age of 32.
Fig. 1.

Mutation and ECG analysis. (A) Mutation analysis confirmed the hERG-FinB mutation in the LQT2 iPSC line, which gave altered DNA cleavage by the SmaI restriction enzyme (lower arrow). (B,C) ECG from leads V1–V3 of the index patient, with a QTc of 437 ms (B), and from the patient’s sister, with the presence of a U-wave following the T-wave; QT(U)c=550 ms.
Characterization of iPSCs
Fibroblasts from an individual with LQT2 were infected with retroviruses encoding for OCT4, SOX2, KLF4 and MYC to generate iPSCs. Morphologically, iPSC colonies exhibited characteristics similar to those of human embryonic stem cells (hESCs), with rounded shape and clear defined edges (Fig. 2A). Both LQT2-specific iPSC lines (UTA.00514.LQT2 and UTA.00525.LQT2) and control iPSC lines expressed endogenous pluripotent markers at the mRNA level, as shown by reverse transcriptase PCR (RT-PCR) (Fig. 2B). The protein expression of pluripotency genes was also demonstrated by immunocytochemical stainings (Fig. 2C) for further confirmation. By contrast, exogenous gene expression was turned off in all iPSC lines by passage six (Fig. 2B). Exogenous gene expression was not detected after cardiac differentiation, demonstrating that these genes were not reactivated in the process (data not shown). The generated iPSC lines were also analyzed for their karyotypes, which were all normal (Fig. 2D).
Fig. 2.

Characterization of iPSCs. (A) Morphology of the iPSC colonies is similar to those of hESCs. The colonies are rather roundish and the edges are well defined and sharp, which is typical for a stem cell colony. (B) Expression of pluripotency markers in LQT2-specific iPSCs is shown by RT-PCR. All the endogenous pluripotency genes studied were turned on in iPSCs by passage 6 (top panel). As a positive control, they were also expressed in hESCs (H7). Expression of Sox2 and very modest expression of Rex1 and Myc was found also in EBs, which were used as a negative control. β-actin served as a loading control. None of the exogenous genes were expressed in iPSCs at passage 11. As a positive control, PCR was also done using the transfected plasmids as templates (bottom panel). RT-PCR results were similar for all the iPSC lines. (C) Immunocytochemical staining of the cells shows that pluripotency markers are expressed also at the protein level. The expression of Nanog, Oct3/4, Sox2, SSEA-4, TRA1-60 and TRA1-81 was similar in all iPSC lines and there were no differences between LQT2-specific and control lines. (D) Karyotypes of all the iPSC lines were analyzed and proved to be normal. (E) Teratomas were made from one LQT2-specific line and two control lines to further confirm the pluripotency of the lines. Tissues from all three germ layers were found in teratomas from every line. (F) EBs were also formed from all the lines to show the pluripotent differentiation capacity. The EB-derived cells from both LQT2-iPSC and all control iPSC lines expressed markers from the three embryonic germ layers.
To confirm the pluripotency of our iPSC lines, an embryoid body (EB) formation assay was performed. The EB-derived cells from LQT2 iPSC and control iPSC lines all expressed markers from the three different embryonic lineages: endoderm, ectoderm and mesoderm (Fig. 2F). Pluripotency of the lines was further confirmed by teratoma formation. Teratomas were made from one LQT2-specific line (UTA.00525.LQT2) and two control lines (UTA.00112.hFF and UTA.01006.WT). In every case, tissues from all three germ layers were found in the teratomas (Fig. 2E).
Cardiomyocyte differentiation and characterization
LQT2-specific iPSCs, control iPSCs and hESCs differentiated into spontaneously beating cells. As shown by RT-PCR, these differentiated control and LQT2 cardiomyocytes expressed cardiac markers: troponin T (TNTT2), ventricular myosin light chain (MLC2V), atrial myosin light chain (MLC2A), connexin-43 (Cx-43), myosin heavy chain β (MHC-β; MYH7), HERG and GATA4 (Fig. 3B). The expression of cardiac troponin T, α-actinin and Cx-43 was also seen at the protein level as evidenced in Fig. 3A. The electrical properties of iPSC- and hESC-derived cardiomyocytes were also studied using microelectrode array (MEA; Fig. 3C,D). There were differences between control and LQT2-specific cells in their field potential durations (FPDs) (Fig. 3C), but both types showed increased chronotrophy when treated with isoprenaline, a β-adrenergic agonist (Fig. 3D), which is the anticipated response and indicates intact β-adrenergic signaling.
Fig. 3.

The expression of cardiac markers in iPSC-derived cardiomyocytes and the electrical properties of the cells. (A) Immunocytochemical staining of different cardiac markers: troponin T and α-actinin are shown in red; green indicates connexin-43 and blue represents DAPI-staining for nuclei. The expression was similar in LQT2-specific and control cells, and there were no line-specific differences in the expression of cardiac proteins. (B) The expression of a larger repertoire of cardiac markers was also studied, with RT-PCR showing that the iPSC-derived cardiac cells manifest cardiac properties. TNTT2, MLC2V, MLC2A, Cx-43, MYH7, GATA4 and HERG were present in the cells at the mRNA level. (C) Electrical properties of the cells were studied with MEA, which revealed the differences between LQT2-specific and control cells. FPD was significantly longer in LQT2-specific cardiomyocytes than in control cardiac cells. However, all lines evince the typical electrical properties of cardiomyocytes. (D) LQT2-specific cardiomyocytes showed increased chronotrophy when challenged with isoprenaline, a β-adrenergic agonist.
Electrophysiological properties of differentiated cardiomyocytes
The differentiation of both control and LQT2-specific iPSCs into cardiomyocyte subtypes was evident from the morphology of the spontaneous action potentials (APs) recorded with the patch-clamp technique. Two types of AP morphology were observed: ventricular-like, which displayed a distinct plateau phase; and atrial-like, which were triangular shaped (Fig. 4A). The AP properties of control-iPSC-derived cardiomyocytes were similar to those of other human ESC and iPSC studies (Table 1) (Gai et al., 2009; Yokoo et al., 2009; Zwi et al., 2009). For most AP parameters there was no significant difference between control and LQT2-iPSC-derived cardiomyocytes (P>0.05; Table 1). However, ventricular-like LQT2-iPSC-derived cardiomyocytes had significantly prolonged AP durations at 50% and 90% repolarization (APD50 and APD90, respectively). The APD50 and APD90 of atrial-like LQT2-iPSC-derived cardiomyocytes, although prolonged, did not reach statistical significance. On the borderline of statistical significance was the slower AP frequency of ventricular-like LQT2-iPSC-derived cardiomyocytes; therefore, further analysis was restricted to a subset of cardiomyocytes to reduce the effect of any rate-dependent APD adaptation.
Fig. 4.

Current-clamp recordings from human iPSC-derived cardiomyocytes. (A) Spontaneous APs from healthy control iPSC-derived (upper APs) and LQT2 patient-derived (lower APs) cardiomyocytes. The dashed line denotes 0 mV. (B) The action potential duration (APD) measured at 50% and 90% repolarization from the AP peak (APD50 and APD90) of spontaneous atrial-like (n=5–6) and ventricular-like APs. For the latter, both the APD50 and APD90 of LQT2 patient-derived cardiomyocytes (n=13) were significantly prolonged compared with those of hESCs (n=7) or control-iPSC origin (n=11). (C) Spontaneous arrhythmogenic activity of an LQT2-iPSC-derived cardiomyocyte.
Table 1.
AP properties of atrial- and ventricular-like cardiomyocytes

Both the APD50 and APD90 of LQT2-iPSC-derived ventricular-like cardiomyocytes were significantly prolonged compared with control-iPSC- and hESC-derived cardiomyocytes (P<0.001; Fig. 4A,B), at an AP frequency of about 1 Hz (P=0.13 between groups). The APD90 was 516.5±26.1 ms in LQT2-specific cardiomyocytes compared with 310.5±19.6 ms in control-iPSC-derived cardiomyocytes or 338.6±19.9 ms in hESC-derived cardiomyocytes. The APD90 did not differ significantly between cardiomyocytes from the two LQT2 iPSC lines (UTA.00514.LQT2 and UTA.00525.LQT2) or between hESC-derived cardiomyocytes and those of control iPSC origin. Collectively, these data indicate that the LQT2-iPSC-derived cardiomyocytes express the disease phenotype, characterized by a prolonged cardiac repolarization phase. Spontaneous arrhythmogenic activity was rare, early after depolarizations (EADs) were observed in only one of 20 LQT2-iPSC-derived cardiomyocytes studied (Fig. 4C) and in no recordings from control iPSC cardiomyocytes (n=20).
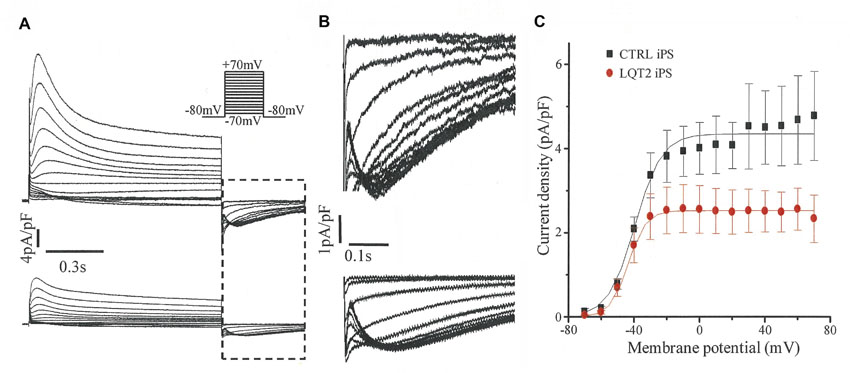
Ventricular-like cardiomyocytes were subjected to further investigations using voltage clamp (Fig. 5). Using the specific inhibitor E-4031, IKr was isolated (Fig. 5A) and its magnitude shown to be markedly reduced in LQT2 iPSC cardiomyocytes compared with that in control iPSC cardiomyocytes (Fig. 5B). Tail IKr density was significantly decreased, by 40–46%, after a depolarizing step to voltages from 0 to +40 mV (P<0.01; Fig. 5C). A similar reduction in tail IKr density (∼40%) was seen in experiments when an alternative method, isotonic cesium conditions (Zhang, 2006), was used to isolate IKr from a different control and LQT2 iPSC line (supplementary material Fig. S1). The reduction in resurgent IKr during repolarization was also demonstrated with the use of a step-ramp voltage protocol as a simplified version of a cardiac AP (Fig. 5D).
Fig. 5.

IKr recorded from iPSC cardiomyocytes with a ventricular-like AP. (A) Example of the isolation of IKr. Whole-cell current, here from a control iPS cardiomyocyte, was recorded in the absence (a) and then presence of 1 μmol/l E-4031 (b), with IKr defined as the subtracted current (a–b), i.e. the E-4031-sensitive current. Current was evoked by a 5-second depolarization from a holding potential of −40 mV as shown in the inset. (B) IKr of a control iPS (black) and LQT2 iPS (red) cardiomyocyte evoked as in A with the time segment between the arrows expanded to show the peak tail currents on return to −40 mV following a step to +20 mV. (C) The peak tail IKr densities of control iPSC (black; n=4) and LQT2 iPSC (red; n=5) cardiomyocytes at membrane potentials from 0 to +40 mV were significantly different (*P<0.01, **P<0.005). (D) IKr currents of control iPSC (black) and LQT2 iPSC (red) cardiomyocytes evoked by a voltage protocol of step to +20 mV for 150 ms and ramp of 120 ms back to the −40 mV holding potential.
The in vitro cardiac FPD on MEA has been shown to correlate with QT interval properties in the ECG, and FPD recordings of beating cell aggregates give insight into the electrical function of myocardial tissue in vitro (Caspi et al., 2008). We observed a negative correlation between FPD and beating rate both in control cardiomyocytes and LQT2-specific cardiomyocytes (Fig. 6A,B). However, the LQT2 cardiomyocytes had a significantly prolonged FPD compared with controls, especially at low beating frequencies (P=0.014; Fig. 6A,B), as determined by nonlinear regression analysis. The two LQT2 iPSC lines behaved the same way and, thus, their data were combined, as were data from the three different control iPSC lines and hESC-derived cardiomyocytes, because they behaved similarly.
Fig. 6.

FPD measured on MEA. (A) The effect of the beating rate on FPD in control and LQT2 cardiomyocytes (CMs). Control and LQT2 cardiomyocytes had a negative correlation with moderately high coefficients of determination (R2). The exponential function gave the best fit as determined by R2 between different fitting functions. The LQT2 cardiomyocytes had significantly more prolonged FPD compared with controls, especially at low beating frequencies, as determined by nonlinear regression analysis (P=0.0136). (B) At beating rates below 50 beats per minute (bpm), the FPD of LQT2 cardiomyocytes differed significantly from control cardiomyocytes (*P<0.05) as determined by t-test. (C) Drug responses of control and LQT2 cardiomyocytes. Sotalol (19 μmol/l) and E-4031 (500 nmol/l for LQT2-specific cells and 700 nmol/l for control cells) was administered to the cardiomyocyte aggregates derived from control iPSCs and LQT2 iPSCs. For both cell lines, baseline and drug conditions for sotalol and E-4031 are shown. Arrows mark the site of pharmacologically induced EADs. With 500 nmol/l E-4031 there were no EADs observed in control cells.
The inotropic response of the iPSC-derived cardiac cells was studied using isoprenaline. A panel of drugs with known QT-prolongation effects, including erythromycin, sotalol and cisapride, and a non-drug compound, E-4031, were investigated. The selective hERG blocker E-4031 increased arrhythmogenicity in control cardiac cells and even more frequently in LQT2 cardiomyocytes (Fig. 6C). Application of sotalol (0.8–19.4 μmol/l), an anti-arrhythmic drug with both β-blocker and class III activity, elicited arrhythmogenicity at the highest tested concentrations only in LQT2 cardiac cells (Fig. 6C). No increased arrhythmogenicity was observed with erythromycin (1.5–16 μmol/l) or cisapride (40–330 nmol/l) in control or LQT2 cardiac cells (data not shown). At baseline, no arrhythmogenicity was observed with control or LQT2 cardiac cells on MEA.
DISCUSSION
In this study we demonstrate that patient-specific iPSCs can be used to model a potentially lethal cardiac arrhythmic disease in vitro. The cells were differentiated into functional cardiomyocytes, which reproduced the phenotypic characteristics of LQT2, including a prolonged repolarization time and increased arrhythmogenicity. Furthermore, at slow beating rates, FPD (QT) was significantly more prolonged in LQT2 cardiomyocytes compared with control cells.
The R176W hERG mutation was reported to have the frequency of 0.5% in apparently healthy individuals (Ackerman et al., 2003) and, according to an epidemiological Finnish study, the mutation is present in about one of 400 Finns (Marjamaa et al., 2009). The majority of these individuals are completely asymptomatic and unaware of their carrier status. This mutation is one of the four founder mutations for LQTS in Finland and these mutations account for almost two-thirds of all established LQTS cases in the country (Fodstad et al., 2004). The QTc interval of LQT2 patients with the R176W mutation is reported to range from 386 to 569 ms, with a mean of 448 ms, whereas the mean for non-carriers was 416 ms (Fodstad et al., 2006). Furthermore, the mutation has also been identified in cases of sudden death (Tester and Ackerman, 2007; Tu et al., 2011). Although our patient had latent LQTS, his family did not. His younger sister died suddenly at the age of 32 when awakened by a telephone and his father died suddenly at the age of 40. The sister was documented to have abnormal QT intervals of up to 582 ms. The symptoms in this family, including palpitations, syncope, and sudden death due to abrupt auditory stimuli during sleep, were typical of LQT2 (Schwartz, 2001; Roden, 2008). In our study, the repolarization time was significantly prolonged in cultured LQT2-specific cardiac cells, compared with control cells, at low beating rates, and this is consistent with the clinical observation that, at slow beating rate, the QT interval is prolonged more in LQT2 patients compared with healthy individuals (Swan et al., 1999).
Although the underlying mechanism of R176W is presently unknown, when heterologously expressed, R176W reduces hERG tail current density by ∼75%, although upon coexpression with wild type the difference in current densities was nullified (Fodstad et al., 2006). The decrease of ∼43% in IKr density observed here in iPS cardiomyocytes from a heterozygous R176W individual might reflect the difference in cellular environment, e.g. the composition of the endogenous IKr channel, which includes both the ubiquitously in vitro expressed hERG1a subunit and hERG1b subunit with its alternatively spliced N-terminus, presence of accessory subunits and/or native interactions. However, it is also possible that this observed discrepancy results from a differential expression of the wild-type and mutant alleles in vitro versus in vivo. In both model systems, R176W does not display a dominant-negative effect, unlike the A614V hERG mutation (Nakajima et al., 1998). iPS cell lines with that particular mutation derived from a severely symptomatic LQT2 patient with recorded TdP have been generated (Itzhaki et al., 2011). This mutation resulted in a decrease in the activating IKr density of 72% with a depolarization step to 0 mV and of 64% in the tail current density following depolarization to +20 mV. Smaller reductions in these values (43% and 40%, respectively) were obtained for the R176W hERG mutation here. This difference in IKr reduction translates to the APD: at 1 Hz the ventricular-like LQT2 iPSC cardiomyocyte APD90 was 166% and ∼200% of control for R176W and A614V, respectively, and in arrhythmogenicity, with EADs rarely observed here (∼5%) but frequently (66%) in the report by Itzhaki and coworkers. Thus, the in vitro results obtained with iPSC cardiomyocytes seems to correspond to differences in expression of the disease, i.e. latent versus overt LQTS. Similarly, Matsa et al. demonstrated that the APD of both atrial-like and ventricular-like iPSC cardiomyocytes was significantly prolonged when derived from an LQT2 patient with episodes of syncope, seizures and TdP (Matsa et al., 2011). Although, in the study by Matsa et al., no spontaneous arrhythmicity was observed, such an effect could be induced pharmaceutically by isoprenalin.
One explanation for the genotype-phenotype discordance in LQTS is the repolarization reserve. This concept proposes that redundant mechanisms are available to bring about normal repolarization; therefore, to elicit the full-blown LQTS phenotype multiple hits might be required to sufficiently reduce the reserve (Roden, 2006). Such ‘hits’ might be the presence of compound mutations, polymorphisms, drug exposure, female gender, hypokalemia or other risk factors (Roden, 2006; Lehtonen et al., 2007). Evidence for the repolarization reserve and its genetic modulation comes from studies of individuals or first-degree relatives of individuals with drug-induced LQTS showing a prolongation of repolarization indices with pharmacological challenge (Kannankeril et al., 2005; Couderc et al., 2009). The unmasking of latent LQTS can occur accidentally (e.g. associated drug-induced TdP) (Lehtonen et al., 2007). In line with the repolarization reserve hypothesis and the clinical data, R176W LQT2 cardiac cells were more sensitive to drug effects that controls. The hERG-channel-specific blocker E-4031 induced arrhythmogenicity in both control and LQT2 cardiac cells, but sotalol induced arrhythmogenic behavior only in LQT2 cardiac cells. Sotalol has been used as a pharmacological challenge and is documented as inducing TdP in a patient carrying a hERG channel mutation (Lehtonen et al., 2007; Couderc et al., 2009). The concentrations of sotalol used in this study are similar to the effective free therapeutic plasma concentration range (1.8–14.7μmol/l) (Redfern et al., 2003). Sotalol probably exacerbates the underlying defect, because the potency of sotalol towards hERG is similar between wild type and variant in transfected cells (Männikkö et al., 2010).
Comparison of single-cell recordings and MEA analysis and ECG findings of the patients revealed an interesting observation. Although single-cell recordings indicated major differences in repolarization time between control- and LQT2-derived cardiac cells (66% increase in AP90 in LQT2 cardiomyocytes), the corresponding differences measured using cell aggregates with the MEA technique were much more moderate (10–20%) and resembled differences in ECG in healthy individuals and LQTS patients (Fodstad et al., 2006). Similar observations can be found in the previously reported iPSC studies on LQTS. AP90 duration was reported to be increased by 50% in LQT1 cardiac cells compared with control cells, whereas the difference in ECG was only in the range of 10–15% (Moretti et al., 2010). In the studies with LQT2 cells, the repolarization times were greatly prolonged already at ECG level (50% or more) and similar differences were observed in their MEA recordings (50% difference between control and LQT2 cardiac cells) (Itzhaki et al., 2011; Matsa et al., 2011). However, in both of these studies, the AP90 duration was increased by 2- to 2.5-fold compared with control repolarization time. It is possible that cell-to-cell contacts in the syncytium result in compensatory mechanisms with a tendency to protect the repolarization system from major deviation from the normal conditions.
Our results on the abnormal electrophysiological properties and increased drug sensitivity of cardiac cells derived from an asymptomatic KCNH2 R176W mutation carrier raise an important issue about LQT2 and also a new challenge, as well as a possibility, for the pharmaceutical industry. LQT2 cell models for severely symptomatic patients (Itzhaki et al., 2011; Matsa et al., 2011) are most useful for studying the pathology of LQT2 and demonstrate in a convincing way that the phenotype of this syndrome can be reproduced in a cell culture model. Our results complement the previous research on LQT2-specific iPSC-derived cardiomyocytes by introducing a cell model for an asymptomatic mutation carrier. According to our findings, even these clinically asymptomatic individuals can possess the inherent electrophysiological abnormality in their cardiac cells. Taking advantage of this large resource of Finnish individuals with the same mutation, it might be possible to evaluate the electrophysiological properties and drug responses of cardiomyocytes from mutation carriers with and without symptoms, and thereby try to identify putative genetic or non-genetic modifiers of LQTS. These types of studies might also assist in the tailoring of individualized drug treatment of these patients. In the future, iPSC technology is likely to be increasingly exploited for drug development and safety studies.
In conclusion, iPSC-derived cardiomyocytes from an individual with LQT2 displayed the disease cardiac phenotype in cell culture conditions even though the individual was relatively asymptomatic. This model provides an additional platform to study the basic pathology of LQTS and to individualize drug treatment in a patient-specific manner. It also provides the means to explore the differences between clinical patients and mutation carriers and to scan the cardiac effects of different drugs on both.
METHODS
The study was approved by the ethical committee of Pirkanmaa Hospital District (R07080).
Cell culture
The LQT2 cells were derived from a 61-year-old man with an R176W mutation of KCNH2 (hERG-FinB) (Fodstad et al., 2004). Primary fibroblasts from a skin biopsy were cultured under fibroblast culturing conditions: Dulbecco’s Modified Eagle’s Medium (DMEM) (Lonza, Basel, Switzerland) containing 10% fetal bovine serum (FBS) (Lonza), 2 mmol/l L-glutamine, 50 U/ml penicillin/streptomycin. 293FT cells (Invitrogen, Carlsbad, CA) were maintained similarly with 1% non-essential amino acids (NEAA) (Cambrex, East Rutherford, NJ). Plat-E (Cell Biolabs, San Diego, CA), irradiated SNL-76/7 (HPA Culture Collections, Salisbury, UK) and mouse embryonic fibroblast (MEF; Millipore, Billerica, MA) cells were cultured without antibiotics. iPSCs and hESCs were maintained in KSR medium: knockout (KO)-DMEM (Invitrogen) containing 20% KO serum replacement (KO-SR, Invitrogen), NEAA, L-glutamine, penicillin/streptomycin, 0.1 mmol/l 2-mercaptoethanol and 4 ng/ml basic fibroblast growth factor (bFGF; R&D Systems, Minneapolis, MN). H7 hESCs (WiCell Research Institute, Madison, WI) and iPSC lines FiPS 6–14 and UTA.00112.hFF (derived from foreskin fibroblasts), UTA.01006.WT and UTA.04602.WT (from adult skin fibroblasts) were used as controls.
Generation of iPSC lines
Patient-specific iPSC lines were established using lentivirus infection followed by retrovirus infection into the primary fibroblasts. The following cells, plasmids and reagents were used: 293FT cells, PlatE cells, pLenti6/UbC/mSlc7a1 vector (Addgene, Cambridge, MA), ViraPower Packaging Mix (Invitrogen), Lipofectamine 2000 (Invitrogen), pMX retroviral vector (hOCT3/4, hSOX2, hKLF4 or hc-MYC; Addgene) and Fugene 6 (Roche Diagnostics, Mannheim, Germany). The protocol used has been described previously (Takahashi et al., 2007). Two LQT2-specific lines were established (UTA.00514.LQT2 and UTA.00525.LQT2) carrying the R176W mutation, which was confirmed by PCR as described previously (Fodstad et al., 2006). The FiPS 6–14 line was derived at the University of Helsinki (provided by Timo Otonkoski) (Rajala et al., 2010). Control iPSC lines from healthy individuals (UTA.01006.WT from a 36-year-old male and UTA.04602.WT from a 55-year-old female) and the UTA.00112.hFF line from human foreskin fibroblasts were established in the same way as the LQT2 lines.
RT-PCR
Total RNA was collected from the iPSC lines at passages 3, 6 and 11 and after cardiac differentiation. For positive pluripotency control, the H7 line was used. RNA from EBs was used as a negative control of pluripotency. RNA was purified with NucleoSpin RNA II kit (Macherey-Nagel, Düren, Germany) and cDNA conversion was performed with a high-capacity cDNA RT kit (Applied Biosystems, Carlsbad, CA). PCR was done with Dynazyme II (Finnzymes Oy, Espoo, Finland) using 1 μl of cDNA as a template and 2 μM primers. As positive controls for exogenous primers, PCR was also done using the transfected plasmids (hOCT3/4, hSOX2, hKLF4 and hc-MYC) as templates. PCR primers for iPSC characterization and detailed reaction conditions have been described previously (Takahashi et al., 2007). Primers for different germ layers and cardiac markers are presented in Table 2. β-actin and GAPDH were used as housekeeping controls.
Table 2.
Primers for RT-PCR of different germ layer markers and cardiac markers.

Immunocytochemistry for pluripotency
iPSCs at passage 8 were fixed with 4% paraformaldehyde (Sigma-Aldrich) and stained with anti-OCT3/4 (1:400; R&D Systems), anti-tumor-related antigen (TRA)1-60 (1:200; Millipore), anti-SOX2, anti-NANOG, anti-stage-specific embryonic antigen (SSEA)4 and anti-TRA1-81 (all 1:200; from Santa Cruz Biotechnology, Santa Cruz, CA). The secondary antibodies (Invitrogen) were Alexa-Fluor-568-donkey-anti-goat-IgG, Alexa-Fluor-568-goat-anti-mouse-IgM or Alexa-Fluor-568-donkey-anti-mouse-IgG.
EB formation
EBs were cultured without feeder cells in EB medium (KO-DMEM with 20% FBS, NEAA, L-glutamine and penicillin/streptomycin) without bFGF for 5 weeks. RNA isolation and reverse transcription from EBs was performed as described above. The expression of markers characteristic of ectoderm, endoderm and mesoderm development in EBs was determined using RT-PCR (primers described in Table 2).
Mutation analysis
The hERG-FinB mutation was assayed with restriction enzyme analysis (Fodstad et al., 2006) by amplifying the genomic DNA with primers for hERG (forward: 5′-ACCACGTGCCTCTCCTCTC-3′, reverse: 5′-GTCGGGGTTGAGGCTGTG-3′) (reagents from Applied Biosystems) and digesting the amplified PCR product with SmaI digestion enzyme (Fermentas GmbH, St Leon-Rot, Germany). The hERG-FinB mutation resulted in deletion of a SmaI-cleavage site. SmaI-cleaved PCR products were detected with gel electrophoresis: products for wild type were 182, 79, 46 and 23 bp and for R176W heterozygote 182, 125, 79, 46 and 23 bp long.
Karyotype analysis
Karyotypes of the cell lines were defined using standard G-banding chromosome analysis by a commercial company (Medix Laboratories, Espoo, Finland) according to standard procedure.
Teratoma formation
iPSCs were injected into nude mice under the testis capsule. Tumor samples were collected 8 weeks after injection and fixed with 4% paraformaldehyde. The sections were stained with hematoxylin and eosin.
Cardiac differentiation and characterization
Cardiomyocyte differentiation was carried out by co-culturing iPSCs and H7 hESCs with END-2 cells (a kind gift from Christine Mummery, Hubrecht Institute, Utrecht, The Netherlands). END-2 cells were cultured as described earlier (Mummery et al., 2003). The beating areas of the cell colonies were mechanically excised and treated with collagenase A (Roche Diagnostics) as described previously (Mummery et al., 2003). Seven days after dissociation, cells were fixed with 4% paraformaldehyde for immunostaining with anti-cardiac-troponin-T (1:2000; Abcam, Cambridge, MA), anti-α-actinin (1:1500; Sigma-Aldrich, St Louis, MO) and anti-connexin-43 (1:1000; Sigma-Aldrich).
Patch-clamp technique
APs were recorded from spontaneously beating dissociated cells using the perforated patch (by amphotericin) configuration of the patch-clamp technique with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA). A coverslip with the adhered cells was placed in the recording chamber and perfused with extracellular solution consisting of (in mmol/l) 143 NaCl, 4 KCl, 1.8 CaCl2, 1.2 MgCl2, 5 glucose, 10 HEPES (pH 7.4 with NaOH; osmolarity adjusted to 301±3 mOsm). Patch pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus, Kent, UK) and had resistances of 1.5–3.5 MO when filled with a solution consisting of (in mmol/l): 122 K-gluconate, 30 KCl, 1 MgCl2, 5 HEPES (pH 7.2 with KOH; osmolarity adjusted to 290±3 mOsm). The final concentration of amphotericin B (solubilized in dimethylsulfoxide) in the pipette was 0.24 mg/ml.
The data was filtered at 2 kHz, and digitized (Digidata 1322A; Molecular Devices) at 10 kHz; data acquisition and analysis was performed with pClamp 9.2 software (Molecular Devices). For some cardiomyocytes, voltage-clamp experiments were also performed to record IKr. Experiments were conducted at 36±1°C.
Field potential recordings
Field potentials of spontaneously beating cardiomyocyte aggregates were recorded with the MEA platform (Multi Channel Systems, Reutlingen, Germany) at 37°C. MEAs were hydrophilized with FBS for 30 minutes, washed with sterile water and coated with 0.1% gelatin for 1 hour. Cardiomyocyte aggregates were plated onto MEAs in EB medium. FPD and beating frequency were determined manually with AxoScope software (Molecular Devices). Isoprenaline (Isuprel; Hospira, Lake Forest, IL), D,L-sotalol (Sigma), erythromycin (Abbott, IL), cisapride (Sigma) and E-4031 (Alomone Labs, Jerusalem, Israel) were diluted in 5% FBS containing EB medium for drug tests. Baseline conditions as well as drug effects were recorded for 2 minutes after a 2-minute stabilization period. Baseline FPDs were measured from 43 control cardiomyocyte aggregates and 30 LQT2 cardiomyocyte aggregates.
TRANSLATIONAL IMPACT.
Clinical issue
Long QT syndrome (LQTS) is a life-threatening cardiac disorder that predisposes individuals to ventricular arrhythmias and sudden death. The syndrome is characterized by a prolonged QT interval (detected by electrocardiography) and can be caused either by genetic defects or as a side effect of certain drugs. LQTS type 2 (LQT2) occurs owing to defective functioning of the hERG potassium channel, and is caused by mutations in the KCNH2 gene (also known as HERG). The drug-induced form of LQTS also occurs owing to altered function of the hERG channel. Because prolongation of QT interval is one of the most common severe side effects of both cardiac and non-cardiac drugs, new drug candidates must be carefully tested for their effects on the hERG channel. However, physiological human cell models to test for this effect were not previously available. The clinical prevalence of LQTS is only ∼1:5000, but its genetic prevalence has been estimated to be much higher (1:250 to 1:2000). Therefore, it is possible that asymptomatic carriers of LQTS-associated mutations are more susceptible to the side effect of certain medications, and are at risk of developing severe symptoms in certain settings.
Results
This study investigates whether an LQTS-related phenotype can be detected in an in vitro model based on cells from an asymptomatic carrier of an LQT2-associated KCNH2 mutation. The authors generate patient-specific induced pluripotent stem cells (iPSCs) from an asymptomatic individual and differentiate them into functional cardiac cells. These cells recapitulate the phenotypic characteristics of LQT2 in vitro, including prolonged repolarization time and increased arrhythmogenicity. Additionally, at slow beating rates, cardiomyocyte aggregates derived from these iPSCs present prolonged field potential duration (QT) compared with control cells. These results are in line with clinical findings that individuals with LQT2 usually display symptoms when heart rate is slow. These results indicate that electrophysiological abnormalities can be detected in iPSC-derived cardiac cells, even when derived from asymptomatic carriers of KCNH2 mutations.
Implications and future directions
This in vitro model of LQT2 offers a new system with which to evaluate electrophysiological properties and drug responses of cardiomyocytes from KCNH2 mutation carriers with and without LQTS symptoms. In addition, the model provides a platform from which to study the basic pathology of LQTS and to identify genetic and non-genetic modifiers that can be considered in designing and developing medications to treat distinct patient groups. In addition, the model provides the means to analyze the cardiac side effects of different drugs in carriers of LQTS-associated mutations.
Statistical methods
Data are given as mean ± s.e.m. or s.d. Comparison of patch-clamp data between LQT2-iPSC and control-iPSC cardiomyocytes was performed using Student’s t-test for independent data. One-way analysis of variance followed by Tukey test was used for comparison of multiple groups. The IKr data was assessed using Student’s t-test for independent data. The difference in FPDs between different populations of spontaneously beating cardiomyocyte aggregates was determined by nonlinear regression analysis using R software. The difference in FPDs between populations when categorized according to beating frequencies was determined by t-test with SPSS software (IBM).
Supplementary Material
Acknowledgments
We thank Merja Lehtinen and Henna Venäläinen for technical support, Christine Mummery for providing END-2 cells, Timo Otonkoski for sharing his knowledge, Annukka Lahtinen for help in the LQTS genotyping of the iPSC lines, Heini Huhtala for help on statistical analysis, and Kenta Nakamura, Chris Schlieve and the Gladstone Stem Cell Core for advice and reagents. We also thank Takayuki Tanaka for help in setting up our iPSC system.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
A.L.L. was involved in iPS cell generation and analysis, cardiomyocyte differentiation and characterization, figure preparation, and writing the manuscript. V.J.K. performed MEA experiments and analysis, wrote parts of the manuscript, was involved in editing the manuscript, and was involved in figure preparation. H.C. designed and performed all patch-clamp experiments and analysis. A.-P.K. provided expertise in electrophysiology. M.P.-M. was involved in cardiac differentiation and cardiomyocyte analysis. E.K. planned and performed cardiac cell differentiation experiments. J.H. designed the MEA experiments. K.K. provided genetic expertise and provided genotyped patients. H.S. provided genetic and cardiological expertise on LQTS. B.R.C. helped design the study. S.Y. provided expertise in iPSC technology. O.S. helped design the study and analyze the results. K.A.-S. was leader of the group, and was involved in the planning and evaluation of the results, and finalizing the manuscript.
FUNDING
This work was supported by the Academy of Finland [grant number 126888]; the Finnish Foundation for Cardiovascular Research; Pirkanmaa Hospital District (EVO); the Finnish Cultural Foundation; Biocenter Finland; and the Sigrid Juselius Foundation.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.008409/-/DC1
REFERENCES
- Ackerman M. J., Tester D. J., Jones G. S., Will M. L., Burrow C. R., Curran M. E. (2003). Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin. Proc. 78, 1479–1487 [DOI] [PubMed] [Google Scholar]
- Carvajal-Vergara X., Sevilla A., D’Souza S. L., Ang Y.-S., Schaniel C., Lee D.-F., Yang L., Kaplan A. D., Adler E. D., Rozov R., et al. (2010). Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465, 808–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi O., Itzhaki I., Arbel G., Kehat I., Gepstien A., Huber I., Satin J., Gepstein L. (2008). In vitro electrophysiological drug testing using human embryonic stem cell derived cardiomyocytes. Stem Cells Dev. 18, 161–172 [DOI] [PubMed] [Google Scholar]
- Chiang C.-E., Roden D. M. (2000). The long QT syndromes: genetic basis and clinical implications. J. Am. Coll. Cardiol. 36, 1–12 [DOI] [PubMed] [Google Scholar]
- Couderc J.-P., Kaab S., Hinterseer M., McNitt S., Xia X., Fossa A., Beckmann B. M., Polonsky S., Zareba W. (2009). Baseline values and sotalol-induced changes of ventricular repolarization duration, heterogeneity, and instability in patients with a history of drug-induced torsades de pointes. J. Clin. Pharmacol. 49, 6–16 [DOI] [PubMed] [Google Scholar]
- Curran M. E., Splawski I., Timothy K. W., Vincen G. M., Green E. D., Keating M. T. (1995). A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803 [DOI] [PubMed] [Google Scholar]
- Fodstad H., Swan H., Laitinen P., Piippo K., Paavonen K., Viitasalo M., Toivonen L., Kontula K. (2004). Four potassium channel mutations account for 73% of the genetic spectrum underlying long-QT syndrome (LQTS) and provide evidence for a strong founder effect in Finland. Ann. Med. 36 Suppl. 1, 53–63 [DOI] [PubMed] [Google Scholar]
- Fodstad H., Bendahhou S., Rougier J. S., Laitinen-Forsblom P. J., Barhanin J., Abriel H., Schild L., Kontula K., Swan H. (2006). Molecular characterization of two founder mutations causing long QT syndrome and identification of compound heterozygous patients. Ann. Med. 38, 294–304 [DOI] [PubMed] [Google Scholar]
- Gai H., Leung E. L.-H., Costantino P. D., Aguila J. R., Nguyen D. M., Fink L. M., Ward D. C., Ma Y. (2009). Generation and characterization of functional cardiomyocytes using induced pluripotent stem cells derived from human fibroblasts. Cell Biol. Int. 33, 1184–1193 [DOI] [PubMed] [Google Scholar]
- Hancox J. C., McPate M. J., El Harchi A., Zhang Y. H. (2008). The hERG potassium channel and hERG screening for drug-induced torsades de pointes. Pharmacol. Ther. 119, 118–132 [DOI] [PubMed] [Google Scholar]
- Hedley P. L., Jørgensen P., Schlamowitz S., Wangari R., Moolman-Smook J., Brink P. A., Kanters J. K., Corfield V. A., Christiansen M. (2009). The genetic basis of long QT and short QT syndromes: a mutation update. Hum. Mutat. 30, 1486–1511 [DOI] [PubMed] [Google Scholar]
- Itzhaki I., Maizels L., Huber I., Zwi-Dantsis L., Caspi O., Winterstern A., Feldman O., Gepstein A., Arbel G., Hammerman H., et al. (2011). Modelling the long QT syndrome with induced pluripotent stem cells. Nature 471, 225–229 [DOI] [PubMed] [Google Scholar]
- Kannankeril P. J., Roden D. M., Norris K. J., Whalen S. P., George J. A. L., Murray K. T. (2005). Genetic susceptibility to acquired long QT syndrome: Pharmacologic challenge in first-degree relatives. Heart Rhythm 2, 134–140 [DOI] [PubMed] [Google Scholar]
- Lee G., Studer L. (2010). Induced pluripotent stem cell technology for the study of human disease. Nat. Meth. 7, 25–27 [DOI] [PubMed] [Google Scholar]
- Lehtonen A., Fodstad H., Laitinen-Forsblom P., Toivonen L., Kontula K., Swan H. (2007). Further evidence of inherited long QT syndrome gene mutations in antiarrhythmic drug-associated torsades de pointes. Heart Rhythm 4, 603–607 [DOI] [PubMed] [Google Scholar]
- Männikkö R., Overend G., Perrey C., Gavaghan C. L., Valentin J. P., Morten J., Armstrong M., Pollard C. E. (2010). Pharmacological and electrophysiological characterization of nine, single nucleotide polymorphisms of the hERG-encoded potassium channel. Br. J. Pharmacol. 159, 102–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marjamaa A., Salomaa V., Newton-Cheh C., Porthan K., Reunanen A., Karanko H., Jula A., Lahermo P. i., Väänänen H., Toivonen L., et al. (2009). High prevalence of four long QT syndrome founder mutations in the Finnish population. Ann. Med. 41, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsa E., Rajamohan D., Dick E., Young L., Mellor I., Staniforth A., Denning C. (2011). Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur. Heart J. 32, 952–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A., Bellin M., Welling A., Jung C. B., Lam J. T., Bott-Flügel L., Dorn T., Goedel A., Höhnke C., Hofmann F., et al. (2010). Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 363, 1397–1409 [DOI] [PubMed] [Google Scholar]
- Mummery C., Ward-van Oostwaard D., Doevendans P., Spijker R., van den Brink S., Hassink R., van der Heyden M., Opthof T., Pera M., de la Riviere A. B., et al. (2003). Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation 107, 2733–2740 [DOI] [PubMed] [Google Scholar]
- Nakajima T., Furukawa T., Tanaka T., Katayama Y., Nagai R., Nakamura Y., Hiraoka M. (1998). Novel mechanism of HERG current suppression in LQT2: shift in voltage dependence of HERG inactivation. Circ. Res. 83, 415–422 [DOI] [PubMed] [Google Scholar]
- Pollard C. E., Valentin J. P., Hammond T. G. (2008). Strategies to reduce the risk of drug-induced QT interval prolongation: a pharmaceutical company perspective. Br. J. Pharmacol. 154, 1538–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priori S., Napolitano C., Schwartz P. (1999). Low penetrance in the long-QT syndrome. Circulation 99, 529–533 [DOI] [PubMed] [Google Scholar]
- Rajala K., Lindroos B., Hussein S. M., Lappalainen R. S., Pekkanen-Mattila M., Inzunza J., Rozell B., Miettinen S., Narkilahti S., Kerkelä E., et al. (2010). A defined and xeno-free culture method enabling the establishment of clinical-grade human embryonic, induced pluripotent and adipose stem cells. PLoS ONE 5, e10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redfern W. S., Carlsson L., Davis A. S., Lynch W. G., MacKenzie I., Palethorpe S., Siegl P. K. S., Strang I., Sullivan A. T., Wallis R., et al. (2003). Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 58, 32–45 [DOI] [PubMed] [Google Scholar]
- Roden D. M. (2004). Drug-Induced Prolongation of the QT Interval. N. Engl. J. Med. 350, 1013–1022 [DOI] [PubMed] [Google Scholar]
- Roden D. M. (2006). Long QT syndrome: reduced repolarization reserve and the genetic link. J. Int. Med. 259, 59–69 [DOI] [PubMed] [Google Scholar]
- Roden D. M. (2008). Long-QT Syndrome. N. Engl. J. Med. 358, 169–176 [DOI] [PubMed] [Google Scholar]
- Schwartz P. J. (2001). Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation 103, 89–95 [DOI] [PubMed] [Google Scholar]
- Swan H., Viitasalo M., Piippo K., Laitinen P., Kontula K., Toivonen L. (1999). Sinus node function and ventricular repolarization during exercise stress test in long QT syndrome patients with KvLQT1 and HERG potassium channel defects. J. Am. Coll. Cardiol. 34, 823–829 [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872 [DOI] [PubMed] [Google Scholar]
- Tester D. J., Ackerman M. J. (2007). Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J. Am. Coll. Cardiol. 49, 240–246 [DOI] [PubMed] [Google Scholar]
- Tu E., Bagnall R. D., Duflou J., Semsarian C. (2011). Post-mortem review and genetic analysis of sudden unexpected death in epilepsy (SUDEP) cases. Brain Pathol. 21, 201–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazawa M., Hsueh B., Jia X., Pasca A. M., Bernstein J. A., Hallmayer J., Dolmetsch R. E. (2011). Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471, 230–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoo N., Baba S., Kaichi S., Niwa A., Mima T., Doi H., Yamanaka S., Nakahata T., Heike T. (2009). The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells. Biochem. Biophys. Res. Commun. 387, 482–488 [DOI] [PubMed] [Google Scholar]
- Yu J., Vodyanik M. A., Smuga-Otto K., Antosiewicz-Bourget J., Frane J. L., Tian S., Nie J., Jonsdottir G. A., Ruotti V., Stewart R., et al. (2007). Induced pluripotent stem cell lines derived from human somatic cells. Science 318, 1917–1920 [DOI] [PubMed] [Google Scholar]
- Zhang S. (2006). Isolation and characterization of IKr in cardiac myocytes by Cs+ permeation. Am. J. Physiol. Heart Circ. Physiol. 290, H1038–H1049 [DOI] [PubMed] [Google Scholar]
- Zwi L., Caspi O., Arbel G., Huber I., Gepstein A., Park I.-H., Gepstein L. (2009). Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation 120, 1513–1523 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}