

Abstract
Activating mutations in the RAS family or BRAF frequently occur in many types of human cancers but are rarely detected in breast tumors. However, activation of the RAS-RAF-MEK-ERK Mitogen-Activated Protein Kinase (MAPK) pathway is commonly observed in human breast cancers, suggesting that other genetic alterations lead to activation of this signaling pathway. To identify breast cancer oncogenes that activate the MAPK pathway, we screened a library of human kinases for their ability to induce anchorage-independent growth in a derivative of immortalized human mammary epithelial cells (HMLE). We identified PAK1 as a kinase that permitted HMLE cells to form anchorage-independent colonies. PAK1 is amplified in several human cancer types, including 33% of breast tumor samples and cancer cell lines. The kinase activity of PAK1 is necessary for PAK1-induced transformation. Moreover, we show that PAK1 simultaneously activates MAPK and MET signaling; the latter via inhibition of Merlin. Disruption of these activities inhibits PAK1-driven anchorage-independent growth. These observations establish PAK1 amplification as an alternative mechanism for MAPK activation in human breast cancer and credential PAK1 as a breast cancer oncogene that coordinately regulates multiple signaling pathways, the cooperation of which leads to malignant transformation.
Keywords: PAK1, transformation, oncogene, MAPK, MET, Merlin
Introduction
The RAS family of small GTPases, HRAS, KRAS and NRAS, are often mutated in human cancers rendering them constitutively active and oncogenic (reviewed in Lau and Haigis 2009). Oncogenic mutations in the RAS genes are common in selected cancer types, including pancreatic, colon and non-small cell lung cancers. RAS activation may occur directly through these oncogenic mutations or indirectly due to activation of RAS regulators or effectors (reviewed in Downward 2003). Activation of growth factor signaling is a predominant mechanism upstream of RAS that leads to its activation. In epithelial cancers, EGFR and ERBB2 – two ErbB family tyrosine kinase receptors, commonly activate RAS oncogenic function (Mendelsohn and Baselga 2000). Similarly, loss-of-function of a RAS-GAP, NF1, also drives RAS downstream signaling (Bollag et al 1996).
Several alternate mechanisms that activate RAS signaling have also been described. The RAS effector pathway PI3K is activated by mutations of the catalytic subunit PIK3CA, amplification of the downstream target AKT or loss-of-function of PTEN (Scheid and Woodgett 2001). Similarly, activating mutations of BRAF occur in 50% of melanomas leading to constitutive activation of MAPK signaling (Davies et al 2002). These two pathways play important roles in RAS-mediated cell transformation since co-inhibition of PI3K and MAPK efficiently suppresses RAS-driven tumor growth (Engelman et al 2008, Sos et al 2009).
Less than 5% of human breast tumors exhibit oncogenic mutations in the RAS genes (Lau and Haigis 2009, Miyakis et al 1998). RAS signaling in breast cancer is more commonly activated by alterations upstream or downstream of RAS. For example, the RAS pathway is activated through ERBB2 amplification in about 20% of breast tumors (Hynes and MacDonald 2009). Such growth factor activation in breast cancer results in a co-activation of RAS effectors PI3K and MAPK (Neve et al 2002). Co-inhibition of these synergistic pathways has proven more effective than single pathway inhibition for suppression of breast tumorigenesis (Hoeflich et al 2009, Mirzoeva et al 2009). Moreover, co-activation of these RAS effectors is common in breast cancer. Activating mutations in PIK3CA are found in 25–30% of breast tumors while loss-of-function of PTEN by mutation or loss of protein activates the PI3K pathway in 5–30% of human breast tumors (Bachman et al 2004, Freihoff et al 1999, Hennessy et al 2005, Miron et al 2010). In contrast, oncogenic mutations in MAPK pathway components have not been reported. Therefore, alternative mechanisms that activate MAPK signaling to promote breast oncogenesis remain to be defined.
To identify genes that activate or substitute for MAPK activation in breast cancer, we performed a kinase-focused screen in a human mammary epithelial cell transformation model that is dependent on oncogenic RAS. Expression of oncogenic HRAS (HRASV12) in human mammary epithelial cells immortalized with the catalytic subunit of telomerase and SV40 early region exhibit anchorage-independent growth and tumorigenesis (Elenbaas et al 2001). In this transformation model, simultaneous expression of myristoylated-AKT1 and an activated allele of MEK1 (MEK1S218D/S222D, MEKDD) can substitute for oncogenic HRAS. Using HMLE cells expressing MEKDD, we previously found IKBKE as a kinase oncogene amplified in 30% of human breast cancers, and CSNK1E as a transforming gene that also acts as a synthetic lethal partner with activated β-catenin (Boehm et al 2007, Kim et al 2010). To identify genes that activate MAPK signaling and cooperate with active PI3K pathway, we screened for oncogenes in HMLE cells expressing myr-AKT1 (HMLEA). We identified p21-activated kinase 1 (PAK1) as an amplified kinase capable of inducing potent mammary cell transformation through the coordinate regulation of MAPK and MET signaling.
Results
Kinome focused gain-of-function screen for kinases that promote anchorage-independent growth of immortalized human mammary epithelial cells
To identify oncogenes that activate the MAPK pathway in mammary epithelial cells, we expressed an open reading frame (ORF) library composed of kinases in HMLEA cells. Specifically, we pooled 597 myristoylation- and Flag-epitope-tagged kinases, introduced them into HMLEA cells and measured anchorage-independent growth (Figure 1A). Macroscopic colonies formed by HMLEA cells expressing kinase pools or controls were counted at 2 wks. We found that pool #9 formed numerous colonies (Figure 1B). When the 27 kinases in pool #9 were individually tested for anchorage-independent growth of HMLEA cells, we found that the PAK1 induced robust colony formation (Figure 1C). We also observed that myristoylation of PAK1 was unnecessary to induce anchorage-independent growth (Supplemental Figure 1A) and, we used non-myristoylated PAK1for subsequent validation and characterization studies. Expression of PAK1 sufficed to induce anchorage-independent growth of HMLEs (Figure 1D). Thus, our observations establish that PAK1 can transform immortalized human mammary epithelial cells, unlike prior studies carried out in breast cancer cell lines, where PAK1 enhanced anchorage-independent colony formation (Vadlamudi et al 2000). Our findings in HMLEA cells also indicate that PAK1 also cooperates with other oncogenic alterations to induce cell transformation.
Figure 1. Human kinase expression screen in HMLEA cells.

A. An ORF library containing 597 Myr-Flag-epitope tagged Kinase ORFs were organized into 22 pools and screened for their ability to promote anchorage-independent growth of HMLEA cells. B. The average number of macroscopic colonies formed by HMLEA cells expressing the pools or controls at 2 wks. Vector - pBP, MEKDD 1:25 - positive control. C. Anchorage-independent growth of HMLEA cells expressing each of the twenty-seven kinases in Pool #9. D. Anchorage-independent growth of HMLE cells expressing vector control or PAK1. ** p-value <0.001.
PAK1 amplifications in human breast cancer
The observation that PAK1 induces cell transformation suggested that PAK1 could function as an oncogene. To assess the frequency of PAK1 amplification in human cancer, we analyzed a large set of human cancer samples in Tumorscape, a dataset that includes whole genome analyses of somatic copy number alterations in a collection of 3131 human samples, including 243 breast tumor samples and cancer cell lines (Beroukhim et al 2010). We found that PAK1 is amplified in 33% of breast samples (Figure 2A) as well as a smaller fraction of non-small cell lung, ovarian, small cell lung, melanoma and esophageal squamous cancers. These findings confirm and extend smaller scale studies that focused on chromosome 11q13 mainly, but not exclusively, in ovarian cancer (Bekri et al 1997, Bostner et al 2007, Brown et al 2008, Chattopadhyay et al 2010, Kim et al 2006, Lambros et al 2005, Schraml et al 2003, Tu et al 2011). Overall, we found that 16% of all cancers in the Tumorscape exhibited amplification of the locus that contains PAK1.
Figure 2. PAK1 is amplified in human cancers.

A. Copy number profile along chromosome 11q of human tumor samples and cancer cell lines that exhibit highest level of PAK1 amplification divided according to cancer type: breast, non-small cell (NSC) lung, ovarian (Ov), small cell lung (SCL), melanoma (Mel) and esophageal squamous (Esq). PAK1 and CCND1 loci are marked. B. FISH of breast cancer cell lines with (SUM52, SUM190) and without (EFM19, SKBR3) PAK1 amplification. Orange fluorescence indicates chromosome 11 centromere reference probe. Green fluorescence indicates PAK1 probe. White bar = 100 μ. C. Tumorscape copy number data is tabulated along with corresponding average number of PAK1 and reference signals from 100 nuclei per cell line. D. mRNA and protein levels of PAK1 in the four breast cancer cell lines and HMLE cells. E. GISTIC analysis of 3131 human cancers along chromosome 11q showing two distinct amplification peaks containing PAK1 or CCND1. The arrow indicates a higher magnification representation of the dotted area to show PAK1 locus (Red).
To confirm that PAK1 amplifications occur in breast cancer, we performed fluorescence in-situ hybridization (FISH) and found normal copy number in breast cancer cell lines that did not show evidence of PAK1 amplification (EFM19, SKBR3). In contrast, when we examined metaphase spreads derived from breast cancer cell lines that exhibit amplification of PAK1 (SUM52, SUM190), we found clear evidence of PAK1 copy number gain (Figure 2B). The average number of PAK1 signals per nucleus was greater than the chromosome 11 centromere reference signals in SUM52 and SUM190 lines (Figure 2C). The PAK1 FISH signal in the four cell lines correlated with the respective PAK1 copy number in Tumorscape. Furthermore, PAK1 copy number in the four cell lines directly correlated with expression both at the RNA and protein levels (Figure 2D).
Using the Genomic Identification of Significant Targets In Cancer (GISTIC) algorithm (Beroukhim et al 2007) in Tumorscape samples, we found that PAK1 lies on an amplification peak on chromosome 11q distinct from a neighboring peak containing CCND1 (Figure 2E). CCND1 is amplified in 42% of all samples in Tumorscape while 43% of those that exhibit PAK1 amplification also carry CCND1 amplification. These observations strongly suggest that amplifications of PAK1 and CCND1 are independent events.
We also analyzed The Cancer Genome Atlas (TCGA) for PAK1 copy number gain in human cancer samples. We found that 30% of breast invasive adenocarcinoma samples showed significant PAK1 amplification (Q-value 8.04E-39). Together, the observations show that PAK1 is amplified in a significant fraction of human breast tumors and tumor-derived cell lines.
We then investigated whether cell lines that harbor PAK1 amplifications required PAK1 expression for proliferation or anchorage-independent colony formation to establish that PAK1 is one target of the second amplicon on chromosome 11q. We failed to find changes in population-doubling times in SUM52 cells expressing PAK1- or lacZ-specific shRNAs or in HMLE cells expressing control vector or PAK1, indicating that PAK1 expression level does not affect proliferation (Figure 3A). In contrast, suppression of PAK1 by two different PAK1-specific shRNAs in SUM52 and SUM190 lines compromised anchorage-independent growth (Figure 3B). Importantly, in cell lines (SKBR3, EFM19) that do not harbor PAK1 amplification, suppression of PAK1 failed to affect anchorage-independent growth (Figure 3C). These observations demonstrate that cell lines that exhibit amplification and overexpression of PAK1 depend on it for anchorage-independent growth, strongly indicating that one target of the second 11q amplicon is PAK1.
Figure 3. PAK1 amplified cell lines are dependent on PAK1 for transformation.

A. Proliferation of SUM52 cells expressing PAK1- or lacZ-specific shRNAs or HMLE cells expressing vector control or PAK1 calculated over 21 or 23 days. B–C. Effects of suppressing PAK1 on anchorage-independent growth of breast cancer cell lines with (B: SUM52, SUM190) or without (C: EFM19, SKBR3) PAK1 amplification. Error bars = 1 SD. Insets indicate PAK1 protein levels. *** p-value <0.0001.
PAK1 kinase activity in anchorage-independent growth
PAK1 regulates signaling pathways through both kinase-dependent and kinase-independent mechanisms (reviewed in Dummler et al 2009). We evaluated the requirement for PAK1 kinase activity in cell transformation by expressing wild-type PAK1 and its previously characterized kinase-deficient mutant, PAK1K299R (Tang et al 1997). We found that wild-type PAK1, but not PAK1K299R, formed robust anchorage-independent colonies in HMLE (Figure 4A).
Figure 4. PAK1 kinase activity is required for anchorage-independent growth.

A. Anchorage-independent growth by HMLE expressing control vector, wild-type or kinase-dead PAK1 (PAK1K299R). Error bars = 1 SD. ** p-value <0.003 (compared to either vector or kinase-dead control). B. Expression level of PAK1 and its phosphorylation status at T423 and S144 in HMLE expressing vector control, PAK1 or PAK1K299R. C. Phosphorylation levels of three PAK1 substrates; RAF1S338, MEK1S298 and MerlinS518. Corresponding total protein levels are shown.
Prior work has shown that the kinase activity of PAK1 can be assessed by the status of phosphorylation at several internal sites. PAK1 phosphorylation at T423 relieves autoinhibitory conformation, allowing auto-phosphorylation at S144 to maintain kinase activity (reviewed in Bokoch 2003). We evaluated the effect of overexpressing PAK1 on PAK1 kinase activity by assessing the phosphorylation status of PAK1 itself as well as its reported phosphorylation targets. Using HMLE cells expressing a control vector, PAK1 or PAK1K299R, we found that wild-type PAK1 but not PAK1K299R was phosphorylated at both T423 and S144 sites (Figure 4B). However, comparison of total and phosphorylated PAK1 levels indicates that a fraction of PAK1 in HMLE-PAK1 cells may remain in an autoinhibitory conformation. Nevertheless, we found that the phosphorylation levels of three PAK1-specific substrates; RAF1S338, MEK1S298 and MerlinS518, were enhanced in HMLE-PAK1 (Figure 4C; Coles and Shaw 2002, Xiao et al 2002, Zang et al 2002, Zang et al 2008). The elevated phosphorylation levels of PAK1 itself as well as its substrates exclusively in HMLE-PAK1 cells confirm that a greater portion of the wild-type PAK1 is in its active conformation and hence, catalytically active, compared to PAK1K299R. These findings establish that the kinase activity of PAK1 is necessary for the transformation of mammary epithelial cells. Moreover, PAK1 induced phosphorylation of RAF1, MEK1 and Merlin suggests that these pathways may contribute to PAK1-driven transformation.
MAPK signaling and PAK1-driven anchorage-independent growth
Phosphorylation of RAF1S338 and MEK1S298 prime the activation of the MAPK pathway by facilitating phosphorylation of MEK1 at its activation loop and subsequent phosphorylation of ERK1/2. PAK1 expression in HMLE not only increased phosphorylation of RAF1S338 and MEK1S298, two critical upstream members of the MAPK pathway, but also increased phosphorylation of the downstream effectors, ERK1/2T202/Y204 (Figure 4C, 5A). Conversely, suppression of PAK1 in SUM52 repressed phosphorylation of RAF1S338 and ERK1/2T202/Y204, thus indicating regulation of the MAPK pathway by PAK1 (Supplemental Figure 2A).
Figure 5. MAPK activation by PAK1 expression is required for anchorage-independent growth.

A. Phosphorylation status of the MAPK pathway downstream effectors ERK1/2T202/Y204 in HMLE expressing a control vector, PAK1 or PAK1K299R. Total ERK1/2 levels are also shown. B. Anchorage-independent colony formation by HMLE-PAK1 and SUM52 expressing RAF1- or lacZ-specific shRNAs. Corresponding RAF1 protein levels are shown. C. Anchorage-independent growth of HMLE-PAK1, HMLER, SUM52 and EFM19 treated with U0126 at the indicated doses. Representative phospho-ERK1/2T202/Y204 levels are shown. D. Anchorage-independent colony formation by HMLER cells expressing PAK1- or lacZ-specific shRNAs. PAK1 protein levels are shown. Error bars = 1 SD. ***p-value <0.0001. **p-value <0.001.
In consonance with this observation, suppressing RAF1 in two PAK1-dependent cell lines, HMLE-PAK1 and SUM52 suppressed phospho-ERK1/2 levels and compromised anchorage-independent growth (Supplemental Figure 2B; Figure 5B). Furthermore, when we treated SUM52 and EFM19 with U0126, a MEK1/2-specific inhibitor (Favata et al 1998), SUM52 formed dramatically fewer colonies, whereas EFM19 cells, which exhibit no PAK1 copy number gain, were largely unaffected (Figure 5C). We also found that U0126 inhibited ERK1/2T202/Y204 phosphorylation as well as the ability of HMLE-PAK1 to form colonies in a dose-dependent manner (Figure 5C). Together, these genetic and pharmacologic studies indicated that the activation of RAF-MEK-ERK MAPK signaling was essential for PAK1-induced anchorage-independent growth.
Prior work has implicated PAK1 in the regulation of RAS signaling. Even in the presence of oncogenic RAS, loss of PAK1 function or inhibition of PAK1 activators inhibited colony formation (Appledorn et al 2010, Tang et al 1997). Similarly, we found that PAK1 suppression by shRNAs partially inhibited anchorage-independent growth of HMLE expressing HRASV12 (HMLER), reinforcing the role of PAK1 in RAS-driven transformation. We however did not observe any difference in total or phosphorylated levels of PAK1 between HMLE and HMLER cells (Figure 5D; Supplemental Figure 3A). Hence, we conclude that PAK1 gain-of-function activates MAPK pathway, a RAS effector, which is necessary for PAK1-driven anchorage-independent growth. Moreover, amplification resulting in overexpression of PAK1 is an alternate mechanism by which MAPK signaling may be activated in human breast tumors without activating mutations in other components of the RAS-RAF-MAPK pathway.
Merlin function in MET signaling and PAK1-mediated anchorage-independent growth
Merlin or Neurofibromatosis type 2 (NF2) is a tumor suppressor gene deleted in a subset of patients with neurofibromatosis and is a known substrate of PAK1 (reviewed in McClatchey 2007). Merlin and PAK1 are involved in an inhibitory loop where Merlin binds to the p21-binding domain of PAK1, inhibiting its activation and suppressing RAS-dependent transformation (Hirokawa et al 2004, Kissil et al 2003). PAK1, on the other hand, phosphorylates Merlin at S518, which induces translocation of Merlin away from the membrane and inhibits its function (Kissil et al 2002, Shaw et al 2001, Xiao et al 2002).
To investigate whether PAK1 regulation of Merlin contributes to mammary cell transformation, we examined the consequences of modulating PAK1 or NF2 expression in human mammary epithelial cells and breast cancer cell lines. PAK1 expression in HMLE cells, under conditions that led to anchorage-independent growth, resulted in increased phosphorylation of MerlinS518 (Figure 4A, 4C). Conversely, suppression of PAK1 in SUM52 not only inhibited anchorage-independent growth but also suppressed MerlinS518 phosphorylation (Figure 3B, 6A). Furthermore, shRNA-mediated suppression of NF2 induced anchorage-independent growth of HMLE, indicating a tumor suppressive function of Merlin in breast cancer (Figure 6B). These observations demonstrate that PAK1 regulates Merlinat an inhibitory phosphorylation site and that suppression of Merlin promotes transformation of immortalized mammary epithelial cells.
Figure 6. PAK1 expression inhibits Merlin function, subsequently upregulating phospho-tyrosine levels.

A. Phosphorylation status of MerlinS518 in SUM52 cells expressing PAK1- or lacZ-specific shRNAs. B. Anchorage-independent colonies formed by HMLE expressing NF2-or lacZ-specific shRNAs. Merlin protein levels are shown. ***p-value = 0.0002, **p-value = 0.0059. C–D. Immunoblots showing overall phospho-tyrosine levels (C) or phospho-tyrosine levels of EGFR at Y1173 and Y1068 (D) in HMLE cells expressing a control vector, PAK1 or PAK1 K299R. E. Luminex assay showing normalized phospho-tyrosine signals of TKs and associated proteins in HMLE-PAK1 (x-axis) and the fold phospho-tyrosine signal increase in HMLE-PAK1 when compared to HMLE-PAK1K299R (y-axis). Each diamond (gray and black) represents individually distinct antibodies. Analytes considered positive (>10) and showing significant (1.5) fold increase in signal are shown as black diamonds. All labeled analytes (black diamonds) scored in lysates harvested in normal growth conditions.
Merlin has recently been shown to negatively regulate EGFR internalization and signaling through membrance sequestration (Curto et al 2007). In particular, Merlin activity at the membrane of confluent MEFs resulted in a substantial down-regulation of overall phospho-tyrosine levels, whereas NF2−/− MEFs exhibited high phospho-tyrosine levels, indicating that Merlin regulates phospho-tyrosine signaling (Curto et al 2007). To investigate the role of Merlin in membrane receptor activity, we compared phospho-tyrosine levels in HMLE cells expressing wild-type PAK1 or the kinase-inactive mutant PAK1K299R. We found that wild-type but not the kinase-inactive mutant of PAK1 induced an increase in total phospho-tyrosine levels (Figure 6C). Based on this observation, we investigated EGFR phosphorylation status in HMLE-PAK1 compared to HMLE expressing either a control vector or PAK1K299R and found only a small increase, if any, in phosphorylation of EGFR at sites that indicate its activation status, Y1173 and Y1068 (Figure 6D; Batzer et al 1994, Yamauchi et al 1997). Additionally, suppression of EGFR in HMLE-PAK1 cells did not alter colony formation, indicating EGFR may not be the appropriate target of Merlin inhibition in this context (Supplemental Figure 4).
Prior work has indicated that NHERF1, the adaptor protein that interacts with Merlin for EGFR inhibition is not specific to EGFR, but may associate with other growth factor receptors (reviewed in Weinman et al 2006). We thus postulated that receptor tyrosine kinases (RTKs) other than EGFR are affected by Merlin inhibition due to PAK1 amplification and overexpression. To identify RTKs regulated by PAK1, we performed a multiplex antibody-based assay that measures activated TKs and associated proteins in HMLE-PAK1 and HMLE-PAK1K299R cells regardless of their mode of activation (Du et al 2009). We found that six TKs and associated proteins exhibited at least a significant 1.5 fold increase in phospho-tyrosine levels in HMLE-PAK1 when compared to HMLE-PAK1K299R (Figure 6E; Du et al 2009). Confirming our prior findings, ERK2, but not EGFR, scored in this assay. Interestingly, we found that three different MET-specific antibodies showed that MET phospho-tyrosine levels were significantly elevated in HMLE cells expressing PAK1 compared to PAK1K299R. Together, these observations strongly suggested that PAK1-mediated inhibition of Merlin function led to activation of MET signaling rather than EGFR activation.
In HMLE-PAK1 cells, we found that MET was phosphorylated at Y1234/1235 and Y1003 – sites associated with MET activation and trafficking (Figure 7A; reviewed in Trusolino et al 2010). We also found that a MET adaptor protein, GAB1 (Weidner et al 1996), and a MET downstream effector molecule, STAT3 (Boccaccio et al 1998), were phosphorylated in HMLE-PAK1, providing strong evidence that MET signaling was activated by PAK1 overexpression (Figure 7A). Moreover, when NF2 was overexpressed in HMLE-PAK1 cells, phosphorylation of PAK1T423 decreased slightly while that of METY1234/1235 was eliminated (Figure 7B). Conversely, when NF2 was suppressed in HMLE cells, phosphorylation of MET was enhanced (Supplemental Figure 5A).
Figure 7. MET signaling activation due to PAK1 expression promotes anchorage-independent growth.

A. Phospho-tyrosine levels of MET at Y1234/1235 and Y1003, its adaptor protein GAB1Y307 and downstream effector STAT3Y705 in HMLE expressing a control vector, PAK1 or PAK1K299R. Corresponding total protein levels are shown. B. Immunoblots of HMLE-PAK1 cells expressing vector control or NF2 showing levels of Merlin, phosphorylated MET at Y1234/1235, phosphorylated PAK1 at T423. Total protein levels are shown. C. Anchorage-independent growth of HMLE-PAK1 and SUM52 after treatment with PHA-665752, a MET inhibitor, at indicated doses. D. Anchorage-independent colonies formed by HMLE-PAK1 or SUM52 expressing MET-, GAB1- or lacZ-specific shRNAs. ***p-value <0.0001, **p-value <0.002, *p-value <0.01.
To determine the contribution of MET signaling to PAK1-induced anchorage-independent growth, we treated HMLE-PAK1 and SUM52 with the MET inhibitor PHA-665752, which inhibits MET signaling through GAB1 (Christensen et al 2003). We found that PHA-665752 inhibited anchorage-independent growth of both HMLE-PAK1 and SUM52 in a dose-dependent manner (Figure 7C; Supplemental Figure 6A).
When we introduced MET- or GAB1-specific shRNAs in HMLE-PAK1 and SUM52, we found that suppression of MET or GAB1 compromised anchorage-independent growth of both cell lines that are dependent on PAK1 for transformation (Figure 7D; Supplemental Figure 6B, 6C). These observations demonstrate that PAK1 regulates MET activity by modulating Merlin and that MET signaling through GAB1 is required for PAK1-driven anchorage-independent growth.
Inhibition or activation of MET signaling in HMLE derivatives by pharmacologic or genetic methods did not alter phospho-ERK1/2 levels (Supplemental Figures 5A, 5B), indicating that MAPK activation and MET regulation through Merlin are independent events regulated by PAK1 (Figure 8).
Figure 8. PAK1 simultaneously activates two distinct signaling pathways to promote transformation.
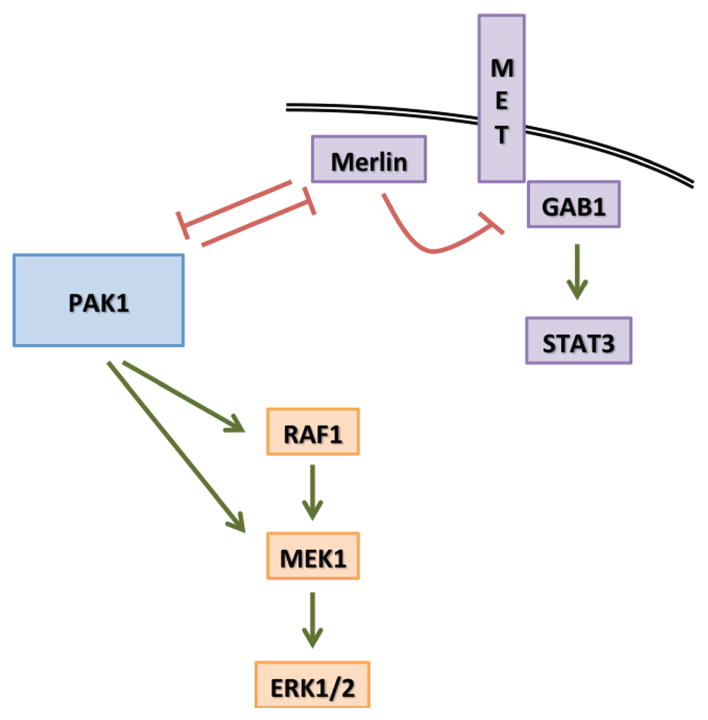
The model depicts PAK1 mechanism of action to activate the RAS-MAPK (Orange) pathway through direct activation of RAF1 and MEK1. The second arm (Purple) shows that PAK1 and Merlin are in an inhibitory loop, which can be pushed by PAK1 amplification and overexpression to inhibit Merlin that leads to MET signaling activation through GAB1.
Discussion
MAPK pathway activation is common in breast cancers (reviewed in Santen et al 2002). In some cases, this activation is driven by amplification of ERBB2. However, a substantial proportion of breast cancers with normal ERBB2 copy number also show activation of MAPK signaling. Here we used an experimental model of mammary epithelial cell transformation to search for kinases that transform these cells in a MAPK-dependent manner
We screened 597 kinase-related ORFs and found PAK1 as a kinase whose expression induced anchorage-independent colony formation in HMLE cells. Based on these observations, we concluded that PAK1 is a transforming kinase, which cooperates with other oncogenic alterations, such as PI3K activation, to enhance transformation.
In parallel, we analyzed two publicly available datasets of human tumor samples and cancer cell lines and found that PAK1 is amplified in 30–33% of human breast tumors. Although prior studies have reported amplifications of 11q13-q14, we used two largest collections of copy number data in human tumors currently available (Tumorscape, TCGA) to confirm that PAK1 is amplified in a substantial portion of breast and other epithelial cancers. We further showed that breast cancer cell lines harboring PAK1 amplification are dependent on its expression for transformation but not proliferation. These observations substantiate PAK1 as the target gene in one of the two 11q13-14 amplicons and further define PAK1 as a bona fide breast cancer oncogene.
Through similar transformation and copy number analyses of group I PAKs, we found that PAK1 and PAK3, but not PAK2, promoted colony formation of HMLE cells. However, we failed to find significant amplification of PAK3 in Tumorscape (Supplemental Figure 1B, data not shown). Hence, among its closest family members, PAK1 shows strongest oncogenic characteristics.
PAK1 is known to regulate several signaling pathways. We have found that PAK1-induced activation of both MAPK and MET signaling are essential for PAK1-driven cell transformation. Although PAK1 also regulates LIMK1/2, the suppression of LIMK1/2 failed to affect anchorage-independent growth (data not shown). Furthermore, PAK1, as an effector of the small GTPases; RAC1 and CDC42, may regulate invasion or metastasis in some cellular contexts. Our observations, however, implicate PAK1 as an oncogene that induces transformation in breast and possibly other cancers. In addition, we found that two signaling pathways – MAPK and Merlin-MET, were coordinately regulated by PAK1 to drive anchorage-independent growth. Using both genetic and pharmacologic manipulations, we showed that anchorage-independent growth induced by PAK1 requires independent activation of MAPK as well as MET signaling. We have also observed that activation of the MAPK pathway by PAK1 is not specific to HMLE or SUM52 cells but also occurs in HMECs only expressing hTERT (HME), in which case, the expression of PAK1 also enhances phosphorylation of ERK1/2 and transforms them (Supplemental Figure 7). Since we and others have found that PAK1 expression is necessary for RAS induced cell transformation, our observations also suggest that amplification of PAK1 is an alternative mechanism of activating MAPK signaling in human breast cancers.
Prior work showed that the tumor suppressor Merlin regulates EGFR signaling. However, we failed to find any evidence using both focused and unbiased assays that PAK1-mediated regulation of Merlin induced alterations in EGFR function. Instead we found that PAK1 expression leads to activation of MET signaling, which was required for anchorage-independent growth. These findings established the contribution of MET signaling to PAK1-dependent transformation and demonstrated that Merlin regulates multiple RTK signaling cascades in a similar manner.
The identification of PAK1 as a breast cancer oncogene provides one mechanism by which the MAPK pathway is activated in breast cancers that do not harbor amplifications of ERRB2 or oncogenic mutations in the RAS-RAF-MEK-ERK pathway. Although ERBB2 expression has been shown to correlate with PAK1 activation, (Arias-Romero et al 2010), indicating that they function in the same pathway, co-amplification of ERBB2 and PAK1 may not entirely be redundant. ERBB2 amplification can lead to activation of other RAS effector pathways in addition to MAPK while PAK1 amplification also activates MET signaling pathway through Merlin inhibition.
Our studies using previously characterized kinase-dead PAK1 mutant allele (K299R) showed that PAK1 kinase activity is required for transformation and establish PAK1 as a target for small molecule inhibition in cancers that harbor PAK1 amplification. Our observations further indicate that breast cancer cell lines such as SUM52 and SUM190 that harbor PAK1 amplification and exhibit corresponding expression levels are best examples of cell lines that show oncogenic addiction to PAK1. Conversely, although HMLER cells did not show overexpression or elevated activity of PAK1, they were dependent on PAK1 for transformation indicating that PAK1 can also behave as an essential gene in RAS-driven cancers. In sum, we present evidence of PAK1 function as a driving oncogene, a cooperating oncogene as well as an essential non-oncogene that mediates transformation of mammary cells through regulation of multiple signaling pathways.
Materials and Methods
Cell lines and reagents
HMLE derivatives were cultured in Clonetics MEGM (Lonza, Basel, Switzerland). SUM52 and SUM190 were obtained from Stephen Ethier and cultured in a 50:50 mixture of MEGM and DMEM/F12 (Invitrogen Carlsbad, CA, USA) 10% inactivated fetal calf serum (IFS, Sigma Aldrich, St. Louis, MO, USA). SKBR3 and EFM19 were cultured in RPMI or RPMI (−) phenol red (Invitrogen), respectively, with 10% IFS. All media contained Penicillin/Streptomycin (Cellgro, Manasas, VA, USA).
pBabe-Puro-Flag-PAK1K299R was derived by site-directed mutagenesis of pBabe-Puro-Flag-PAK1 using QuikChange®IIXL (Stratagene, Santa Clara, CA, USA). Primer sequences are shown in Supplemental Table 1. Retroviral and lentiviral production was carried out as previously described (Boehm et al 2005, Moffat et al 2006). Infections were modified to incorporate spins at 1178 × g (2250 rpm) for 30 min at 25°C. U0126 (EMDBiosciences, Gibbstown, NJ, USA) and PHA-665752 (Tocris, Ellisville, MO, USA) were reconstituted in DMSO and added to cells while seeding in soft agar. ShRNAs were obtained from The RNAi Consortium (TRC) at the Broad Institute; the corresponding reference numbers are as follows: shPAK1#1-TRCN0000195500, shPAK1#2-TRCN0000025258, shNF2#1-TRCN0000039974, shNF2#2-TRCN0000010397, shRAF1#1-TRCN0000195502, shRAF1#2-TRCN0000196969, shMET#1-TRCN0000199327, shMET#2-TRCN0000009850, shGAB1#1-TRCN0000074283, shGAB1#2-TRCN0000074285.
Pooled human kinase ORF screen
The second-generation pBabe-Puro-Myr-Flag kinase open reading frame (ORF) library was developed by the Center for Cancer Systems Biology at the Dana-Farber Cancer Institute and TRC (Johannessen et al 2010). This ORF library consists of 597 kinase-related ORFs that were pooled at equal DNA amounts in random order into 22 pools, each containing 25–27 ORFs. HMLEA was infected with virus produced from the DNA pools. Cells were then selected for puromycin resistance at 1 μg/ml. pBabe-Puro-MEKDD diluted 1:25 with pBabe-Puro, and the parental vector pBabe-Puro were used as positive and negative controls, respectively. HMLEA expressing the pools or controls were seeded in 0.3% Noble agar (Sigma Aldrich) in 6-well plates, 6 replicate wells/pool, and assayed for colony formation. Bottom agar consisted of DMEM with 0.6% Noble agar, 8% IFS and Antibiotic-Antimycotic (Invitrogen). Colony formation was assessed after 2 wks when images of each well were taken at 6.25X magnification on Olympus SZX9 microscope with Olympus Qcolor 3 camera using Magnafire or Q Capture and analyzed with ImageJ software. Macroscopic colonies (>100 sq.pixels, or 0.004 μ) with the circularity of 0.5–1 were counted. Median colony number of the pools was calculated and those that scored 1.5 standard deviations (SD) above median were selected for further study. Colonies >50 sq.pixels (0.002 μ) with the circularity of 0.8–1 were counted in all colony formation assays post-screening.
Immunoblotting
For immunoblot analyses, samples were harvested in RIPA (Cell Signaling Technology – CST, Boston, MA, USA) or CLB (CST) with 1 mM Phenylmethanesulfonyl fluoride (PMSF, Sigma), protease inhibitor (Roche, Manheim, Germany) and phosphatase inhibitor (EMD). Samples were run in 4–12% bis-tris gels (Invitrogen) in MOPS buffer (Invitrogen). Gels were transferred using the iBlot system (Invitrogen). For drug inhibition analysis by immunoblot, cells were treated overnight with DMSO, U0126 or PHA-665752 at the indicated concentrations with constant total volume of DMSO with or without drug. Antibodies used in the study were obtained from CST except for anti-EGFR, which was a gift from Jeonghee Cho, anti-Merlin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and 4G10 (Millipore, Temecula, CA, USA),
Quantitative RT-PCR
RNA was harvested using QiaShredder and RNeasy (Qiagen, Valencia, CA, USA). cDNA was prepared with the help of Advantage RT-for-PCR (Clonetech, Mountain View, CA, USA). qPCR was carried out using SYBR (Applied Biosystems, Beverly, MA, USA). The primers used are listed in Supplemental Table 1.
Proliferation assay
SUM52 expressing shRNAs against PAK1 or lacZ or HMLEs expressing vector control or PAK1 were plated in triplicate in a 6-well plate. Cells were counted the next day (Day 1) as baseline and then counted and replated for 21 d or 23 d at indicated time points to calculate population-doubling times.
Fluorescence in-situ hybridization (FISH)
FISH was carried out as previously described (Knoll and Lichter 2005). Human chromosome 11 cep probe and BAC probe 11-381E5 (PAK1) were used. Hybridization signals were viewed on an Olympus BX-51 fluorescent microscope system.
Luminex assay
Lysates were harvested in normal growth conditions in MEGM. The Luminex assay was carried out for tyrosine kinases (TKs) and associated proteins as previously described (Du et al 2009). Raw data was normalized by subtracting antibody and sample backgrounds. One or more antibodies (analytes) against a specific TK or associated protein may be present. Average normalized signal of triplicates for each of the 244 analytes was calculated. Normalized signals of 10 or higher in HMLE-PAK1 and HMLE-PAK1K299R were considered positive. Normalized signals for analytes in HMLE-PAK1 were divided by corresponding signals in HMLE-PAK1K299R and those with signal fold difference of 1.5 or higher were considered significant.
Copy number analysis
Tumorscape was analyzed as described (Beroukhim et al 2010). GISTIC profile for Tumorscape was created with all 3131 tumor samples and cancer cell lines including 243 breast samples. GISTIC profile for TCGA breast invasive adenocarcinoma was created with 507 cancer samples. Copy number profiles were created for each cancer type with up to 80 samples that show PAK1 amplification. Breast samples with a score of 0.8 (log2 copy number ratio) or higher in Tumorscape were taken into consideration for the PAK1-CCND1 co-amplification study.
Supplementary Material
Acknowledgments
Financial Support: This work was supported in part by a Department of Defense Breast Cancer Research Program Pre-doctoral Fellowship (W81XWH-08-1-0767, Y.S.), grants from the U.S. National Cancer Institute (R33 CA128625, R01 CA130988, W.C.H.), and a Cancer Center Support Grant # NIH 5 P30 CA06516 (Cytogenetics Core).
We thank Nicole A. Spardy, Christine L. Nguyen and Yaara C. Zwang for critical reading of the manuscript. We also thank the Dana-Farber/Harvard Cancer Center Cytogenetics Core for their assistance with FISH studies.
Footnotes
Conflicts of interest: W.C.H., K.P., J.J.Z., and R.B. are consultants for Novartis Pharmaceuticals.
Supplementary information is available at Oncogene’s website.
References
- Appledorn DM, Dao KH, O’Reilly S, Maher VM, McCormick JJ. Rac1 and Cdc42 are regulators of HRasV12-transformation and angiogenic factors in human fibroblasts. BMC Cancer. 2010;10:13. doi: 10.1186/1471-2407-10-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias-Romero LE, Villamar-Cruz O, Pacheco A, Kosoff R, Huang M, Muthuswamy SK, et al. A Rac-Pak signaling pathway is essential for ErbB2-mediated transformation of human breast epithelial cancer cells. Oncogene. 2010;29:5839–5849. doi: 10.1038/onc.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3:772–775. doi: 10.4161/cbt.3.8.994. [DOI] [PubMed] [Google Scholar]
- Batzer AG, Rotin D, Urena JM, Skolnik EY, Schlessinger J. Hierarchy of binding sites for Grb2 and Shc on the epidermal growth factor receptor. Mol Cell Biol. 1994;14:5192–5201. doi: 10.1128/mcb.14.8.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekri S, Adelaide J, Merscher S, Grosgeorge J, Caroli-Bosc F, Perucca-Lostanlen D, et al. Detailed map of a region commonly amplified at 11q13-->q14 in human breast carcinoma. Cytogenet Cell Genet. 1997;79:125–131. doi: 10.1159/000134699. [DOI] [PubMed] [Google Scholar]
- Beroukhim R, Getz G, Nghiemphu L, Barretina J, Hsueh T, Linhart D, et al. Assessing the significance of chromosomal aberrations in cancer: methodology and application to glioma. Proc Natl Acad Sci U S A. 2007;104:20007–20012. doi: 10.1073/pnas.0710052104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaccio C, Ando M, Tamagnone L, Bardelli A, Michieli P, Battistini C, et al. Induction of epithelial tubules by growth factor HGF depends on the STAT pathway. Nature. 1998;391:285–288. doi: 10.1038/34657. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Hession MT, Bulmer SE, Hahn WC. Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol. 2005;25:6464–6474. doi: 10.1128/MCB.25.15.6464-6474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm JS, Zhao JJ, Yao J, Kim SY, Firestein R, Dunn IF, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996;12:144–148. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- Bostner J, Ahnstrom Waltersson M, Fornander T, Skoog L, Nordenskjold B, Stal O. Amplification of CCND1 and PAK1 as predictors of recurrence and tamoxifen resistance in postmenopausal breast cancer. Oncogene. 2007;26:6997–7005. doi: 10.1038/sj.onc.1210506. [DOI] [PubMed] [Google Scholar]
- Brown LA, Kalloger SE, Miller MA, Shih Ie M, McKinney SE, Santos JL, et al. Amplification of 11q13 in ovarian carcinoma. Genes Chromosomes Cancer. 2008;47:481–489. doi: 10.1002/gcc.20549. [DOI] [PubMed] [Google Scholar]
- Chattopadhyay I, Singh A, Phukan R, Purkayastha J, Kataki A, Mahanta J, et al. Genome-wide analysis of chromosomal alterations in patients with esophageal squamous cell carcinoma exposed to tobacco and betel quid from high-risk area in India. Mutat Res. 2010;696:130–138. doi: 10.1016/j.mrgentox.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Christensen JG, Schreck R, Burrows J, Kuruganti P, Chan E, Le P, et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003;63:7345–7355. [PubMed] [Google Scholar]
- Coles LC, Shaw PE. PAK1 primes MEK1 for phosphorylation by Raf-1 kinase during cross-cascade activation of the ERK pathway. Oncogene. 2002;21:2236–2244. doi: 10.1038/sj.onc.1205302. [DOI] [PubMed] [Google Scholar]
- Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. J Cell Biol. 2007;177:893–903. doi: 10.1083/jcb.200703010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Du J, Bernasconi P, Clauser KR, Mani DR, Finn SP, Beroukhim R, et al. Bead-based profiling of tyrosine kinase phosphorylation identifies SRC as a potential target for glioblastoma therapy. Nat Biotechnol. 2009;27:77–83. doi: 10.1038/nbt.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63. doi: 10.1007/s10555-008-9168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenbaas B, Spirio L, Koerner F, Fleming MD, Zimonjic DB, Donaher JL, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Freihoff D, Kempe A, Beste B, Wappenschmidt B, Kreyer E, Hayashi Y, et al. Exclusion of a major role for the PTEN tumour-suppressor gene in breast carcinomas. Br J Cancer. 1999;79:754–758. doi: 10.1038/sj.bjc.6690121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- Hirokawa Y, Tikoo A, Huynh J, Utermark T, Hanemann CO, Giovannini M, et al. A clue to the therapy of neurofibromatosis type 2: NF2/merlin is a PAK1 inhibitor. Cancer J. 2004;10:20–26. doi: 10.1097/00130404-200401000-00006. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, O’Brien C, Boyd Z, Cavet G, Guerrero S, Jung K, et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin Cancer Res. 2009;15:4649–4664. doi: 10.1158/1078-0432.CCR-09-0317. [DOI] [PubMed] [Google Scholar]
- Hynes NE, MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009;21:177–184. doi: 10.1016/j.ceb.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DH, Muto M, Kuwahara Y, Nakanishi Y, Watanabe H, Aoyagi K, et al. Array-based comparative genomic hybridization of circulating esophageal tumor cells. Oncol Rep. 2006;16:1053–1059. [PubMed] [Google Scholar]
- Kim SY, Dunn IF, Firestein R, Gupta P, Wardwell L, Repich K, et al. CK1epsilon is required for breast cancers dependent on beta-catenin activity. PLoS One. 2010;5:e8979. doi: 10.1371/journal.pone.0008979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kissil JL, Johnson KC, Eckman MS, Jacks T. Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. J Biol Chem. 2002;277:10394–10399. doi: 10.1074/jbc.M200083200. [DOI] [PubMed] [Google Scholar]
- Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell. 2003;12:841–849. doi: 10.1016/s1097-2765(03)00382-4. [DOI] [PubMed] [Google Scholar]
- Knoll JH, Lichter P. In situ hybridization to metaphase chromosomes and interphase nuclei. Curr Protoc Hum Genet. 2005;Chapter 4(Unit 4):3. doi: 10.1002/0471142905.hg0403s45. [DOI] [PubMed] [Google Scholar]
- Lambros MB, Fiegler H, Jones A, Gorman P, Roylance RR, Carter NP, et al. Analysis of ovarian cancer cell lines using array-based comparative genomic hybridization. J Pathol. 2005;205:29–40. doi: 10.1002/path.1681. [DOI] [PubMed] [Google Scholar]
- Lau KS, Haigis KM. Non-redundancy within the RAS oncogene family: insights into mutational disparities in cancer. Mol Cells. 2009;28:315–320. doi: 10.1007/s10059-009-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey AI. Neurofibromatosis. Annu Rev Pathol. 2007;2:191–216. doi: 10.1146/annurev.pathol.2.010506.091940. [DOI] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- Miron A, Varadi M, Carrasco D, Li H, Luongo L, Kim HJ, et al. PIK3CA mutations in in situ and invasive breast carcinomas. Cancer Res. 2010;70:5674–5678. doi: 10.1158/0008-5472.CAN-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva OK, Das D, Heiser LM, Bhattacharya S, Siwak D, Gendelman R, et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009;69:565–572. doi: 10.1158/0008-5472.CAN-08-3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakis S, Sourvinos G, Spandidos DA. Differential expression and mutation of the ras family genes in human breast cancer. Biochem Biophys Res Commun. 1998;251:609–612. doi: 10.1006/bbrc.1998.9527. [DOI] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- Neve RM, Holbro T, Hynes NE. Distinct roles for phosphoinositide 3-kinase, mitogen-activated protein kinase and p38 MAPK in mediating cell cycle progression of breast cancer cells. Oncogene. 2002;21:4567–4576. doi: 10.1038/sj.onc.1205555. [DOI] [PubMed] [Google Scholar]
- Santen RJ, Song RX, McPherson R, Kumar R, Adam L, Jeng MH, et al. The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002;80:239–256. doi: 10.1016/s0960-0760(01)00189-3. [DOI] [PubMed] [Google Scholar]
- Scheid MP, Woodgett JR. Phosphatidylinositol 3′ kinase signaling in mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2001;6:83–99. doi: 10.1023/a:1009520616247. [DOI] [PubMed] [Google Scholar]
- Schraml P, Schwerdtfeger G, Burkhalter F, Raggi A, Schmidt D, Ruffalo T, et al. Combined array comparative genomic hybridization and tissue microarray analysis suggest PAK1 at 11q13.5-q14 as a critical oncogene target in ovarian carcinoma. Am J Pathol. 2003;163:985–992. doi: 10.1016/S0002-9440(10)63458-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Paez JG, Curto M, Yaktine A, Pruitt WM, Saotome I, et al. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Dev Cell. 2001;1:63–72. doi: 10.1016/s1534-5807(01)00009-0. [DOI] [PubMed] [Google Scholar]
- Sos ML, Fischer S, Ullrich R, Peifer M, Heuckmann JM, Koker M, et al. Identifying genotype-dependent efficacy of single and combined PI3K- and MAPK-pathway inhibition in cancer. Proc Natl Acad Sci U S A. 2009;106:18351–18356. doi: 10.1073/pnas.0907325106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Chen Z, Ambrose D, Liu J, Gibbs JB, Chernoff J, et al. Kinase-deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol Cell Biol. 1997;17:4454–4464. doi: 10.1128/mcb.17.8.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nat Rev Mol Cell Biol. 2010;11:834–848. doi: 10.1038/nrm3012. [DOI] [PubMed] [Google Scholar]
- Tu HF, Chang KW, Chiang WF, Liu CJ, Yu EH, Liu ST, et al. The frequent co-expression of the oncogenes PIK3CA and PAK1 in oral carcinomas. Oral Oncol. 2011 doi: 10.1016/j.oraloncology.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Adam L, Wang RA, Mandal M, Nguyen D, Sahin A, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–36244. doi: 10.1074/jbc.M002138200. [DOI] [PubMed] [Google Scholar]
- Weidner KM, Di Cesare S, Sachs M, Brinkmann V, Behrens J, Birchmeier W. Interaction between Gab1 and the c-Met receptor tyrosine kinase is responsible for epithelial morphogenesis. Nature. 1996;384:173–176. doi: 10.1038/384173a0. [DOI] [PubMed] [Google Scholar]
- Weinman EJ, Hall RA, Friedman PA, Liu-Chen LY, Shenolikar S. The association of NHERF adaptor proteins with g protein-coupled receptors and receptor tyrosine kinases. Annu Rev Physiol. 2006;68:491–505. doi: 10.1146/annurev.physiol.68.040104.131050. [DOI] [PubMed] [Google Scholar]
- Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem. 2002;277:883–886. doi: 10.1074/jbc.C100553200. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Ueki K, Tobe K, Tamemoto H, Sekine N, Wada M, et al. Tyrosine phosphorylation of the EGF receptor by the kinase Jak2 is induced by growth hormone. Nature. 1997;390:91–96. doi: 10.1038/36369. [DOI] [PubMed] [Google Scholar]
- Zang M, Hayne C, Luo Z. Interaction between active Pak1 and Raf-1 is necessary for phosphorylation and activation of Raf-1. J Biol Chem. 2002;277:4395–4405. doi: 10.1074/jbc.M110000200. [DOI] [PubMed] [Google Scholar]
- Zang M, Gong J, Luo L, Zhou J, Xiang X, Huang W, et al. Characterization of Ser338 phosphorylation for Raf-1 activation. J Biol Chem. 2008;283:31429–31437. doi: 10.1074/jbc.M802855200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.