

SUMMARY
Analysis of zebrafish mutants that demonstrate abnormal locomotive behavior can elucidate the molecular requirements for neural network function and provide new models of human disease. Here, we show that zebrafish quetschkommode (que) mutant larvae exhibit a progressive locomotor defect that culminates in unusual nose-to-tail compressions and an inability to swim. Correspondingly, extracellular peripheral nerve recordings show that que mutants demonstrate abnormal locomotor output to the axial muscles used for swimming. Using positional cloning and candidate gene analysis, we reveal that a point mutation disrupts the gene encoding dihydrolipoamide branched-chain transacylase E2 (Dbt), a component of a mitochondrial enzyme complex, to generate the que phenotype. In humans, mutation of the DBT gene causes maple syrup urine disease (MSUD), a disorder of branched-chain amino acid metabolism that can result in mental retardation, severe dystonia, profound neurological damage and death. que mutants harbor abnormal amino acid levels, similar to MSUD patients and consistent with an error in branched-chain amino acid metabolism. que mutants also contain markedly reduced levels of the neurotransmitter glutamate within the brain and spinal cord, which probably contributes to their abnormal spinal cord locomotor output and aberrant motility behavior, a trait that probably represents severe dystonia in larval zebrafish. Taken together, these data illustrate how defects in branched-chain amino acid metabolism can disrupt nervous system development and/or function, and establish zebrafish que mutants as a model to better understand MSUD.
INTRODUCTION
Maple syrup urine disease (MSUD) is an inherited metabolic disorder of branched-chain amino acids (BCAA): isoleucine, leucine and valine (Strauss and Morton, 2003; Chuang et al., 2006; Chuang et al., 2008). It demonstrates an autosomal recessive pattern of inheritance, and affects ∼1 in every 185,000 children worldwide. However, much higher incidence rates are observed in Old Order Mennonite communities, with a ratio of 1:200 children, owing to founder effects. The first step of BCAA metabolism consists of a reversible transamination reaction to yield α-keto acids. The second step is oxidative decarboxylation of the α-keto acids by the mitochondrial branched-chain α-keto acid dehydrogenase (BCKD) complex. Mutation in any of the four genes encoding the three catalytic components of the BCKD complex has been shown to cause MSUD. Affected individuals accumulate BCAAs and α-keto acids in tissues and plasma, which cause the urine and bodily secretions to smell like maple syrup (burned sugar), hence the name of the disorder. In the most severe or ‘classic’ form of MSUD, the elevated BCAAs and α-keto acids can have a devastating impact on the central nervous system (CNS). If not treated, toxic build up of BCAAs and α-keto acids cause severe dystonia, coma, cerebral edema, dysmyelination and death within a couple weeks after birth. Treatment for MSUD typically consists of severe dietary restriction of BCAAs (Strauss et al., 2010). However, even with a carefully monitored diet, secondary illnesses can lead to metabolic crisis and neurological damage, which is thought to generate the mental retardation and psychiatric problems that are often observed. An alternative treatment for MSUD is elective liver transplantation, which reduces plasma levels of BCAAs and α-keto acids and spares the CNS from severe injury (Strauss et al., 2006). Unfortunately, the scarcity of livers for transplantation, surgical risks and lifetime usage of immunosuppressants limit this treatment option. A better understanding of the mechanisms that cause the neuropathology of MSUD and development of new therapeutic drugs could greatly benefit affected individuals as well as yield new insight into CNS metabolism.
To investigate the pathophysiology of MSUD, a mouse model of the classic form of the disease was created by deleting the gene encoding the E2 subunit of the BCKD complex (Homanics et al., 2006). A model of a less severe form of the disease, intermediate MSUD, was also developed. These mice recapitulate several aspects of MSUD, including elevated levels of BCAAs and α-keto acids in tissues and plasma, severe neurological impairment, and decreased phenotypic severity in response to liver cell transplantation (Skvorak et al., 2009a; Skvorak et al., 2009b; Zinnanti et al., 2009). However, the neuropathological mechanisms of MSUD remain poorly understood. Additional animal models could provide a complementary tool to study the mechanisms of this disease that generate CNS injury.
Developing zebrafish provide an excellent system to investigate the cellular and molecular mechanisms of MSUD. Although BCAA metabolism has yet to be described in this system, larval zebrafish offer several advantageous features, including small size, rapid development external to the mother, optical transparency, large clutch sizes and organ systems that are broadly similar to those in mammals. These features make zebrafish larvae readily amenable to a wide variety of behavioral, genetic, imaging, physiological and pharmacological approaches. The identification of zebrafish mutants with impaired BCKD complex function hold promise as a particularly useful tool to investigate the effects of disrupted BCAA metabolism.
Zebrafish perform a characteristic sequence of motor behaviors during development, which reflects different stages of locomotor network formation (Kimmel et al., 1974; Saint-Amant and Drapeau, 1998; Downes and Granato, 2006; McKeown et al., 2009). These behaviors were used as the basis for extensive mutagenesis screens to identify mutants with specific defects in embryonic motility (Granato et al., 1996). Mutants that demonstrated similar, abnormal behavior were grouped into phenotypic classes, including the accordion class. Starting around 21 hours post-fertilization (hpf), wild-type embryos demonstrate smooth, alternating tail coils. By contrast, accordion class mutants compress along the rostro-caudal axis and relax, like an accordion. Quetschkommode (que) mutants were grouped into the accordion class but, outside of the initial study identifying these mutants, it has not been characterized nor has the identity of the mutated gene been reported.
In this study, we reveal that que mutants contain a mutation that disrupts the E2 subunit of the BCKD complex. Correspondingly, we present evidence that que mutants accumulate high concentrations of BCAAs. que larvae also contain reduced levels of the neurotransmitter glutamate, which probably contributes to the aberrant CNS function and abnormal behavior observed in this mutant. Combined, these data identify the que mutant as a vertebrate model of MSUD, which holds promise as a powerful tool to better understand this disease and identify therapeutic compounds.
RESULTS
que mutants exhibit abnormal rostro-caudal compressions in response to touch
A single allele of que (ti274) was identified from a previously performed mutagenesis screen (Granato et al., 1996; Zottoli and Faber, 2000). The mutation is recessive, and homozygous mutants first demonstrate abnormal rostro-caudal compressions at around 72 hpf (data not shown). The abnormal behavior becomes more robust by 96 hpf. At this time point, high-speed video analysis shows that wild-type larvae respond to touch by first performing a large-amplitude body bend in which the nose touches the tip of the tail, a so-called C-start or C-bend (defined here as greater than 110°), followed by lower-amplitude body undulations to swim away (Fig. 1A; supplementary material Movie 1) (Eaton et al., 1977; Zottoli and Faber, 2000). By contrast, que mutants do not perform the initial C-bend or lower-amplitude body undulations, and instead demonstrate rostro-caudal shortening (Fig. 1B; supplementary material Movie 2). This behavior, called accordion behavior (Granato et al., 1996), is due to abnormal coordination of axial left-right muscle contractions. Other mutants have been shown to demonstrate accordion behavior due to defects in either CNS function, such as bandoneon (beo), or muscle relaxation, such as accordion (acc) (Gleason et al., 2004; Hirata et al., 2004; Hirata et al., 2005; Olson et al., 2010). We used kinematic analysis to compare the swimming behavior of wild-type, beo, acc and que larvae to each other to determine whether we could distinguish defects in CNS function from defects in muscle relaxation and classify que mutants. No clear trend emerged; however, que mutants consistently show the most dramatic disruption of swimming behavior compared with acc or beo mutants. Although acc and beo mutants demonstrate abnormal swimming behavior, C-bends are often observed (Fig. 1C–E). By contrast, que mutants very rarely execute large-amplitude body bends (Fig. 1F). que mutants continue to perform accordion behavior 5–6 days post-fertilization (dpf). They fail to inflate the swim bladder, which would enable them to feed, and eventually die around 7 dpf.
Fig. 1.

que mutants exhibit abnormal swimming behavior at 96 hpf. (A,B) Selected frames from high-speed video recordings are shown with times indicated in milliseconds. (A) A wild-type larva demonstrates a normal C-bend (A4, asterisk) in response to a touch stimulus, followed by smaller-amplitude body undulations to clear the field (A5-A12). (B) A que mutant demonstrates abnormal rostro-caudal shortening and it fails to escape. (C–F) Kinematic traces are shown, with zero degrees indicating a straight body and positive and negative angles representing body bends in opposite directions. Time is shown in seconds. Ten representative traces are shown for each phenotype. (C) Wild-type embryos typically perform a C-bend (defined here as greater than 110°; asterisks) followed by smaller-amplitude body undulations. (D) bandoneon (beo) mutants, which contain a CNS defect and demonstrate behavior similar to que, sometimes perform a C-bend followed by abnormal body bends. (E) accordion (acc) mutants, which contain a muscle relaxation defect and also demonstrate behavior similar to que, sometimes perform a C-bend but fail to perform smaller-amplitude body bends. (F) que mutants rarely perform a C-bend and demonstrate few smaller-amplitude body undulations.
que mutants demonstrate abnormal motor output
To better examine whether que mutants harbor a defect in CNS function as opposed to a defect in muscle relaxation, we analyzed fictive locomotor output from the spinal cord by performing extracellular peripheral nerve recordings. Zebrafish larvae demonstrate bouts of motor output in response to touch (Fig. 2A,B). These bouts are composed of tightly coordinated bursts that alternate rapidly between the left and right sides and orchestrate the axial muscle contractions that constitute swimming. In wild-type larvae, we observed rapid alternations in locomotor output between the left and right sides, with little overlap in bursting activity (6.5±6%, n=5; Fig. 2C), as described previously (Masino and Fetcho, 2005). In que mutants, although the coordination of left-right locomotor activity was similar to wild-type siblings (compare Fig. 2C with 2D), the amount of overlap between left-right bursting activity was significantly increased (19.1±11.3%, n=7, t=−2.3, P<0.05). This increase in activity overlap is consistent with the abnormal coordination of left-right muscle contractions performed by que mutants. These data do not rule out the possibility that que mutants contain a defect in muscle relaxation; however, they do indicate that abnormal motor output from the CNS at least contributes to the behavior of this mutant.
Fig. 2.

The spinal locomotor output is altered in que mutants. (A,B) Representative single extracellular peripheral nerve recordings from (A) wild type and (B) a que mutant. que mutants produce a greater number of bouts following a gentle tap to the head (arrow) than do wild type. (C,D) Representative paired (left-right) extracellular peripheral nerve recordings in which (C) wild type fish demonstrate tightly coordinated bursting activity with little overlap. By contrast, (D) que mutants generate greater bursting overlap. Gray bars denote burst activity in the top trace and extend to the bottom trace for ease of comparison.
To further characterize potential differences in locomotor output between wild-type and que mutant siblings, we examined a range of bout and burst properties related to rhythmic locomotor activity during fictive swimming (Masino and Fetcho, 2005). Although most of these properties were not significantly different between wild-type and mutant larvae (Table 1), que mutants generated a significantly greater number of bouts following touch stimulus applied to the head than did wild-type larvae [11.6±6.2 bouts and 1.9±0.9 bouts, respectively (t=3.5, P<0.01, n=5)]. These results suggest that, compared with wild type, there are subtle, yet significant, changes in the locomotor circuit that underlie swimming in que mutant larvae and participate in generating the mutant behavioral phenotype.
Table 1.
Comparison of bout and burst properties related to fictive locomotor activity in wild type and que mutants

The que gene encodes dihydrolipoamide branched-chain transacylase E2 (Dbt)
To determine the molecular identity of the que gene, we used a positional cloning strategy. Using a three-generation map cross panel, we screened pools of genomic DNA from wild-type siblings and homozygous mutants with a panel of simple sequence length polymorphism (SSLP) markers. que mapped to chromosome 22, which confirmed previous low-resolution mapping results (Geisler et al., 2007). We then used DNA extracted from single embryos and single nucleotide polymorphism (SNP) markers to refine the map position to a 0.36 cM interval between the markers ENSDART109865 and wu:f63d09 (Fig. 3A). Extensive genome database analysis and sequencing of nearby candidate genes led us to dihydrolipoamide branched chain transacylase E2 (dbt), which encodes a subunit of the BCKD complex, which is required for BCAA metabolism. Zebrafish Dbt is a predicted 493 amino acids in length, and it is ∼78.2% identical to the human protein (data not shown). Mutations in the human DBT gene are known to cause MSUD, which can result in severe dystonia and death if not treated. Given that que mutants demonstrate abnormal behavior and nervous system function consistent with the severe dystonia observed in humans, we sequenced the dbt gene. Sequence analysis of the dbt gene from que homozygotes revealed a single nucleotide substitution in the splice donor site of exon 6 compared with wild type. The guanine of the intron side of this splice site is changed to an adenine (Fig. 3B). To determine whether this change affects the splicing of intron 6, as would be predicted, we performed reverse transcriptase (RT)-PCR using one primer in exon 6 and one primer in exon 7 (Fig. 3C). We found that RNA extracts from wild-type larvae were spliced according to prediction; however, RNA extracts from homozygous mutants revealed a larger transcript that contained the entire 86 base pairs of intron 6, indicating that it was spliced incorrectly (data not shown). This intron alters the sequence downstream of Lys268 and contains four stop codons, which would prematurely truncate the Dbt protein by 224 amino acids. dbt contains an acetyl transferase domain, essential for its function (Chuang et al., 2008), which would be largely absent from the que mutant protein. Interestingly, in humans, a mutation that prematurely truncates the DBT protein at the orthologous position (Lys257) was reported in an individual with the most severe or ‘classic’ form of MSUD (Herring et al., 1992; Chuang et al., 2008). These data indicate that the que mutation is a loss-of-function allele that diminishes or abolishes BCKD complex function.
Fig. 3.

Theque gene encodes dihydrolipoamide branched-chain transacylase E2 (Dbt), a subunit of the BCKD complex. (A) que maps to a 0.36 cM interval on chromosome 22. Molecular markers are shown at the top and the number of recombinants out of the number of meiotic events are shown at bottom. dbt was positioned within the zero recombinant interval. (B) Chromatogram sequence traces of dbt from wild-type, hetero-and homozygous mutant larvae. The nucleotide substitution at the exon-intron boundary (G to A) can be observed in hetero- and homozygotes. (C) A schematic of the dbt protein is shown with the boundaries between exons indicated as vertical lines. Protein domains, including the acetyl transferase domain, are also shown. Below, the wild-type splice pattern is illustrated, with protein-coding exon 6, the sequence at the splice site, the intervening intron, and protein-coding exon 7 depicted. The que mutant splicing pattern is also illustrated, including the nucleotide substitution, which results in a failure to remove the intron. The intron contains four stop codons (asterisks). RT-PCR results using mRNA from wild type, que mutants and –RT controls are also shown using primers targeted towards exon 6 and exon 7. A larger DNA product, containing intron sequence, can be observed using mRNA isolated from que mutants. (D,E) Ten kinematic traces are shown for embryos injected with (D) the control morpholino or (E) a dbt translation-blocking morpholino. Embryos injected with the control morpholino perform C-bends (asterisk) and normal swimming behavior. dbt morphant embryos demonstrate abnormal swimming behavior and few large amplitude body bends, similar to que mutants.
To further confirm the molecular identity of que, we injected wild-type embryos with either a standard control morpholino or a morpholino designed to block translation of dbt. Embryos were injected at the one- to four-cell stage and monitored over the course of development. Embryos injected with the standard control morpholino exhibited mostly normal behavior throughout the course of development (97.2% of surviving larvae, n=107; Fig. 3D). Notably, 37.5% (n=144) of surviving larvae injected with the morpholino designed to target dbt demonstrated clear rostro-caudal compressions and fewer large-amplitude body bends at 96 hpf, similar to que mutants (Fig. 3E). It is important to note that morpholinos are known to lose effectiveness at ∼4–5 dpf owing to turnover, which probably explains why not all embryos injected with the dbt morpholino demonstrated the robust accordion behavior performed by que mutants (Bill et al., 2009). We also attempted to perform rescue experiments in mutant embryos by injecting mRNA encoding dbt at the one- to four-cell stage and analyzing motility behavior at 4 dpf. We did not observe rescue (data not shown); however, mRNA is known to lose effectiveness ∼2 dpf owing to turnover. The que behavioral phenotype is not apparent until 3–4 dpf, which indicates that Dbt is required at this stage of development and precludes mRNA rescue. Regardless, the mapping data, nature of the que mutation, aberrant mRNA splicing observed in mutants, and morpholino phenocopy all argue that the que gene encodes Dbt.
dbt mRNA becomes enriched in the brain and gut organs during development
We next examined the spatial and temporal expression of dbt in developing zebrafish. RT-PCR revealed that dbt mRNA was present at all time points examined from 6 hpf to 120 hpf (Fig. 4A). In situ hybridization also confirmed early expression. dbt was detected at the two-cell stage, indicating it is a maternally deposited mRNA (Fig. 4B). The spatial expression of dbt is initially widespread through 24 hpf (Fig. 4C,D); however, its expression pattern over the next few days of development becomes enriched in the brain and organs in the gut, such as liver and intestine (Fig. 4E–H). These data suggest that dbt plays an important role in BCAA metabolism through function in these tissues. The prominent expression within the brain, in particular, suggests that dbt is important for CNS function. Intriguingly, the expression pattern of dbt, with progressive enrichment in the brain and gut organs over the course of development, is reminiscent of another mitochondrial protein that is important for CNS function, Opa3 (Pei et al., 2010).
Fig. 4.

dbt becomes enriched in the brain and organs in the gut across development. (A) RT-PCR results are shown using mRNA isolated at different time points during zebrafish development. dbt was detected at all stages examined. –RT and β-actin controls are also shown. (B–H) In situ hybridization shows broad dbt expression at (B) the two-cell stage, (C) 12 hpf and (D) 24 hpf. The inset of a cross-section (the black bracket indicates the approximate level of the cross-section) reveals robust dbt expression in muscle. dbt expression become enriched in the brain and gut at (E) 48 hpf and (F) 96 hpf. (G) Sagittal sections of stained 96 hpf embryos illustrate expression in the brain (arrowhead) and gut. The white bracket indicates a portion of the region shown at higher magnification in H. Above the left edge of the white bracket, dbt expression in the liver can be observed, whereas above the right edge of the white bracket, dbt expression in the intestine is revealed.
que mutants harbor elevated levels of BCAAs
In mammalian systems, impaired DBT function, as demonstrated by MSUD-affected individuals, results in elevated levels of BCAAs (Strauss and Morton, 2003; Chuang et al., 2006; Chuang et al., 2008). Because dbt is disrupted in que mutants, we investigated their free amino acid profiles. Owing to the small size of larval zebrafish, a homogenate of 50 whole animals was used for each assay. We compared the free amino acid levels of wild-type and que mutant larvae at 96 hpf. Strikingly, que larvae harbor elevated levels of BCAAs (Fig. 5A,B). Isoleucine, leucine and valine concentrations were 788%, 1006% and 688% (n=3, P<0.01) of those of wild type, respectively. que mutants also showed a marked decrease in free glutamine levels, at 24% of wild-type (n=3, P<0.01). In addition, statistically significant decreases were observed in the levels of a wide variety of free amino acids, including aspartate (16%), GABA (32%) and serine (28% of wild type; all n=3, P<0.01); and alanine (44%), glutamate (38%), glycine (41%), methionine (49%) and threonine (60% of wild type; all n=3, P<0.05). To rule out the possibility that abnormal motor behavior itself alters free amino acid levels, we examined beo mutants, which contain a mutation in the glycine receptor β2 subunit and exhibit abnormal behavior similar to que mutants (Fig. 5C) (Hirata et al., 2005). The free amino acid levels in beo mutants (n=1) were similar to that of wild-type controls, indicating that accordion behavior alone does not substantially alter free amino acid concentrations. Combined, these data provide strong evidence that mutation of the que gene results in an error in amino acid metabolism, yielding a prominent accumulation of BCAAs.
Fig. 5.
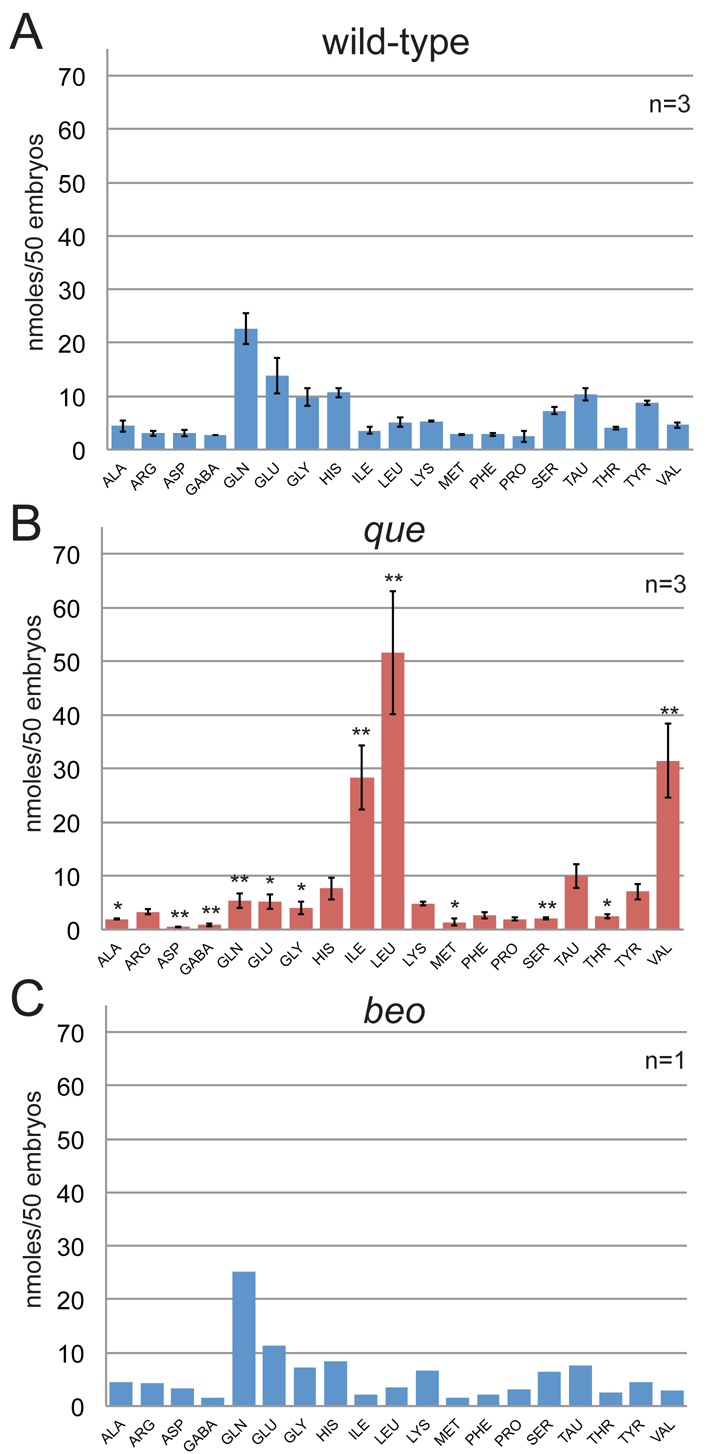
The free amino acid profile of que mutants shows elevated levels of BCAAs at 96 hpf. The amino acids are referred to by their three-letter code, except for GABA. Each experiment contained a homogenate of 50 larvae. The error bars indicate standard error. (A) The free amino acid profile for wild-type larvae (n=3). (B) The free amino acid profile of que mutant larvae reveals a dramatic accumulation of BCAAs: isoleucine, leucine and valine. Other amino acid levels were reduced. * Significant difference from wild-type at P<0.05; ** significant difference from wild-type at P<0.01 (n=3). (C) The free amino acid profile of beo, a zebrafish mutant that demonstrates abnormal behavior owing to a CNS defect, indicates that abnormal behavior alone does not markedly alter free amino acid levels (n=1).
Glutamate levels are reduced in the brain of que mutant larvae
Although the neuropathology of MSUD is not well understood, reduced concentrations of neurotransmitters, including glutamate, were observed in the intermediate MSUD mouse model (Zinnanti et al., 2009). Neurotransmitter depletion was found to correlate with abnormal motor behavior and a highly abnormal posture consisting of recumbency and stiff, extended limbs. Given that our analysis of the free amino acids levels in que mutants showed decreased concentrations of free glutamate and these mutants demonstrate abnormal CNS function and motor behavior, we examined the distribution of glutamate using an antibody. As a control for antibody penetration and overall tissue morphology, we also stained using an acetylated tubulin antibody. Antibody penetration and general morphology of the brain of que mutants seemed similar to wild type at 96 hpf (compare Fig. 6A with 6D). By contrast, glutamate levels were markedly reduced in que mutant larvae (n=5 embryos, 12 sections, P<0.01; compare Fig. 6B with 6E, Fig. 6G), which probably contributes to the abnormal nervous system function and behavior observed by this stage of development.
Fig. 6.

que mutants contain a reduced concentration of glutamate in the brain. (A–F) Cross-sectional views of the hindbrain of 96 hpf larvae are shown. Immunohistochemistry using antibodies against acetylated tubulin, which predominantly labels axon tracts, reveals the overall structure of the brain and demonstrates tissue penetration of the antibodies. Staining using an antibody against L-glutamate illustrates the distribution of this neurotransmitter. (A) Labeling with the anti-acetylated tubulin reveals the axon tracts and overall structure of the hindbrain of wild-type larvae. (B) The hindbrain of wild-type larvae contains a broad distribution of L-glutamate. (C) The merged images show several L-glutamate-positive cells surrounded by anti-acetylated tubulin labeling. (D) The overall structure of the hindbrain of que mutants revealed by anti-acetylated tubulin appears similar to the hindbrain wild-type larvae. (E) The fluorescence intensity of labeling with the L-glutamate antibody is greatly reduced compared with wild type when imaged using the same microscope settings. However, increasing the gain of the confocal microscope shows more faint L-glutamate staining (inset). (F) The merged images show little L-glutamate staining compared with acetylated tubulin labeling. (G) The graph shows a significant reduction in L-glutamate staining intensity in que mutants normalized to acetylated tubulin staining. The fluorescent intensity values are the analog-to-digital converter values of the entire frame (n=5 embryos, 12 sections, **P<0.01). Very similar results were obtained when a region of interest was selected to encompass a smaller, designated portion of the brain.
DISCUSSION
In this study, we revealed that the dbt gene plays an essential role in developing zebrafish. The molecular nature of the que mutation, phenocopy through antisense morpholino injection, and the profile of free amino acid concentrations indicate that loss of dbt function result in abnormal amino acid metabolism and a dramatic accumulation of BCAAs. We determined that the dbt gene becomes enriched in the brain and organs in the gut during zebrafish development, and that impaired dbt function results in reduced levels of glutamate in the brain. Because glutamate is crucial for CNS function, the reduced levels of this neurotransmitter probably promote the abnormal spinal cord output and accordion behavior demonstrated by que mutants.
dbt is required for brain function in zebrafish
The findings from this study, as well as observations in rodent and human systems, suggest a model for how mutation of the zebrafish dbt gene leads to abnormal swimming. In mammalian systems, dbt is required for the second step of BCAA metabolism, and its impairment leads to elevated levels of BCAAs and α-keto acids in plasma and tissue (Chuang et al., 2006; Chuang et al., 2008). Elevated BCAAs in the plasma, in particular leucine, are thought to out-compete other amino acids at the blood-brain barrier, which results in neurotransmitter deficiencies, growth restrictions, cytotoxic edema, myelin disruption, and impaired energy metabolism throughout the CNS (Zinnanti et al., 2009). α-keto acid toxicity has also been proposed to directly disrupt CNS function. Intracranial injection of α-ketovaleric acid, which is derived from valine, has been shown to elicit seizures in rats, whereas administration of other α-keto acids had no behavioral effect (Coitinho et al., 2001). In individuals with MSUD, reducing the concentrations of BCAAs and α-keto acids in the plasma by liver transplantation can protect CNS function and development (Strauss et al., 2006).
We propose that very similar mechanisms regulate BCAA metabolism in zebrafish (Fig. 7). Throughout the first 5 days of zebrafish development, the embryo consumes the presumptive equivalent of a high-protein diet in mammals by absorbing BCAA-containing proteins from the yolk (Link et al., 2006; Tay et al., 2006). During the earliest stages of development, within the first few hours post-fertilization, the metabolic needs of the embryo are largely met by maternally derived mitochondria and mRNA (Mendelsohn and Gitlin, 2008; Zhang et al., 2008; Abrams and Mullins, 2009). However, as embryogenesis proceeds, BCAA metabolism increasingly relies upon zygotic transcription. In wild-type embryos, BCKD complex function in the liver, other gut organs and the brain itself, protect the CNS from BCAA toxicity, similar to mammalian systems. According to this model, BCKD complex function supports appropriate import of amino acids into the CNS, robust metabolic generation of neurotransmitters, which are essential to support the coordinated CNS output that generates vigorous swimming behavior. In que mutants, our data indicate that mutation of dbt disrupts BCKD complex function to cause the toxic accumulation of BCAAs and, probably, α-keto acids. This error probably causes abnormal retention, metabolism and import of amino acids into the CNS, reduced levels of glutamate and other neurotransmitters, abnormal CNS function, and accordion behavior (Fig. 7). It will be interesting to use transgenic approaches to determine whether restoring gene function in the liver of que mutants preserves normal CNS function, as shown in mammals (Strauss et al., 2006; Skvorak et al., 2009a; Skvorak et al., 2009b). Driving gene expression in other organs not yet explored in mammals can also be investigated, which might indicate new therapeutic options for individuals with MSUD.
Fig. 7.

A working model of how mutation of dbt results in abnormal, accordion behavior. Similar to mammalian systems, we propose that wild-type zebrafish regulate metabolism of BCAAS via the BCKD complex. Many of these metabolic or molecular steps (black arrows) might occur in organs in the gut (such as the liver and intestine) but also the CNS. Amino acids (AA), such as glutamine, are transported across the blood-brain barrier and used to generate glutamate, GABA and other neurotransmitters (NT). These neurotransmitters are required for coordinated nervous system output to orchestrate swimming behavior. In que mutants, we propose that impaired BCKD function results in the accumulation of BCAA and α-keto acids. This yields reduced retention and metabolism, and reduced transport of other amino acids (white arrows) across the blood-brain barrier, yielding diminished neurotransmitter synthesis. The abnormal levels of neurotransmitters contribute to aberrant nervous system output and abnormal, accordion behavior. It is also possible that elevated concentrations of α-keto acids directly disrupt neural circuits to cause accordion behavior.
que mutants are a new animal model of MSUD
que larvae harbor a mutation in zebrafish dbt, which results in elevated BCAA levels, similar to both the mouse models of MSUD and affected humans. In the mouse model of intermediate MSUD, elevated levels of BCAAs have been shown to correlate with progressive disruption of CNS function and concomitant defects in motor behavior that culminate in severe dystonia (Silberman et al., 1961; Morton et al., 2002; Zinnanti et al., 2009). Similarly, severe dystonia has been reported in MSUD-affected individuals during acute metabolic decompensation (Silberman et al., 1961; Morton et al., 2002; Zinnanti et al., 2009). que mutants demonstrate a progressive defect in motor behavior that culminates in abnormal CNS function and accordion behavior. Accordion behavior is probably the expression of severe dystonia in developing zebrafish. We and others have previously shown that zebrafish mutants that exhibit accordion behavior contain mutations in genes known to control movement and muscle tone in mammalian systems (Downes and Granato, 2004; Gleason et al., 2004; Hirata et al., 2004; Hirata et al., 2005; Wang et al., 2008; Olson et al., 2010).
The findings from this study indicate that que mutants are a new animal model of MSUD. One aspect of MSUD is the distinct, maple syrup smell of bodily secretions of affected individuals. We did not detect any distinct odor of que mutants (data not shown); however, this is probably due to the small size and minute amounts of secretions produced by larval zebrafish. Nevertheless, que larvae seem to recapitulate molecular, biochemical, cellular and behavioral aspects of MSUD. Because larval zebrafish contain a smaller nervous system than do mammalian systems, with fewer numbers of cells, que mutants provide a promising system to better characterize the progression of CNS injury in response to BCAA toxicity. Moreover, the small size, aquatic nature, development that is external to the mother and the ability to obtain large numbers of zebrafish embryos make them amenable to small-molecule screens (Zon and Peterson, 2010). The behavioral phenotype of que mutants is robust and easily quantifiable; therefore, que mutants could be developed into a high-throughput system to screen libraries of compounds to identify small molecules that improve swimming behavior. Compounds that improve the behavioral phenotype of que mutants could be candidate therapeutics for individuals with MSUD.
Other aspects of MSUD can also be investigated using larval zebrafish. Gene targeting approaches can be readily employed, such as morpholino injection or zinc-finger nuclease technology, to model MSUD caused by disruption of other BCKD complex subunits (Ekker, 2008; Bill et al., 2009). These technologies can also be used to examine the in vivo role of BCKD regulatory proteins, such as the BCKD phosphatase or kinase. Genetic modifier screens can also be performed using the que mutant to search for genes that can compensate for disruptions in BCAA metabolism. Taken together, these approaches can provide a promising platform to better understand CNS metabolism and develop new therapies to combat MSUD.
METHODS
Zebrafish maintenance and breeding
All animal protocols were approved by the Institutional Animal Care and Use Committees (IACUC) at the University of Massachusetts and the University of Minnesota. Zebrafish were raised and maintained according to standard procedures. Developing zebrafish were kept at 28.5°C in E3 media and staged according to morphological criteria (Kimmel et al., 1995; Parichy et al., 2009). Experiments were performed using queti274, beoap21 and acctq206 mutant alleles maintained on a mixed Tübingen (Tü) or tub longfin (TLF) genetic background.
Behavioral analysis
To characterize swimming behavior, light-touch stimuli were applied to the head of larvae using a 1 mm insect pin. The response was recorded using a high-speed video camera (Fastec Imaging, San Diego, CA), recording 500–1000 frames per second, mounted to a 35 mm lens (Nikon, Melville, NY). The head-to-tail angle for each frame was measured using automated software developed by G.B.D.’s laboratory (Kelly Anne McKeown and Sandy Whittlesey unpublished). Briefly, pixel density analysis was used to identify three landmarks along the larval body: the tip of the nose, the border between the yolk ball and yolk extension, and the tip of the tail. These three points form an angle, and these angles were plotted over time using Microsoft Excel.
Electrophysiological recordings
Zebrafish larvae at 4 dpf were anesthetized with 0.02% Tricaine-S (Western Chemical) in extracellular recording solution (Legendre and Korn, 1994; Drapeau et al., 1999; Masino and Fetcho, 2005) and paralyzed with 0.1% (w/v) α-bungatoxoin (Sigma), which significantly reduced or abolished postsynaptic muscle activity based on patch recordings from muscle fibers (Masino and Fetcho, 2005). The extracellular solution was superfused continuously at 22–26°C. Larvae were pinned in a dorsoventral position to a Sylgard-lined glass-bottom Petri dish and the skin was removed. Extracellular suction electrode recording techniques were used to monitor the activity of peripheral nerves during fictive behavior (Masino and Fetcho, 2005). Activity occurred spontaneously but was also initiated by gently applying a touch stimulus to the head with a tungsten pin controlled and positioned by a manual micromanipulator (MX130, Siskiyou, Grants Pass, OR). The tip of the extracellular suction electrode (∼15 μm tip diameter) was positioned at the dorsoventral midline of a myotomal cleft where the skin had been removed. All extracellular recordings were restricted to between body segments 7 and 15. A MultiClamp 700B (Molecular Devices, Sunnyvale, CA) amplifier was used to monitor extracellular voltage in current-clamp mode at a gain of 1000 (Rf=50 MO) with the low- and high-frequency cut-off at 100 and 4000 Hz, respectively. Recordings were sampled at 10 kHz. Extracellular recordings were digitized using a digitizing board (DigiData series 1440A, Molecular Devices, Sunnyvale, CA) acquired using pClamp 10 software and rectified offline.
Analysis of peripheral nerve activity
A program written in MATLAB (Mathworks, Natick, MA) was used to analyze the data. Estimates of mean burst frequency were determined from a Fourier transform, such that the initial estimate of mean burst frequency was the frequency at which the Fourier transform magnitude peaked over a frequency band from 0.1 to 5 Hz. The rectified voltage recordings were smoothed with a Gaussian-weighted moving average with 99% of the weight concentrated over an interval whose width was one-quarter of the reciprocal of the estimated burst frequency (‘quarter-width’).
The occurrence times of rhythmic bursts in the smoothed voltages were determined with an algorithm that searched for local peaks and troughs over quarter-width intervals while forcing adjacent peaks and troughs to be separated by at least quarter-width, and furthermore forcing peaks and troughs to alternate. With the peaks and troughs defined, the individual ‘burst sections’ were then defined as the interval between adjacent troughs. To determine the start of individual bursts, the burst-onset was defined as the time at which the smoothed waveform rose from the first trough to 10% of the way to the next peak. Similarly, burst-termination was defined as the time at which the smoothed waveform fell from the peak by 90% of the vertical distance to the next trough. Next, the analysis program was used to determine the bout and burst properties for each voltage trace in a manner similar to that used by Masino and Fetcho (Masino and Fetcho, 2005). Finally, the amount of overlap in activity between alternating (left-right) bursts was measured as the proportion of the total burst time occupied by the simultaneous activity in the paired (left-right) extracellular recordings (Fig. 2C,D, overlap indicated by gray bars). The means and standard deviations for each parameter were then determined.
Chromosomal mapping and sequence analysis
Crosses between fish heterozygous for the que allele and WIK fish were used to generate a three-generation map cross panel. F2 que mutant embryos and wild-type siblings were collected, sorted based upon the 96 hpf phenotype and stored in methanol at −20°C. DNA was extracted from more than 833 mutant larvae, and SSLP markers and SNP markers were obtained and generated against genes to refine the mapping interval. Exons and intron-exon boundaries of candidate genes were sequenced (Genewiz, South Plainfield, NY) from wild type, que siblings and homozygous mutants.
Morpholino analysis
Wild-type zebrafish embryos were pressure injected at the one- to four-cell stage with 12 ng of morpholino designed to block translation of dbt or the standard control morpholino (Gene Tools, Phinomath, OR). This amount of morpholino was selected based upon dose-response experiments in which higher doses were found to generate morphological defects and/or lethality. The sequence of the translation-blocking morpholino was 5′-CGCACAG-TAATGACCGCCGCCATCT-3′. Underlined residues indicate the start codon. The control morpholino sequence was 5′-CCTCTTAC-CTCAGTTACAATTTATA-3′. The embryos were raised at 28.5°C, and locomotive behavior was examined across development. Kinematic analysis, as described above, was performed at 96 hpf.
RT-PCR
RT-PCR was used to analyze mRNA splicing in mutants as well as examine expression during development. Primers designed against dbt protein-coding exon 6 (5′-ATCAAACTAAGCGAAG-TTGTCGG-3′) and exon 7 (5′-GCGCAACCGGACCAAC-3′) were used to amplify cDNA from wild-type and homozygous que mutant larvae. The primers used to amplify β-actin were 5′-CACACCGTGCCCATCTATGA-3′ and 5′ -AGGATCTTCATCAGGTAGTCTGTCAG-3′. The RNAs were reverse transcribed using the Omniscript kit (Qiagen, Venlo, The Netherlands). The dbt PCR products were sequenced for confirmation (Genewiz, South Plainfield, NY). RT-PCR reactions were performed multiple times to decrease the likelihood of amplification artifacts.
TRANSLATIONAL IMPACT.
Clinical issue
Maple syrup urine disease (MSUD) is an inherited disorder that results in disrupted metabolism of branched-chain amino acids (isoleucine, leucine and valine), resulting in the toxic accumulation of these amino acids and their byproducts. This disease can have a devastating impact on the central nervous system (CNS), resulting in mental retardation, severe dystonia, coma or death if not treated. Although mouse models of the disease have been developed and some disease genes are known, the cellular and molecular mechanisms that promote brain injury in individuals with MSUD are not well understood.
Results
In this paper, the authors characterize a zebrafish mutant called quetschkommode (que) that exhibits defects in motor behavior. The mutation is found in the dihydrolipoamide branched chain transacylase E2 (dbt) gene, a homolog of the human DBT gene, which can cause MSUD when mutated. In addition to abnormal behavior, que fish are shown to have disrupted metabolism of branched-chain amino acids and aberrant CNS function, mirroring features of human MSUD.
Implications and future directions
These data reveal the que zebrafish mutant to be a new animal model of MSUD. Because zebrafish offer a number of advantages for cellular, molecular, pharmacological and genetic analysis, the que mutant provides a unique tool to deepen our understanding of and develop new therapeutic options for this disease.
Whole-mount in situ hybridization
Antisense digoxigenin probes were generated against dbt using cDNA (Genbank ID BC090917) acquired from Open Biosystems (Huntsville, AL). Whole-mount, colorimetric in situ hybridization was performed using established protocols (Thisse and Thisse, 2008) and examined using a compound microscope (Zeiss, Thornwood, NY) attached to a digital camera (Zeiss, Thornwood, NY). Cross-sections were generated by hand sectioning in situ hybridization stained embryos with a razor blade attached to a surgical blade holder. To generate sagittal sections, in situ hybridization stained embryos were embedded in 1.5% Agar and 5% sucrose. Blocks were kept in 30% sucrose solution overnight. The next day blocks were cut into 20 mm sections using a cryostat (Leica, Buffalo Grove, IL).
Amino acid quantification
For each amino acid quantification experiment 50 96-hpf larvae were sorted based upon the locomotor phenotype, were flash frozen in liquid nitrogen and stored at −80°C. The samples were homogenized and precipitated with 0.1 M lithium citrate, 3.3% 5-sulphosalicylic acid. The samples were sonicated for 10 minutes, then centrifuged at 4600 g for 20 minutes. The supernatant was then applied to VivaSpin500 size exclusion columns (Sartorium, Germany) and centrifuged at 15,000 g for 4 hours. The flow-through was stored at −80°C then sent to the University of California Davis California Genome and Proteomics Center to resolve free amino acid concentrations.
Antibody staining
The tissue was prepared by embedding 96 hpf larvae as described for in situ hybridization. Double immunostaining was performed using standard protocols (similar to Downes and Granato) (Downes and Granato, 2004). The primary antibodies were rabbit anti-L-glutamate (1:100 dilution, ab9440; Abcam, Cambridge, MA) and mouse anti-acetylated tubulin (1:200 dilution, T6793; Sigma, St Louis, MO). Secondary antibodies were Alexa Fluor 488 goat anti rabbit (1:1000 dilution, A11034; Molecular Probes/Sigma, St Louis, MO) and Alexa Fluor 594 goat anti mouse (1:1000 dilution, A11005; Molecular Probes, Sigma, St Louis, MO). Images were acquired using a confocal microscope (Nikon, Melville, NY).
The fluorescent intensity for acetylated tubulin and L-glutamate antibody staining was quantified using the EZ Viewer program (Nikon, Melville, NY) by collecting entire frames (10,1283 μm2) or selecting a region of interest above the notochord (3060 μm2) for both channels. The numbers used for quantification are the analog-to-digital converter (ADC) values of L-glutamate normalized to acetylated tubulin.
Supplementary Material
Acknowledgments
The authors thank Wuhong Pei and Benjamin Feldman at the National Human Genome Research Institute for helpful advice about amino acid quantification, and members of the Downes laboratory and University of Massachusetts Amherst zebrafish community for thoughtful discussion.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
M.A.M. and G.B.D. conceived and designed the experiments. T.F. and A.M.L. performed the experiments. T.F., A.M.L., M.A.M. and G.B.D. analyzed the data. M.A.M. and G.B.D. contributed reagents/materials/analysis tools. T.F., M.A.M. and G.B.D. wrote the paper.
FUNDING
This work was supported by grants from the Minnesota Medical Foundation [3806-9227-07, 4052-9238-11]; the Office of the Dean of the Graduate School of the University of Minnesota [Grant-in-Aid of Research, Artistry and Scholarship] (to M.A.M.); the Baden-Württemberg Stiftung gGmbH (to T.F.); and the National Institute of Health [R01-NS65054 (to M.A.M.), T32-DA022616 (to A.M.L.), K01-NS057409 to (G.B.D.)].
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.008383/-/DC1
REFERENCES
- Abrams E. W., Mullins M. C. (2009). Early zebrafish development: it’s in the maternal genes. Curr. Opin. Genet. Dev. 19, 396–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bill B. R., Petzold A. M., Clark K. J., Schimmenti L. A., Ekker S. C. (2009). A primer for morpholino use in zebrafish. Zebrafish 6, 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang D. T., Chuang J. L., Wynn R. M. (2006). Lessons from genetic disorders of branched-chain amino acid metabolism. J. Nutr. 136 Suppl. 1, 243S–249S [DOI] [PubMed] [Google Scholar]
- Chuang D. T., Wynn R. M., Shih V. E. (2008). Maple syrup urine disease (branched-chain ketoaciduria). In The Online Metabolic And Molecular Bases Of Inherited Diseases (ed. Valle D., Beaudet A. L., Vogelstein B., Kinzler K. W., Antonarakis S. E., Ballabio A.), Chapter 87, pp. 1–42 New York: McGraw-Hill [Google Scholar]
- Coitinho A. S., de Mello C. F., Lima T. T., de Bastiani J., Fighera M. R., Wajner M. (2001). Pharmacological evidence that alpha-ketoisovaleric acid induces convulsions through GABAergic and glutamatergic mechanisms in rats. Brain Res. 894, 68–73 [DOI] [PubMed] [Google Scholar]
- Downes G. B., Granato M. (2004). Acetylcholinesterase function is dispensable for sensory neurite growth but is critical for neuromuscular synapse stability. Dev. Biol. 270, 232–245 [DOI] [PubMed] [Google Scholar]
- Downes G. B., Granato M. (2006). Supraspinal input is dispensable to generate glycine-mediated locomotive behaviors in the zebrafish embryo. J. Neurobiol. 66, 437–451 [DOI] [PubMed] [Google Scholar]
- Drapeau P., Ali D. W., Buss R. R., Saint-Amant L. (1999). In vivo recording from identifiable neurons of the locomotor network in the developing zebrafish. J. Neurosci. Methods 88, 1–13 [DOI] [PubMed] [Google Scholar]
- Eaton R. C., Farley R. D., Kimmel C. B., Schabtach E. (1977). Functional development in the Mauthner cell system of embryos and larvae of the zebra fish. J. Neurobiol. 8, 151–172 [DOI] [PubMed] [Google Scholar]
- Ekker S. C. (2008). Zinc finger-based knockout punches for zebrafish genes. Zebrafish 5, 121–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler R., Rauch G. J., Geiger-Rudolph S., Albrecht A., van Bebber F., Berger A., Busch-Nentwich E., Dahm R., Dekens M. P., Dooley C., et al. (2007). Large-scale mapping of mutations affecting zebrafish development. BMC Genomics 8, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleason M. R., Armisen R., Verdecia M. A., Sirotkin H., Brehm P., Mandel G. (2004). A mutation in serca underlies motility dysfunction in accordion zebrafish. Dev. Biol. 276, 441–451 [DOI] [PubMed] [Google Scholar]
- Granato M., van Eeden F. J., Schach U., Trowe T., Brand M., Furutani-Seiki M., Haffter P., Hammerschmidt M., Heisenberg C. P., Jiang Y. J., et al. (1996). Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 123, 399–413 [DOI] [PubMed] [Google Scholar]
- Herring W. J., McKean M., Dracopoli N., Danner D. J. (1992). Branched chain acyltransferase absence due to an Alu-based genomic deletion allele and an exon skipping allele in a compound heterozygote proband expressing maple syrup urine disease. Biochim. Biophys. Acta 1138, 236–242 [DOI] [PubMed] [Google Scholar]
- Hirata H., Saint-Amant L., Waterbury J., Cui W., Zhou W., Li Q., Goldman D., Granato M., Kuwada J. Y. (2004). Aaccordion, a zebrafish behavioral mutant, has a muscle relaxation defect due to a mutation in the ATPase Ca2+ pump SERCA1. Development 131, 5457–5468 [DOI] [PubMed] [Google Scholar]
- Hirata H., Saint-Amant L., Downes G. B., Cui W. W., Zhou W., Granato M., Kuwada J. Y. (2005). Zebrafish bandoneon mutants display behavioral defects due to a mutation in the glycine receptor beta-subunit. Proc. Natl. Acad. Sci. USA 102, 8345–8350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homanics G. E., Skvorak K., Ferguson C., Watkins S., Paul H. S. (2006). Production and characterization of murine models of classic and intermediate maple syrup urine disease. BMC Med. Genetics 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel C. B., Patterson J., Kimmel R. O. (1974). The development and behavioral characteristics of the startle response in the zebra fish. Dev. Psychobiol. 7, 47–60 [DOI] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995). Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- Legendre P., Korn H. (1994). Glycinergic inhibitory synaptic currents and related receptor channels in the zebrafish brain. Eur. J. Neurosci. 6, 1544–1557 [DOI] [PubMed] [Google Scholar]
- Link V., Shevchenko A., Heisenberg C. P. (2006). Proteomics of early zebrafish embryos. BMC Dev. Biol. 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masino M. A., Fetcho J. R. (2005). Fictive swimming motor patterns in wildtype and mutant larval zebrafish. J. Neurophysiol. 93, 3177–3188 [DOI] [PubMed] [Google Scholar]
- McKeown K. A., Downes G. B., Hutson L. D. (2009). Modular laboratory exercises to analyze the development of zebrafish motor behavior. Zebrafish 6, 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn B. A., Gitlin J. D. (2008). Coordination of development and metabolism in the pre-midblastula transition zebrafish embryo. Dev. Dyn. 237, 1789–1798 [DOI] [PubMed] [Google Scholar]
- Morton D. H., Strauss K. A., Robinson D. L., Puffenberger E. G., Kelley R. I. (2002). Diagnosis and treatment of maple syrup disease: a study of 36 patients. Pediatrics 109, 999–1008 [DOI] [PubMed] [Google Scholar]
- Olson B. D., Sgourdou P., Downes G. B. (2010). Analysis of a zebrafish behavioral mutant reveals a dominant mutation in atp2a1/SERCA1. Genesis 48, 354–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parichy D. M., Elizondo M. R., Mills M. G., Gordon T. N., Engeszer R. E. (2009). Normal table of postembryonic zebrafish development: staging by externally visible anatomy of the living fish. Dev. Dyn. 238, 2975–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei W., Kratz L. E., Bernardini I., Sood R., Yokogawa T., Dorward H., Ciccone C., Kelley R. I., Anikster Y., Burgess H. A., et al. (2010). A model of Costeff Syndrome reveals metabolic and protective functions of mitochondrial OPA3. Development 137, 2587–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint-Amant L., Drapeau P. (1998). Time course of the development of motor behaviors in the zebrafish embryo. J. Neurobiol. 37, 622–632 [DOI] [PubMed] [Google Scholar]
- Silberman J., Dancis J., Feigin I. (1961). Neuropathological observations in maple syrup urine disease: branched-chain ketoaciduria. Arch. Neurol. 5, 351–363 [DOI] [PubMed] [Google Scholar]
- Skvorak K. J., Hager E. J., Arning E., Bottiglieri T., Paul H. S., Strom S. C., Homanics G. E., Sun Q., Jansen E. E., Jakobs C., et al. (2009a). Hepatocyte transplantation (HTx) corrects selected neurometabolic abnormalities in murine intermediate maple syrup urine disease (iMSUD). Biochim. Biophys. Acta 1792, 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skvorak K. J., Paul H. S., Dorko K., Marongiu F., Ellis E., Chace D., Ferguson C., Gibson K. M., Homanics G. E., Strom S. C. (2009b). Hepatocyte transplantation improves phenotype and extends survival in a murine model of intermediate maple syrup urine disease. Mol. Ther. 17, 1266–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss K. A., Morton D. H. (2003). Branched-chain ketoacyl dehydrogenase deficiency: maple syrup disease. Curr. Treat. Options Neurol. 5, 329–341 [DOI] [PubMed] [Google Scholar]
- Strauss K. A., Mazariegos G. V., Sindhi R., Squires R., Finegold D. N., Vockley G., Robinson D. L., Hendrickson C., Virji M., Cropcho L., et al. (2006). Elective liver transplantation for the treatment of classical maple syrup urine disease. Am. J. Transplant. 6, 557–564 [DOI] [PubMed] [Google Scholar]
- Strauss K. A., Wardley B., Robinson D., Hendrickson C., Rider N. L., Puffenberger E. G., Shelmer D., Moser A. B., Morton D. H. (2010). Classical maple syrup urine disease and brain development: principles of management and formula design. Mol. Genet. Metab. 99, 333–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay T. L., Lin Q., Seow T. K., Tan K. H., Hew C. L., Gong Z. (2006). Proteomic analysis of protein profiles during early development of the zebrafish, Danio rerio. Proteomics 6, 3176–3188 [DOI] [PubMed] [Google Scholar]
- Thisse C., Thisse B. (2008). High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat. Protoc. 3, 59–69 [DOI] [PubMed] [Google Scholar]
- Wang M., Wen H., Brehm P. (2008). Function of neuromuscular synapses in the zebrafish choline-acetyltransferase mutant bajan. J. Neurophysiol. 100, 1995–2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Z., Ouyang Y. C., Hou Y., Schatten H., Chen D. Y., Sun Q. Y. (2008). Mitochondrial behavior during oogenesis in zebrafish: a confocal microscopy analysis. Dev. Growth Differ. 50, 189–201 [DOI] [PubMed] [Google Scholar]
- Zinnanti W. J., Lazovic J., Griffin K., Skvorak K. J., Paul H. S., Homanics G. E., Bewley M. C., Cheng K. C., Lanoue K. F., Flanagan J. M. (2009). Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain 132, 903–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zon L. I., Peterson R. (2010). The new age of chemical screening in zebrafish. Zebrafish 7, 1. [DOI] [PubMed] [Google Scholar]
- Zottoli S. J., Faber D. S. (2000). The Mauthner cell: what has it taught us? Neuroscientist 6, 26–38 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.