

Abstract
Glycogen is a complex glucose polymer found in a variety of tissues, including brain, where it is localized primarily in astrocytes. The small quantity found in brain compared to e.g., liver has led to the understanding that brain glycogen is merely used during hypoglycemia or ischemia. In this review evidence is brought forward highlighting what has been an emerging understanding in brain energy metabolism: that glycogen is more than just a convenient way to store energy for use in emergencies—it is a highly dynamic molecule with versatile implications in brain function, i.e., synaptic activity and memory formation. In line with the great spatiotemporal complexity of the brain and thereof derived focus on the basis for ensuring the availability of the right amount of energy at the right time and place, we here encourage a closer look into the molecular and subcellular mechanisms underlying glycogen metabolism. Based on (1) the compartmentation of the interconnected second messenger pathways controlling glycogen metabolism (calcium and cAMP), (2) alterations in the subcellular location of glycogen-associated enzymes and proteins induced by the metabolic status and (3) a sequential component in the intermolecular mechanisms of glycogen metabolism, we suggest that glycogen metabolism in astrocytes is compartmentalized at the subcellular level. As a consequence, the meaning and importance of conventional terms used to describe glycogen metabolism (e.g., turnover) is challenged. Overall, this review represents an overview of contemporary knowledge about brain glycogen and its metabolism and function. However, it also has a sharp focus on what we do not know, which is perhaps even more important for the future quest of uncovering the roles of glycogen in brain physiology and pathology.
Keywords: glycogen shunt, glycogen phosphorylase, glycogen synthase, glutamate, neuron, astrocyte, microdomain, noradrenaline
Introduction
Glycogen, a glucose polymer consisting of many thousands of glucose units connected by a mixture of α 1–4 and α 1–6 glycosidic bonds, is normally considered to function as a glucose store. Certainly, in the liver, which contains a very large amount of glycogen (approx. 100–500 μ mol/g), its function as a glucose reservoir securing a constant blood glucose level under a variety of conditions is well recognized. In the brain, however, the glycogen content is limited averaging 3–12 μ mol/g tissue and its role as an energy source during aglycemia is restricted to a few minutes and hence, it is unlikely that the major functional role of glycogen would be accounted for as an energy source during such conditions (Brown, 2004). It is much more likely that glycogen serves a dynamic role during normal brain function and recent studies have provided evidence that this may indeed be the case (Brown et al., 2005; Gibbs et al., 2006; Schousboe et al., 2007, 2010; Dienel et al., 2007; Sickmann et al., 2009, 2012; Walls et al., 2009; Suzuki et al., 2011). In addition to the fact that brain glycogen is found predominantly in astrocytes (Cataldo and Broadwell, 1986), it has been shown that the content of glycogen is closely correlated to astrocytic differentiation since the highest amount was found in well differentiated cells expressing other astrocytic markers such as S100β and GFAP (Brunet et al., 2010).
Glycogen metabolism, the glycogen shunt and glycogen turnover
Glycogen synthesis requires formation of UDP-glucose from glucose-1-phosphate and UTP catalyzed by UDP-glucose pyrophosphorylase, followed by formation of a new α 1–4 glycoside linkage catalyzed by glycogen synthase (GS). Formation of α 1–6 glycoside bonds, and thereby branching, is catalyzed by the branching enzyme. The degradation of glycogen leading to formation of glucose-1-phosphate is catalyzed by the concerted actions of glycogen phosphorylase (GP) and debranching enzyme. The enzyme phosphoglucomutase ensures a rapid equilibrium between glucose-1-phosphate and the glycolytic intermediate glucose-6-phosphate. The overall sum of these reactions is a conversion of glucose to glycogen via glucose-6-phosphate and glucose-1-phosphate and subsequently the recovery of glucose-6-phosphate. This series of reactions, i.e., the metabolism of glucose via glycogen, is referred to as the glycogen shunt (Walls et al., 2009). The energy yield via glycolysis using glycogen rather than glucose to initiate the sequence of reactions might seem to be increased by 50% (from 2 mol ATP to 3) but, as obvious from the laws of thermodynamics, storage of glucose in form of glycogen and subsequent recovery of glucose is not energy neutral. Since glucose entering the cell is phosphorylated by hexokinase as the first step of glycolysis as well as glycogenesis, it is useful to calculate the overall energy yield starting from glucose-6-phosphate: degradation of one mol glucose-6-phosphate to pyruvate via glycolysis yields three mol ATP. However, since glycogen synthesis costs one ATP-equivalent (in form of UTP) per glucose-6-phosphate, the overall energy yield for glycolysis of glycogen-derived glucose-6-phosphate is only two ATP per mol. If viewed only in light of short-term energy gain, this might seem like a waste of energy. However, it amounts to a clear benefit for long-term stability of the cellular energy state, considering that glycogen is the only available storage form of glucose, which is not only the fastest ATP source but also the only cytosolic one. Thus, glycogen synthesis would probably be better described as a luxury that is affordable in times of ample energy supply. A luxury that cells like neurons, which are in constantly high energy demand, cannot provide for themselves, hence their predominant lack of glycogen.
It is of considerable interest to be able to investigate the functional role of the glycogen shunt. In order to do so, specific pharmacological tools are required and inhibition of GP has turned out to constitute an important avenue in this respect. Nevertheless, it should be emphasized that the synthesis and degradation of individual glycogen granules must be separated temporally and/or spatially due to opposite regulation of GS and GP by reversible phosphorylation as well as the presence of allosteric modulators in the microenvironment encircling the glycogen granule (see Sections “Regulation of Glycogen Phosphorylase,” “Regulation of Glycogen Synthase,” and “Glycogen Metabolism at the Subcellular Level”). The terms “glycogen shunt” as well as the more vaguely defined, although commonly employed, “glycogen turnover” have been used to describe aspects of glycogen metabolism at the general level of the cell culture or tissue being investigated. As will be discussed in Section “Glycogen Metabolism at the Subcellular Level,” the depiction at the subcellular level should also be considered.
Structure and size of the glycogen molecule
According to the widely accepted Whelan model (Gunja-Smith et al., 1970), glycogen is a spherical polymer of glucose arranged in concentric tiers consisting of the branched B-chains and the un-branched A-chains comprising the inner layers and the outermost layer, respectively. The fully synthesized glycogen molecule is normally referred to as β-glycogen. In the end, each glycogen granule is confined to 12 tiers because of the glucose density of the outermost tier which causes steric hindrance in the interaction between the metabolizing enzymes and the glycogen granule (Melendez-Hevia et al., 1993). Considering the structure of the glycogen molecule there is a clear correlation to its role as a highly efficient energy store: by way of design, evolution has rendered glycogen optimal for storing the largest amount of glucose in the smallest possible volume with minimum effect on osmolarity. Furthermore, the greatest attainable number of non-reducing ends is made available for GP before a branch point thereby enabling maximal speed of glucose release (Melendez-Hevia et al., 1993).
Using electron microscopy, glycogen in the central nervous system (CNS) has been visualized as electron dense granules of 10–40 nm diameter (Cataldo and Broadwell, 1986; Wender et al., 2000). This is in accordance with values obtained from resting muscle of 10–44 nm with a mean of 25 nm (Marchand et al., 2002 and references therein). It has been estimated that each tier of a glycogen molecule adds 3.8 nm to the diameter (Goldsmith et al., 1982), which means that the average glycogen molecule would contain between 7 and 8 tiers, i.e., the average glycogen molecule is not fully synthesized β-glycogen. This is somewhat surprising since the glucose content of each additional layer is doubled compared to the previous, which means that the majority of the storage capacity of glycogen is not utilized in an average particle. Assuming a maximal size of 55,000 glycosyl units arranged into 12 tiers and that 34.6% of the glycosyl units in a glycogen molecule are contained in the outermost tier (Melendez-Hevia et al., 1993), an 8 tier molecule contains approximately 10,000 glycosyl units which is less than 20% of a fully synthesized molecule (Graham et al., 2010). However, if the amount of glucose normally contained in glycogen in a given cell would gather into fully synthesized β-glycogen, a concomitant reduction in the number of granules to less than 20% of the normal would ensue thereby putatively disrupting subcellular glycogen availability. The mechanisms behind molecular size restriction and the functional consequence of intermediate-sized glycogen granules compared to maximal-sized with regard to the regulation of glycogen metabolism is further discussed in Section “Restriction of Granule Size.”
It is commonly cited that the four outermost tiers of β-glycogen, regularly referred to as macroglycogen, are the dynamic and thereby metabolically active part of the molecule (Lomako et al., 1991), whereas the innermost 8 tiers, termed proglycogen, are merely a scaffold for the functional molecule (Elsner et al., 2002). Since an average glycogen molecule is within the size of proglycogen, the larger part of glycogen in astrocytes and muscle should not be metabolized under normal circumstances, which is obviously not correct. Moreover, in muscle, the categorization of glycogen molecules into pro- and macroglycogen has been proven to be a non-functional definition, originally derived from their different solubility in acid (reviewed in Lomako et al., 2004), since the size frequency distribution is normal as opposed to bimodal, rendering the existence of two sizewise distinct pools of glycogen unlikely (Marchand et al., 2002).
Regulation of glycogen phosphorylase
The rate limiting enzyme in the degradation of glycogen, GP, is converted from the less active GPb conformation, into the more active GPa, through phosphorylation catalyzed by phosphorylase kinase (Figure 1). In muscle, phosphorylase kinase has been shown to be activated allosterically by calcium through binding to calmodulin, an integral subunit of this enzyme (Cohen et al., 1978). In addition, phosphorylase kinase can be activated by protein kinase A (PKA) in response to increased cAMP concentrations. Although not strictly tissue specific, phosphorylase kinase exists in muscle, liver, and brain isoforms (Proux et al., 1974) and the brain isoform seems to possess similar properties to the muscle isoform (reviewed in Psarra and Sotiroudis, 1996). However, sequencing of cDNA has revealed an alternative mRNA splicing confined to the brain isoform, resulting in deletion of a major part of the cAMP-dependent phosphorylation site on the β-subunit (Harmann et al., 1991). The functional importance of this deletion is still not clarified although it is in line with the lower importance of regulation via phosphorylation of the brain isoform of GP (see Section “Isoforms of Glycogen Phosphorylase”). Naturally, regulation of GP through reversible phosphorylation is not a straightforward canonical pathway but an interconnected network. The calcium level not only influences phosphorylase kinase directly but it can potentially also affect cAMP levels via regulation of adenylate cyclase (AC). Four of the nine transmembrane AC isoforms are sensitive to calcium: AC 1 and AC 8 are stimulated in a calmodulin dependent manner while AC 5 and AC 6 are inhibited directly by calcium. Moreover, some isoforms of phosphodiesterase are activated by calcium. Accordingly, depending on the cellular distribution of enzymatic isoforms calcium can affect both generation and degradation of cAMP thereby potentially modulating the phosphorylation cascade that activates GP.
Figure 1.

Regulation of glycogen metabolism. The cAMP level is increased by adenylate cyclase and decreased by phosphodiesterase. Depending on the isoforms of these two enzymes, calcium can be either inhibiting or activating. An increased cAMP level leads to activation of protein kinase A, which in turn activates phosphorylase kinase. Phosphorylation by phosphorylase kinase, which requires calcium for activation, stimulates glycogen phosphorylase. Glycogen phosphorylase is also allosterically inhibited by ATP and activated by AMP, which accumulates during ATP depletion. Glycogen synthase is deactivated through phosphorylation by protein kinase A, phosphorylase kinase and glycogen synthase kinase 3. Glycogen synthase is activated by protein phosphatase 1, which inhibits phosphorylase kinase and glycogen phosphorylase. Glucose entering the cell is phosphorylated by hexokinase to glucose-6-phosphate and subsequently enters either glycolysis or the glycogen shunt, the latter constituting metabolism of glucose via glycogen. In glycogenesis one ATP equivalent in form of UTP is used by UDP-glucose pyrophosphorylase. Thus, glycogenolysis and subsequent glycolysis of glucose-6-phosphate entail a net gain of two ATP, whereas glycolysis of glucose-6-phosphate derived directly from glucose produces three ATP.
The inactivation of GP by dephosphorylation is catalyzed by phosphoprotein phosphatase 1 (PP1) which dephosphorylates both GP and phosphorylase kinase (reviewed by Cohen, 1989).
In addition to reversible phosphorylation, GP is also highly regulated allosterically. The enzyme is activated by AMP and inhibited by ATP (Figure 1) and glucose-6-phosphate. The regulation of GP by the AMP/ATP ratio reflects the role of glycogen breakdown in providing fuel for the cell in times of local energy deprivation. As with all enzymes regulated by the AMP/ATP ratio, modulation by AMP is a more sensitive regulation, due to the low levels of this nucleotide. The cAMP concentration may also be directly linked to the AMP/ATP ratio: ATP is the substrate of ACs and while global cellular ATP concentrations (millimolar levels) are far too high to play a regulatory role in AC activity, ATP microdomains surrounding AC could potentially reach micromolar levels close to the Km values of ACs.
The two kinds of GP regulation can be viewed as reflecting temporal and spatial differences in energy demanding states: As opposed to the rising AMP concentration that is mainly a result of declining energy levels inside the cell, calcium and cAMP are second messengers triggered by signals from outside the cell, mediating in this case a future energy need as a consequence of cell activation. As an allosteric modulator AMP is a direct activator of GP, while the phosphorylation cascade consists of a sequence of reactions. Thus, in terms of reactions needed to stimulate glycogenolysis, decrease in AMP could be viewed as a fast route as opposed to a slower activation by the cAMP-triggered phosphorylation cascade.
Isoforms of glycogen phosphorylase
GP exists in different isoforms: the liver isoform, the muscle isoform, and the brain isoform. With the exception of liver-GP, expression of the isozymes is not strictly tissue specific: muscle-GP is expressed predominantly in skeletal muscle but also, in lower concentrations, in heart muscle and brain. While the highest concentration of brain-GP is found in astrocytes, it is also expressed in heart muscle and even at very low concentrations in skeletal muscle (Schmid et al., 2009). Additionally, brain-GP is the predominant isoform in fetal tissue and only during maturation expression of the liver and muscle isoforms starts. Tissue specific isozyme expression patterns in rat have been shown to stabilize at different time points depending on the organ, ranging from 19th day of gestation for heart muscle to post natal day 9 for brain (Sato et al., 1976; Richter et al., 1983).
In vitro studies of the two isoforms expressed in astrocytes, brain-GP and muscle-GP, show differences in regulation: the muscle isoform seems to be strongly activated by phosphorylation and less so by AMP, while the brain isoform shows weak response to phosphorylation. Moreover, phosphorylated brain-GP is only fully activated in the presence of AMP. Binding of AMP to either muscle-GP or brain-GP increases the binding affinity for glycogen, although this Km change is only physiologically relevant in the brain due to the lower levels of glycogen found in brain compared to muscle. It has been speculated that these differences in regulation reflect the relative importance of extracellular and intracellular control of energy metabolism in different tissues (Crerar et al., 1995).
Regulation of glycogen synthase
Glycogen degradation is held in balance with glycogen synthesis. The key enzyme for glycogenesis is GS, which catalyzes the transfer of a glucose residue from UDP-glucose to a glycogen chain. Analogous to GP, GS exists in three isoforms and it has been shown that muscle and brain GS (and also GP) share greater identity than either do with the corresponding liver isozymes (Newgard et al., 1988; Pellegri et al., 1996).
Reversible phosphorylation and allosteric regulation
Like GP, GS is regulated allosterically (mainly by glucose-6-phosphate) and by reversible phosphorylation, but contrary to GP the dephosphorylated form of GS constitutes the active state. The regulation of GS by covalent modifications is highly complex and consists of sequences of hierarchal phosphorylations on at least nine different sites catalyzed by a range of kinases including PKA, phosphorylase kinase and glycogen synthase kinase 3 (reviewed in Roach, 2002; Brown, 2004). AMP-activated protein kinase (AMPK) has also been shown to directly phosphorylate GS in vitro (Carling and Hardie, 1989). The relevance of this finding in the in vivo situation, at least in muscle, however, has been questioned (Roach, 2002). Conversely, GS is activated by PP1 which is a key regulator of the balance between glycogenesis and glycogenolysis as it not only activates GS, but also deactivates GP and phosphorylase kinase.
Regulation via subcellular distribution
Changes in the subcellular distribution of GS are an additional regulatory mechanism that has been demonstrated in peripheral tissues. For instance, in cultured hepatocytes, GS reversibly translocates during a switch in metabolic state (e.g., high extracellular glucose) and accumulates at the cell periphery, presumably bound to the glycogen granule, in contrast to being evenly distributed in the cytosol (Garcia-Rocha et al., 2001). Similarly, the targeting of GS in human muscle shifted from a glycogen-enriched membrane fraction (i.e., subsarcolemmal and intermyofibrillar spaces) to the cytoskeleton (intramyofibrillar space) in response to decreasing glycogen levels (Nielsen et al., 2001). Extending these findings, the subcellular location to which the translocation of GS is targeted in muscle has been shown to be dependent on the specific site(s) of phosphorylation on GS (Prats et al., 2009). Moreover, although less studied, GP, and laforin (a dual-specificity phosphatase known to be associated with the glycogen granule) have been reported to alter their intracellular distribution pattern in response to the metabolic status of the cell (reviewed in Graham et al., 2010) as well as glucokinase in hepatocytes (reviewed in Ferrer et al., 2003). The question of whether other enzymes involved in glycogen metabolism demonstrate a similar translocation pattern is still unanswered. As an example, branching enzyme could exhibit a distribution pattern analogous to that of GS, since changing the relative ratio of the activity of these two enzymes could entail a different degree of branching and, thereby, a suboptimal structure of the glycogen molecule (Melendez-Hevia et al., 1993). Nevertheless, whether translocation of GS and other enzymes translates into targeting to the glycogen granule at specific locations and, most importantly, if similar processes occur in the brain, remains to be confirmed experimentally. In essence, translocation of enzymes in response to alterations in energy status demonstrates that glycogen metabolism is highly dynamic and capable of adapting to both the immediate and long-term energy needs of the cell at the level of specific subcellular domains.
Role of glycogen targeting proteins in the regulation of glycogen metabolism
It has been thoroughly established in muscle and liver that glycogen targeting proteins are an important factor in the regulation of glycogen metabolism. Glycogen-associated enzymes like GS, GP, PP1, or phosphorylase kinase either bind glycogen targeting proteins or contain them in the form of regulatory subunits. However, the expression and particularly the function of glycogen targeting proteins in the brain have received very limited attention. Although it would be reasonable to assume that the physical interaction between glycogen and its associated proteins share features across tissues, only two of the described four types of PP1 regulating subunits, PPP1R5 (also commonly known as GM or RGi) and PPP1R6, have been shown to be widely distributed in human tissue, including the brain (Doherty et al., 1996; Armstrong et al., 1997). The presence of the murine analog of PPP1R5, protein targeting glycogen, in cultured astrocytes from mice was established by Allaman et al. (2000), who also demonstrated that the expression of protein targeting glycogen was induced by the neurotransmitters noradrenaline and vasoactive intestinal peptide. The cellular stress sensor AMPK is activated by a rising AMP/ATP ratio and phosphorylates a vast assortment of target proteins such as the glycogen targeting proteins, changing targeting of enzymes to glycogen and triggering a switch from glycogenesis to glycogenolysis. PP1 is targeted to GS by various glycogen-targeting regulatory subunits including protein targeting glycogen. Activity of protein targeting glycogen is downregulated by the laforin-malin complex, consisting of the dual-specificity phosphatase laforin and the E3-ubiquitin ligase malin. In this step, AMPK catalyzes the phosphorylation of protein targeting glycogen, thus accelerating its laforin/malin dependent ubiquitination and subsequent degradation. Furthermore, AMPK increases laforin-malin interaction (Vernia et al., 2009b).
Restriction of granule size
The brain expresses at least two of the three glycogen targeting subunits of PP1 contained in muscle, and the average size and size frequency distribution of glycogen granules are comparable between the two tissues (Wender et al., 2000; Marchand et al., 2002). Hence, it is reasonable to draw parallels from the well-studied muscle to the brain regarding the mechanisms behind restriction of the molecular size as well as the functional consequence of this restriction.
When replenishing muscle glycogen both in vitro and in vivo new glycogen molecules are preferentially synthesized initially and only subsequently an increase in the mean diameter of the granules is observed (Elsner et al., 2002; Marchand et al., 2007). Since a given number of glucose molecules incorporated into glycogen would yield addition of an identical number of non-reducing ends whether the synthesis of new small molecules or big molecules were prioritized, no advantage with regard to the maximal achievable rate of synthesis and degradation would be obtained in either case assuming that this rate is independent of the granule size. However, Marchand et al. (2007) observe that there is an inverse correlation between the diameter of the glycogen molecule and the rate of net synthesis in muscle recovering from prolonged exercise. The authors speculate that this could be due to deterioration of the interactions between glycogen and its associated proteins when the surface area and density of the outermost tier of the molecule is amplified, although no specific mechanisms are discussed. If this is the case, the preferential synthesis of smaller molecules would entail an elevated rate of synthesis initially and, hence, that the rate of replenishment should correlate to the degree of depletion of the glycogen stores. The slower rate of synthesis of larger glycogen molecules also correlates well with the earlier finding by the same group: even during resting conditions the mean diameter of the glycogen molecules rarely reach the maximal attainable size (Marchand et al., 2002). Whether glycogen degradation is also detained by increasing granule size was not investigated. Graham et al. (2010) suggested that once the glycogen granule has grown to an intermediate size, the interactions and regulation would begin to favor GP over GS activation, although the mechanism remains unclear. However, this suggestion is in line with the thought that increased surface area combined with altered glucose density in the “working area” of the enzymes could modify the protein interactions at the surface of the glycogen molecule and thereby reduce granule growth. On the other hand, restriction of granule growth could also be conducted through alterations in the glycogen-surface protein interactions unrelated to changes in the molecular size. For instance, Worby et al. (2008) presented evidence that malin ubiquitinates protein targeting glycogen in a laforin-dependent manner which marks protein targeting glycogen for proteasome-dependent degradation thereby halting glycogen synthesis, and hence, putatively confining the size of the glycogen molecule.
Speculative delineation of protein-polymer interactions during glycogen metabolism
Targeting the enzymes of glycogen metabolism to the granule as a whole would not be very useful, since they can only act at the end of a glycogen chain, so binding somewhere in the middle would be quite useless. There is, however, an example of a similar biological situation that has been described and researched for the last 40 years: DNA binding proteins like DNA-polymerase or transcriptional repressors have to find their specific binding site on a long DNA strand. In 1970 it was discovered that the association rate for these proteins and their specific binding sites are surprisingly high, which could not be explained by standard kinetic models (Riggs et al., 1970). A few years before this discovery, Adam and Delbrück (1968) had developed an idea that might explain enhancement of reaction rates in biological systems: In their model reduction in dimensions of diffusion from three dimensions in solution, to two dimensions in membranes and finally one dimension along a polymer chain would dramatically increase the speed of reactions. To greatly simplify the idea, one can imagine that the reduction of dimensions limits the possible directions in which a molecule can move, while leaving the force behind the movement the same, thus increasing the chances that the molecule will move in the “right direction” to encounter its reaction partner (for detailed explanation and calculations see Richter and Eigen, 1974). This would increase reaction rates of diffusion-controlled processes immensely and provide an attractive explanation for too high association rates of DNA binding proteins: instead of having to find their specific binding sites by three-dimensional diffusion, the proteins are thought to unspecifically bind a DNA strand and then “slide along” the strand until they arrive at their specific binding place. Evidence for this idea has accumulated over many years through careful study of reaction rates and recently several groups have managed to visualize one-dimensional diffusion along DNA strands (reviewed in Gorman and Greene, 2008).
DNA binding proteins sliding along strands could be a model for kinetics at the glycogen granule: enzymes are targeted to a glucose chain and by one-dimensional diffusion move along the chain until they reach the non-reducing end. This would greatly enhance the efficiency of the targeting process and accelerate glycogen metabolism by diminishing diffusion as a rate limiting factor.
Although equating DNA and glycogen binding proteins is highly intriguing, it also provokes critical questions and exposes our lacking knowledge of the dynamic polymer-protein and protein-protein interactions crucial for processes occurring at the non-reducing ends at the surface of the glycogen granule. For instance; is it possible that an approximately 200 kD protein as GP, possibly attached to one or more associated proteins, can slide on a glucose chain that might be severely sterically hindered? If not, where are the enzymes attached while synthesizing or degrading glycogen and how does this affect the kinetics of glycogen metabolism? Or, do the interactions of protein targeting glycogen/GS/GP etc., with glycogen change when the chains are elongated and shortened—or even branched and debranched? Despite an emerging body of studies identifying glycogen targeting proteins, glycogen-associated enzymes and their interactions, the answers to these and related questions are still largely unknown.
Role of cAMP and calcium in regulating brain glycogenolysis
In muscle, calcium released from the endoplasmic reticulum constitutes the initial coupling between contraction and glycogenolysis. This is in contrast to the noradrenaline-induced activation of PKA and thereby phosphorylase kinase via cAMP which is triggered later in a bout of exercise. In liver, though, cAMP is prevailing in the covalent regulation of glycogenolysis (Brown, 2004; Greenberg et al., 2006). In the brain, allosteric activation by AMP appears to be predominant in the regulation of brain-GP whereas muscle-GP is more susceptible to reversible phosphorylation. However, the relative importance of allosteric versus reversible phosphorylation and calcium mediated versus cAMP induced glycogenolysis as well as the functional significance of this regulation in the brain is still rather elusive. The second messenger pathways involved in the glycogenolytic response to noradrenaline is probably one of the most studied in brain and might present an indication of the relative importance of calcium and cAMP in this process.
Two decades ago Subbarao and Hertz (1990) dissected the glycogenolytic response to noradrenaline in cultured murine astrocytes. It was concluded that glycogenolysis was mediated through the concerted actions of β- and α2-adrenergic receptors, while stimulating the α1-receptor had no effect. Interestingly, when blocking the response to noradrenaline with subtype selective antagonists, the glycogenolytic effect of β- and α2-adreneric receptors appeared at least additive, suggesting that both cAMP and calcium, although not through the α1-receptor, could be involved in regulating glycogen degradation in the brain. This, however, was not significant when attempting to selectively stimulate β- and α2-receptors alone and in combination, which might be due to the fact that the employed α2-agonist clonidine is only a partial agonist (see for instance Limon-Boulez et al., 2001). Using mouse cortical slices, Ververken et al. (1982) also found that glycogenolysis induced by noradrenaline was elicited through stimulation of the cAMP-coupled β-receptor, although calcium played a crucial role since the removal of extracellular calcium abolished the response to noradrenaline. Moreover, they observed that the activity of GP increased faster than the concomitant cAMP rise when stimulating with noradrenaline, implying the additional involvement of another signaling pathway. That both β- and α1-receptor agonists stimulate glycogenolysis in cultured murine cortical astrocytes was shown by Sorg and Magistretti (1991), at the same time corroborating and contradicting previous findings by Ververken et al. (1982) and Subbarao and Hertz (1990). It should be noted though, that the discrepancies might be due to differences in the utilized cell culture systems/tissue preparation, since the response of glycogen metabolism to noradrenaline changes across development (days in vitro) and differ between species (O'Dowd et al., 1995). Overall it seems that the involvement of the β-adrenergic receptor (and thereby cAMP) in the glycogenolytic response in astrocytes to noradrenaline is consistently being emphasized. Although the mechanism by which calcium elicits its role in this context is not unequivocally described, its involvement in glycogenolysis is obvious but the extent and relative importance compared to cAMP awaits further clarification.
We are not aware of any in vivo studies conducted to elucidate the relative influence of various regulatory pathways of glycogenolysis in the brain. However, the relative importance of allosteric activation versus phosphorylation in the insulin-induced regulation of GS in muscle has recently been examined using an elegant approach: genetic modification of GS at a site crucial for interaction with glucose-6-phosphate was conducted in a mouse model rendering the enzyme insensitive to allosteric activation. This alteration resulted in a markedly reduced muscle glycogen level and an 80% decrease in muscle glycogen synthesis induced by insulin, clearly demonstrating the importance of allosteric activation in this pathway (Bouskila et al., 2010). Similar studies dealing with brain isoforms of GS, GP and phosphorylase kinase would be of significant value in determining the relative significance of the outlined regulatory mechanisms for brain glycogen metabolism in vivo.
Glycogen metabolism at the subcellular level
The involvement and importance of brain glycogen as a source of energy and as precursor for neurotransmitters in physiological processes (e.g., synaptic activity and memory formation) has been investigated during the last decade (see for instance Gibbs et al., 2006, 2007; Suzuki et al., 2011; Sickmann et al., 2012 and Sections “Energy Substrates Supporting Synaptic Activity” and “Glycogen may be Used to Maintain Glutamatergic Neurotransmission”). Although emphasis has been put on elucidating the mechanisms behind these associations, the subcellular localization of involved pathways has largely been ignored. However, and as outlined above, the regulation of glycogen metabolism is a complex interplay between a vast array of enzymes, their modulators, and associated proteins, so that the spatiotemporal localization of these and the glycogen granules most likely plays a crucial role.
To our knowledge the existence of glycogen microdomains in astrocytes has not been shown directly, although indirect evidence has accumulated over the past decades. For instance, in the aforementioned experiments with noradrenaline in cultured astrocytes (Subbarao and Hertz, 1990), the maximal effect of stimulating either β- or α2-adreneric receptors alone or using noradrenaline was only 30–60% glycogen degradation, signifying the presence of at least two pools of glycogen; one that is sensitive to noradrenergic stimulation and one that is not. While this could also be a result of agonist-induced receptor desensitization, an in vivo study in rats showed that depletion of noradrenalin decreases the glycogenolytic response to extreme energy demand (Harik et al., 1982). In addition to this, the intermolecular mechanisms of glycogen metabolism have been examined in a range of glycogen-containing tissues, including brain (Devos and Hers, 1979, 1980a,b; Chee et al., 1983; Brainard et al., 1989; Calder and Geddes, 1992; Elsner et al., 2002). In brief, these studies have examined whether glycogen granules at the level of a tissue or cell culture are metabolized simultaneously (in phase) or sequentially (out of phase). Collectively they support the existence of glycogen microdomains, since they provide evidence that a sequential component is involved in glycogen synthesis and degradation, i.e., glycogen molecules are not metabolized as one pool, but rather as several pools, possibly even one for each glycogen granule.
Prerequisite for the existence of glycogen microdomains is of course compartmentation of the regulatory pathways controlling glycogen metabolism. In this regard, close localization of glycogen and its associated enzymes creates a distinct regulatory advantage: as explained in Sections “Regulation of Glycogen Phosphorylase” and “Regulation of Glycogen Synthase” the balance of glycogenesis and glycogenolysis is achieved to a large extent by having the same signals activating one pathway while deactivating the other. Naturally, this can only work when the signal has access to the key-enzymes of both cascades at the same time. Considering the ubiquitous nature of the signaling molecules involved in regulating glycogen metabolism, a need for generation of specificity through formation of spatial microdomains is obvious. For one of the second messengers involved this has already been demonstrated: A-kinase anchoring proteins (AKAPs) are scaffolding proteins sequestering ACs, AC activators, phosphodiesterases, PKA, downstream targets of PKA, and other proteins. Due to this compartmentation, rises in the cAMP concentration are fast, short-term and spatially restricted. Moreover, by bringing targets of PKA close to cAMP generation, AKAPs generate specificity of a very unspecific signaling pathway: instead of activating a vast array of cellular targets, PKA is limited in its interaction partners (reviewed by Carnegie et al., 2009). Furthermore, it has been established that calcium, another modulator of glycogen metabolism, can reside together with cAMP in one microdomain (Willoughby et al., 2010). Therefore, compartmentation facilitated by AKAPs and glycogen targeting proteins can be viewed as restricting signaling spatially and temporally and thus creating putative regulatory microdomains of glycogen metabolism.
In muscle, functionally distinct subcellular domains of glycogen have already been described and quantified in human biopsies by employing electron transmission microscopy (Marchand et al., 2002, 2007; Nielsen et al., 2010). Furthermore, and most interestingly, a correlation between the amount of glycogen situated within the myofibrillar space (as opposed to the glycogen pools in between the tightly packed myofibrils and in the subsarcolemmal space) and the release rate of calcium from the sarcoplasmatic reticulum has been established (Ortenblad et al., 2011). The observed functional correlation is probably due to the existence of a strong binding between glycogen with associated proteins and the sarcoplasmatic reticulum membrane. This association is highly dynamic, varying with the metabolic status of the muscle cell (reviewed in Ortenblad et al., 2011). The existence of a coupling between glycogen availability in a spatially confined domain and fundamental functionality of a tissue (here sarcoplasmatic reticulum function and thereby contraction of the muscle) underlines the importance of having readily accessible glycogen in the correct subcellular compartment. Although muscle and brain cannot be compared directly, the great spatiotemporal heterogeneity of the brain combined with a constant energy demand and the need for mechanisms to ensure the right amount of energy at the right place and time render it highly likely that similar correlations may also play an important role in normal brain function.
In this context it is worth considering the actual meaning as well as importance of examining adherence to the so-called last-in-first-out principle, i.e., if the glucose unit that was incorporated into the glycogen molecule most recently is also the first one to be liberated upon degradation (see for instance Elsner et al., 2002). Imagine that stimuli A and B induce glycogenesis and glycogenolysis, respectively. If A and B have their primary effects within the same subcellular location, one might observe obedience to the last-in-first-out principle, although this would also depend on the intermolecular mechanisms of glycogen metabolism. On the other hand, if A and B stimulate glycogen synthesis and degradation at two distinct subcellular sites, a different conclusion might be reached. Since regulation of the rate of synthesis of the individual glycogen molecule is based on the size of the granule (as discussed in Section “Restriction of Granule Size”), and the size of each granule in turn depends on the relative ratio of glycogenic and glycogenolytic stimuli within a subcellular domain, we here hypothesized that glycogen molecules within a distinct subcellular location are synthesized or degraded simultaneously (in phase). Although depending on the utilized stimuli, this would result in glycogen synthesis and/or degradation appearing to occur sequentially (out of phase) when observing at the overall level of for instance a cell culture, as has been described in brain and other glycogen containing tissues (Devos and Hers, 1979, 1980a,b; Chee et al., 1983; Brainard et al., 1989; Calder and Geddes, 1992; Elsner et al., 2002).
Glycogen phosphorylase isoforms in hereditary diseases
As discussed earlier, astrocytes not only express the brain isoform of GP but also the muscle isoform. Hereditary deficiencies in glycogen metabolism indicate that expression of two GP isozymes in astrocytes is more than just an issue of redundancy: McArdle's disease is a hereditary myopathy caused by a deficiency in muscle-GP. The disease is characterized by painful exercise-induced cramps and early fatigue as well as weakness after exercise (Lucia et al., 2008). Patients show no symptoms connected to brain or heart, probably since these tissues also express brain-GP. However, if this double expression was purely redundant, mutations of brain-GP could be equally balanced out by the expression of muscle-GP. However, brain-GP is the only one of the three isoforms with no known loss-of-function mutations, indicating that deficiency in this enzyme would be lethal.
The fact that brain-GP seems to be the isoform most important for survival is not necessarily connected to its function in the brain, since it is the predominant isoform in fetal tissues and deficiency would be lethal in early development simply because of lack of the other isozymes. Nevertheless, another disease points to a special role of brain-GP in the fully developed brain: Lafora disease is a neurodegenerative disease resulting in progressive myoclonus epilepsy and death (Ganesh et al., 2006). Due to a defect in regulation of glycogen synthesis, insoluble polyglucans, so-called Lafora bodies, accumulate in neurons. Lafora disease is caused by mutations of laforin and malin, leading to inactivity or reduced activity of laforin/malin (Vernia et al., 2009a). In neurons, which express GS but not GP, the laforin/malin complex catalyzes degradation of hyperphosphorylated GS, thus effectively preventing synthesis of glycogen under normal conditions. Deficiency in either laforin or malin leads to activation of neuronal GS, causing Lafora body accumulation and apoptosis (Vilchez et al., 2007). Apart from neurons, Lafora bodies accumulate in heart, liver, muscle, and other tissues. In contrast to neurons these tissues do express glycogenolytic enzymes, yet they appear to be unable to degrade the polyglucans effectively. However, astrocytes, the only cell type expressing predominantly brain-GP, show no such accumulations, which could be due to a functional or regulatory difference in this isozyme. Since rising AMP concentrations activate AMPK, which in turn activates laforin/malin, this leads to degradation of protein targeting glycogen (Figure 2). Lack of protein targeting glycogen causes inactive PP1, resulting in active GP and inactive GS. In addition to reducing GP inhibition by this pathway, AMP also directly activates GP. In Lafora disease, however, rising AMP levels could no longer affect PP1 via protein targeting glycogen, so that GS would be constantly active and GP inhibited. However, as explained in Section “Isoforms of Glycogen Phosphorylase,” there are regulatory differences between muscle-GP and brain-GP: while muscle-GP is mostly activated by phosphorylation, brain-GP responds more strongly to allosteric activation by AMP. Therefore, increased activity of PP1 would affect muscle-GP greatly while brain-GP could still be activated by the rising AMP concentration. Thus, one could speculate that the expression pattern of GP isoforms is responsible for the lack of Lafora bodies in astrocytes compared to muscle and other tissues.
Figure 2.

Regulatory differences of GP isoforms as a possible explanation for Lafora body distribution. An increased AMP concentration leads to activation of AMP-activated protein kinase, which activates the laforin-malin complex, causing degradation of protein targeting glycogen. Since protein targeting glycogen is needed to activate protein phosphatase 1, which in turn would activate glycogen synthase and inhibit glycogen phosphorylase, this would lead to degradation of glycogen by glycogen phosphorylase. In addition, glycogen phosphorylase is allosterically activated by an increased AMP level, further shifting the balance in favor of glycogen breakdown. The main astrocytic isoform of glycogen phosphorylase (brain-GP) is activated mostly allosterically and less by phosphorylation, in contrast to the muscle isoform (muscle-GP). This regulatory difference could explain why lack of laforin/malin activity in Lafora disease leads to formation of Lafora bodies in muscle and other tissues, but not in astrocytes.
Inhibitors of glycogen phosphorylase
Isofagomine and DAB
Metabolism of liver glycogen plays a major role in the homeostatic regulation of blood glucose and therefore, strategies aimed at developing treatments for diabetes have involved development of pharmacological tools to manipulate glycogen degradation in the liver. This led to the synthesis of isofagomine which turned out to be a potent inhibitor of liver GP (Lundgren et al., 1996). Isofagomine was subsequently characterized as an inhibitor of GP in homogenates of cultured mouse cerebral cortical astrocytes as well as mouse brain and it was found to potently inhibit this enzyme with an IC50 value of about 1 μ M (Waagepetersen et al., 2000). It was, however, observed that much higher concentrations of isofagomine were needed to completely inhibit glycogen degradation in intact astrocytes indicating poor passage of the astrocytic plasma membrane (Waagepetersen et al., 2000). Unfortunately, isofagomine production was discontinued and as an alternative 1,4-dideoxy-1,4-imino-D-arabinitol (DAB) was characterized as an inhibitor of astrocytic GP (Walls et al., 2008). This compound turned out to be somewhat difficult to use mainly due to poor membrane penetration. However, employing a preincubation period of 1 h, DAB at concentrations between 300 and 1000 μ M inhibited GP almost completely and it was concluded that this compound can be used to evaluate the glycogen shunt activity (Walls et al., 2008).
CP-316,819
In addition to isofagomine and DAB another phosphorylase inhibitor CP-316,819 ([R-R*,S*]-5-chloro-N-[2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmethyl)propyl]-1H-indole-2-carboxamide) has recently been utilized to investigate the functional role of brain glycogen (Dienel et al., 2007; Suh et al., 2007; Sickmann et al., 2009, 2010, 2012). The interesting property of the CP compound is that in contrast to isofagomine and DAB, CP-316,819 only has an inhibitory effect on GP if glucose is present (Suh et al., 2007). This means that if given to animals or cultured astrocytes under normo-glycemic conditions glycogen stores will be increased but if given under hypoglycemic conditions the lack of an inhibitory action on GP will allow the utilization of glycogen. In keeping with this, rats treated with CP-316,819 and subsequently exposed to hypoglycemia exhibited less neuronal damage than controls not receiving CP-316,819 (Suh et al., 2007).
Role of glycogen during hypoglycemia and ischemia
Hypoglycemia leads to a reduction of the supply of glucose to the brain but the flux of glucose from blood to brain is not zero. Moreover, it has been shown by Suda et al. (1990) that both synaptic activity and glucose utilization are reduced during hypoglycemia. Hence, it has been estimated that the glycogen store normally available in the brain would be able to support the energy metabolism for about one and a half hours during hypoglycemia (Gruetter, 2003). During ischemia in which no oxidative metabolism takes place much less ATP can be generated by the concerted action of glycogenolysis and glycolysis (see Schousboe et al., 2010) and as a consequence the brain glycogen store will be depleted within a few minutes of ischemia (Lowry et al., 1964; Swanson et al., 1989). Hence, glycogen in the brain should not be considered as a reserve for energy production but is more likely a dynamic molecule which may serve as an energy substrate under physiological conditions (Swanson, 1992; Swanson and Choi, 1993; Brown et al., 2005; Schousboe et al., 2007, 2010; Hertz et al., 2007; Suzuki et al., 2011). Recent studies utilizing the pharmacological tools discussed in Section “Inhibitors of Glycogen Phosphorylase” to manipulate glycogen metabolism have provided evidence that this may indeed be the case.
Energy substrates supporting synaptic activity
Although neurotransmission is mediated by a large number of neurotransmitters, the vast majority of synapses utilize either glutamate or GABA as the transmitter substance (Nichols, 1994). In relation to energy metabolism it should be taken into account that the ratio between excitatory, glutamatergic neurons and the inhibitory, GABAergic neurons is close to nine (Attwell and Laughlin, 2001). Therefore, it is likely that in terms of energy cost related to synaptic activity one needs to emphasize the cost of glutamatergic synaptic activity (Attwell and Laughlin, 2001). Actually, analyzing the energy use related to neural activity in cerebellum Howarth et al. (2010) reached the conclusion that excitatory neurons utilize three times as much energy as inhibitory neurons. In this context it is important to realize that by far the majority of the energy expenditure is associated with the postsynaptic actions of glutamate and only a small fraction is directly related to glutamate recycling, i.e., release and cellular uptake (Attwell and Laughlin, 2001). Nevertheless, it is important to understand how glycogen may play a role as an energy source during neurotransmission. Glycogen in astrocytes is degraded to pyruvate, which is partly used for oxidative metabolism and partly for lactate production (Walls et al., 2009). It has been demonstrated that astrocytes release lactate generated from glycogen in vitro (Dringen et al., 1993) and evidence has now been obtained that this may also be the case in vivo (Suzuki et al., 2011). Therefore, astrocytes may provide neurons with lactate derived from glycogen and in order to adequately discuss glycogen as an energy source for neurotransmission, it may be pertinent to evaluate glucose and lactate as energy generating molecules in relation to glutamate release and uptake. Using cultured cerebellar granule cells as a model system for glutamatergic neurons, Bak et al. (2006) investigated the ability of glucose and lactate to sustain glutamatergic activity. Using 13C-labeled glucose and lactate it was shown that lactate could be used as an energy substrate during resting conditions but during stimulation induced by repetitive exposure of the neurons to the glutamate receptor agonist N-methyl-D-aspartate (NMDA) in the presence of 15 mM K+ to relieve the Mg2+ block, glucose supported the additional energy demand (Figure 3). Moreover, using threo-benzyloxyaspartate (TBOA) to block glutamate transporters it could be shown that glucose is an essential energy substrate to fuel the machinery for glutamate transport, i.e., the maintenance of an intact Na+-gradient across the plasma membrane (Figure 4). On the basis of these experiments it appears that glucose is an important energy source for the maintenance of glutamatergic activity. In line with this, it was recently suggested, based on mathematical modeling of human data, that rapid glycogen mobilization in astrocytes reduces glucose uptake into astrocytes allowing more glucose to be taken up by neurons (DiNuzzo et al., 2010, 2011). The neuronal preference for glucose during activation may also be important in light of the demonstration that glycolytically derived ATP by the enzymes glyceraldehyde 3-phosphate dehydrogenase/3-phosphoglycerate kinase and pyruvate kinase is essential for vesicular transport of glutamate (Ikemoto et al., 2003; Ishida et al., 2009). It should be mentioned that other authors have suggested that lactate may be preferred over glucose as an energy substrate for neurons (Bouzier-Sore et al., 2003; Schurr, 2006; Pellerin and Magistretti, 2011; Suzuki et al., 2011). Indeed, there is much controversy regarding the differential roles of glucose and brain-derived lactate as energy substrates for neurons and astrocytes; however, this is beyond the scope of this review and the reader is referred to recent reviews by Dienel (2011) and Pellerin and Magistretti (2011). In this context it should be mentioned that glycogen degradation does not exclusively yield glucose-1-phosphate: Since GP can only catalyze cleavage of α-1,4- linkages, it cannot degrade glycogen completely. GP degrades a glycogen branch until four glucose residues remain. Glycogen debranching enzyme catalyzes further digestion in two steps: first, three of the remaining residues are transferred onto a nearby branch, where they are once again target for GP. In a second step debranching enzyme cleaves off the α-1,6- linked glucose residue at the branching point. Consequently, about one tenth of the glucose contained in glycogen is liberated as free glucose during glycogenolysis. Since glucose transporters are bidirectional, these glucose molecules could be released by the cell, unless they are phosphorylated by hexokinase. Thus, depending on localization of glycogen in relation to hexokinase, astrocytes would in theory be able to release glycogen-derived glucose for neuronal use.
Figure 3.

Glucose but not lactate supports synaptic activity. Cultured (glutamatergic) cerebellar neurons increase the utilization of glucose but not lactate during synaptic activity as evidenced by increased labeling in glutamate (expressed as percent molecular carbon labeling, MCL; see below) from 13C-labeled glucose but not 13C-labeled lactate when subjected to pulses of the glutamate receptor agonist NMDA (data from Bak et al., 2006). Pulses of NMDA induce depolarization and pulsatile release of neurotransmitter glutamate in these cultures (Bak et al., 2003). Thus, cultured neurons rely on oxidative metabolism of glucose but not lactate when engaged in neurotransmission activity. The experiments were performed by superfusing the cultured cerebellar neurons in the presence of either [U-13C]glucose (2.5 mM; Glc in bold letters) and lactate (1 mM) or [U-13C]lactate (1 mM; Lac in bold letters) and glucose (2.5 mM) and repetitive depolarization (black bars) was induced by pulses of NMDA (300 μ M) plus potassium (15 mM). The data represents the molecular carbon labeling (MCL) in percent (see Bak et al., 2006 for a definition of MCL) of glutamate ± SD MCL (%) is a measure of labeling in the entire glutamate pool, e.g., a MCL of 20% means that 20% of all carbon atoms in the glutamate pool are 13C. Thus, an increase in MCL of glutamate means that the labeled substrate was metabolized to pyruvate and further oxidized in the TCA cycle to a higher extent. Glucose is metabolized to pyruvate via glycolysis and lactate via the lactate dehydrogenase-catalyzed reaction. Glutamate reflects the labeling in the TCA cycle intermediates, since glutamate is labeled via its cognate keto acid α-ketoglutarate, an intermediate in the TCA cycle. For a thorough discussion of this, please see Bak et al. (2006). Statistically significant differences between the control (gray bars) and those that were repetitively depolarized are indicated by asterisks and determined using an unpaired, two-tailed Student's t-test. *P < 0.05.
Figure 4.

Glucose but not lactate supports neurotransmitter homeostasis. The ability of cultured (glutamatergic) cerebellar neurons to perform re-uptake of release D-[3H]aspartate (pre-loaded marker of glutamate release) during superfusion is diminished in the presence of lactate as compared to the presence of glucose (data from Bak et al., 2006). This is evidenced by an increase in the apparent pulsatile NMDA-induced release of D-[3H]aspartate in the presence of lactate; however, since this increase was abolished by the additional presence of the glutamate transport blocker TBOA, it most likely reflects a diminished ability to take up a fraction of the released D-[3H]aspartate. Experimentally, cultured cerebellar neurons were superfused and the depolarization-coupled release of preloaded D-[3H]aspartate evoked by 30 s pulses of NMDA (300 μ M) plus potassium (15 mM) was quantified (see Bak et al., 2006 for details). The effect of TBOA (100 μ M) on the release in the presence of glucose (2.5 mM, black and open bars) or lactate (1 mM, hatched and gray bars) was investigated. The release in the presence of glucose alone was used as a reference and normalized to 100 (black bar). The results are averages ± SD. Statistically significant differences were calculated using One-Way ANOVA followed by a Tukey–Kramer post hoc test. *Significantly different from the other three conditions (P < 0.01).
Glycogen may be used to maintain glutamatergic neurotransmission
It was suggested by Shulman et al. (2001) that activity of the glycogen shunt may be elevated during brain activation. The availability of specific pharmacological agents to interfere with glycogen metabolism has provided the necessary tools to directly study the role of the glycogen shunt. As delineated by Walls et al. (2009) the glycogen shunt refers to the sequence of reactions allowing glucose to be incorporated into a glycogen molecule and subsequently be released from glycogen as glucose-1-phosphate which upon conversion to glucose-6-phosphate enters the glycolytic pathway. Thus, this is an alternative to the direct introduction of glucose into the glycolytic pathway via its conversion to glucose-6-phosphate.
It is now evident from studies performed both in vitro and in vivo that several brain regions are dependent upon glycogen metabolism to support neurotransmission during physiological conditions. In vivo glycogen degradation was necessary for memory consolidation in young chickens and recently glycogen was shown to be crucial for long-term memory formation and for the maintenance of long-term potentiation of synaptic strength in living rats (Gibbs et al., 2006; Suzuki et al., 2011). Moreover, it has been shown that glycogen supports neuronal function in the optic nerve as well as in cerebral cortical and hippocampal slice preparations in the absence of other energy substrates (Swanson and Choi, 1993; Ransom and Fern, 1997; Wender et al., 2000; Brown et al., 2005; Suh et al., 2007; Suzuki et al., 2011). The fact that inhibition of the monocarboxylate transporters (MCTs) abolishes the effect of glycogen suggests that lactate derived from glycogen is involved in the maintenance of neurotransmission. This aspect of astrocytic glycogen supporting neuronal signaling has recently been shown to include the cerebellar region. Using the GP inhibitor CP-316,819 we have shown that glycogen is needed for maintenance of a low extracellular glutamate concentration, i.e., the balance of release and uptake, both of which are energy-demanding processes (Sickmann et al., 2009; Schousboe et al., 2010). It should be noted that these experiments were performed in the presence of glucose, which appears to be preferred by neurons over lactate as an energy substrate during intense stimulation (Bak et al., 2006, 2009). However, neurons do metabolize lactate to a significant extent although this is not correlated with neuronal activation (Bak et al., 2009). As delineated in Section “Regulation of Glycogen Phosphorylase”, glycogen mobilization is mediated by activation of the degrading enzyme GP either via phosphorylation involving a series of intracellular reactions or via allosteric modulation governed by AMP. Since the predominant part of the energy consuming processes associated with neurotransmission is confined to the neuronal compartment, involvement of glycogen in sustaining neuronal expenses requires a neuronal signal that can initiate glycogen degradation via cAMP and/or calcium and most likely a subsequent transfer of glycogen-derived lactate from astrocytes to neurons for oxidative metabolism to proceed.
The fact that glycogen degradation is also regulated by an increase in AMP in the cellular microenvironment suggests that glycogen is used for energy production within astrocytes. The clearance of extracellular glutamate is partly mediated by sodium dependent high affinity transporters in the astrocytic membrane, which rely upon Na+/K+-ATPase activity for restoration of the intracellular Na+ concentration. Thus, abruption of astrocytic energy metabolism may lead to impaired glutamate uptake or reversal of the transporter causing high extracellular glutamate concentrations affecting neurotransmission and possibly causing excitotoxic neurodegeneration Schousboe et al., 2010, 2011. Hence, neuronal failure may be established as a consequence of impairments in astrocytic energy homeostasis suggesting that the energy generated by glycogen metabolism may not need to be transferred to the neuronal compartment in order to sustain neurotransmission. It has recently been investigated to what extent the glycogen shunt may be involved in generation of energy needed to sustain glutamate transport in astrocytes. Using DAB to inhibit GP, Sickmann et al. (2009) reported that even in the presence of glucose, inhibition of glycogen degradation caused a decrease in D-aspartate uptake. It thus appears that even under conditions where the energy production via glucose metabolism should be adequate an intact glycogen shunt is required for maintenance of optimal glutamate transport capacity. This conclusion was also reached on the basis of results obtained using a different experimental approach in which the capacity for glutamate uptake was indirectly monitored quantifying the amount of homoexchange of preloaded D-[3H]aspartate induced with pulses of 1 mM D-aspartate during superfusion of astrocytes (Schousboe et al., 2010). Using this strategy, DAB in the presence of glucose reduced the capacity of the cells to re-accumulate released D-[3H]aspartate during exposure to 1 mM D-aspartate.
The importance of glycogen shunt activity was confirmed in cultured astrocytes investigating the metabolism of [1,6-13C]glucose in the absence and presence of the GP inhibitor DAB. Subsequent determination of 13C labeling in lactate and glutamate, reflecting glycolysis and TCA cycle metabolism, respectively, demonstrated that under non-stimulated conditions inhibition of GP led to an enhanced glycolysis, i.e., a supercompensation (Walls et al., 2009) analogous to that observed in the rat brain in vivo following sensory stimulation (Dienel et al., 2007). Moreover, the supercompensation was even further enhanced when the metabolic demand was increased by exposure of the astrocytes to 250 μ M D-aspartate to activate glutamate transporters and subsequent Na+/K+-ATPase activity (Walls et al., 2009).
Altogether, these results unequivocally show that the glycogen shunt is of critical importance for regulation of glucose metabolism in astrocytes, a conclusion, which was also reached by Hertz et al. (2007, 2010) using different approaches.
Noradrenaline affects glucose metabolism via the glycogen shunt
Noradrenaline has previously been shown to stimulate glycogen synthesis and degradation in cultured astrocytes by elevating intracellular cAMP and calcium levels (Subbarao and Hertz, 1990; Obel et al., 2012). Using noradrenaline in combination with DAB to inhibit GP in primary cultures of astrocytes, Walls et al. (2009) could show that under such experimental condition 40% of the overall glucose metabolism proceeded through the glycogen shunt. In addition to its effects on glycogen metabolism noradrenaline has been demonstrated to increase glucose uptake (Hutchinson et al., 2007) and activity of the enzymes pyruvate dehydrogenase and α-ketoglutarate dehydrogenase leading to an increase in TCA cycle activity (Subbarao and Hertz, 1991; Chen and Hertz, 1999). Here, we present new data demonstrating that astrocytic glucose metabolism is enhanced in primary cultured astrocytes exposed to noradrenaline. The cells were superfused (1 ml/min) with a medium containing 2.5 mM glucose in the absence or presence of noradrenaline (100 μ M) for 15, 30, 45, or 60 min. Tracer amounts of 2-deoxy-[3H]glucose were added for the last 15 min of each experiment. At the end of the superfusion period the cells were extracted with ethanol to allow determination of intracellular radioactivity representing both uptake and subsequent metabolism of glucose. Figure 5 shows that exposure to noradrenaline almost doubles glucose uptake and metabolism. In addition, glucose metabolism via glycolysis and the TCA cycle was investigated by superfusion of cultured astrocytes with medium containing [1,6-13C]glucose (see Walls et al., 2009 for details). Subsequent mass spectrometric analyses of the 13C labeling in lactate and glutamate, reflecting glycolysis and TCA cycle metabolism, respectively, revealed that exposure to 100 μ M noradrenaline increases metabolism via both of these pathways (Figures 6A,B). The stimulatory effect of noradrenaline on [3-13C]lactate synthesis from [1,6-13C]glucose was observed prior to the effect on [4-13C]glutamate production, demonstrating that the noradrenaline-induced stimulation of glycolysis happens faster than stimulation of pyruvate metabolism via pyruvate dehydrogenase and the TCA cycle.
Figure 5.

2-Deoxy-[3H]glucose transport and metabolism is accelerated in cultured astrocytes exposed to norepinephrine. Astrocyte cultures were superfused with medium containing 2.5 mM of glucose in the absence (control; white bars) or presence of 100 μ M norepinephrine (NE; blacks bar). The cultures were superfused for 15, 30, 45, or 60 min (n = 3–4) and during the last 15 min tracer amounts of 2-deoxy-[3H]glucose (2DG) were added to the superfusion medium. The astrocytes were extracted with ethanol and the extent of 2DG uptake was determined by measuring radioactivity in cell extracts (counts per minute; CPM). The 2DG uptake reflects both glucose transport and subsequent metabolism. The presence of NE gave rise to an increase in 2DG uptake in all time periods compared to controls. No significant differences were observed between time periods within controls. However, in astrocytes exposed to NE, the 2DG uptake was significantly higher from 30 to 45 min compared to all other time periods during NE exposed conditions. * and # indicate statistically significant differences between control and NE exposed astrocytes, and amongst NE exposed astrocytes, respectively (One-Way ANOVA followed by Bonferroni post hoc test, P < 0.05).
Figure 6.
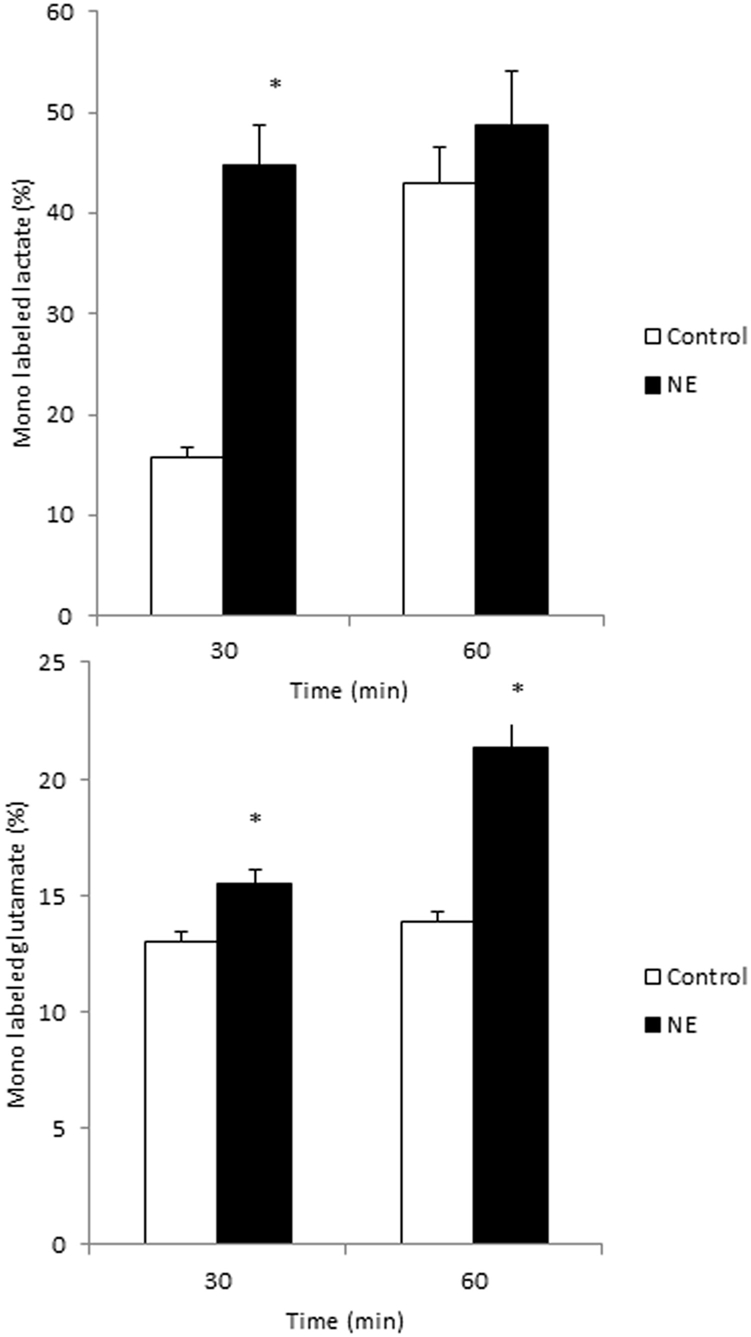
Glycolysis and TCA cycle metabolism are augmented in cultured astrocytes exposed to norepinephrine. Cultured astrocytes were superfused for 60 min with medium containing 2.5 mM of [1,6-13C]glucose in the absence (control; white bars) or presence of 100 μ M norepinephrine (NE; black bars). The percent 13C enrichment in lactate, reflecting glycolytic activity (A) and glutamate, reflecting TCA cycle metabolism of acetyl-coA derived from glucose (B) was monitored employing liquid chromatography—mass spectrometric (LC-MS) analyses. Statistically significant differences between controls and NE treated astrocytes were determined using an unpaired Student's T-test (P < 0.05) and are indicated by an asterisk (*). Data modified from Walls et al. (2009).
Concluding remarks
In this review recent evidence has been brought forward highlighting what has been an emerging understanding in the field of brain energy metabolism: that brain glycogen is more than just a convenient way for the astrocyte to store energy for use in emergencies—it is in fact a dynamic molecule with versatile implications in normal brain function. However, although further studies of the role of glycogen in various physiological processes of the brain will be of utmost importance, we here encourage a closer look into the molecular and subcellular mechanisms underlying glycogen metabolism. In this context, studies conducted in liver and muscle have proven to be a significant source of inspiration for mechanistic details and prospect experimental approaches.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Financial support for this work was obtained from the Danish Medical Research council (grants number 09-066319 and 10-094362 and 09-063393).
References
- Adam G., Delbrück M. (1968). “Reduction of dimensionality in biological diffusion processes,” in Structural Chemistry and Molecular Biology, eds Rich A., Davidson N. (New York: W. H. Freeman and Co; ), 198–215 [Google Scholar]
- Allaman I., Pellerin L., Magistretti P. J. (2000). Protein targeting to glycogen mRNA expression is stimulated by noradrenaline in mouse cortical astrocytes. Glia 30, 382–391 [PubMed] [Google Scholar]
- Armstrong C. G., Browne G. J., Cohen P., Cohen P. T. (1997). PPP1R6, a novel member of the family of glycogen-targeting subunits of protein phosphatase 1. FEBS Lett. 418, 210–214 [DOI] [PubMed] [Google Scholar]
- Attwell D., Laughlin S. B. (2001). An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow Metab. 21, 1133–1145 10.1097/00004647-200110000-00001 [DOI] [PubMed] [Google Scholar]
- Bak L. K., Schousboe A., Sonnewald U., Waagepetersen H. S. (2006). Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J. Cereb. Blood Flow Metab. 26, 1285–1297 10.1038/sj.jcbfm.9600281 [DOI] [PubMed] [Google Scholar]
- Bak L. K., Schousboe A., Waagepetersen H. S. (2003). Characterization of depolarization-coupled release of glutamate from cultured mouse cerebellar granule cells using DL-threo-beta-benzyloxyaspartate (DL-TBOA) to distinguish between the vesicular and cytoplasmic pools. Neurochem. Int. 43, 417–424 10.1016/S0197-0186(03)00030-5 [DOI] [PubMed] [Google Scholar]
- Bak L. K., Walls A. B., Schousboe A., Ring A., Sonnewald U., Waagepetersen H. S. (2009). Neuronal glucose but not lactate utilization is positively correlated with NMDA-induced neurotransmission and fluctuations in cytosolic Ca2+ levels. J. Neurochem. 109(Suppl. 1), 87–93 10.1111/j.1471-4159.2009.05943.x [DOI] [PubMed] [Google Scholar]
- Bouskila M., Hunter R. W., Ibrahim A. F., Delattre L., Peggie M., van Diepen J. A., Voshol P. J., Jensen J., Sakamoto K. (2010). Allosteric regulation of glycogen synthase controls glycogen synthesis in muscle. Cell Metab. 12, 456–466 10.1016/j.cmet.2010.10.006 [DOI] [PubMed] [Google Scholar]
- Bouzier-Sore A. K., Voisin P., Canioni P., Magistretti P. J., Pellerin L. (2003). Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J. Cereb. Blood Flow Metab. 23, 1298–1306 10.1097/01.WCB.0000091761.61714.25 [DOI] [PubMed] [Google Scholar]
- Brainard J. R., Hutson J. Y., Hoekenga D. E., Lenhoff R. (1989). Ordered synthesis and mobilization of glycogen in the perfused heart. Biochemistry 28, 9766–9772 [DOI] [PubMed] [Google Scholar]
- Brown A. M. (2004). Brain glycogen re-awakened. J. Neurochem. 89, 537–552 10.1111/j.1471-4159.2004.02421.x [DOI] [PubMed] [Google Scholar]
- Brown A. M., Sickmann H. M., Fosgerau K., Lund T. M., Schousboe A., Waagepetersen H. S., Ransom B. R. (2005). Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J. Neurosci. Res. 79, 74–80 10.1002/jnr.20335 [DOI] [PubMed] [Google Scholar]
- Brunet J. F., Allaman I., Magistretti P. J., Pellerin L. (2010). Glycogen metabolism as a marker of astrocyte differentiation. J. Cereb. Blood Flow Metab. 30, 51–55 10.1038/jcbfm.2009.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calder P. C., Geddes R. (1992). Heterogeneity of glycogen synthesis upon refeeding following starvation. Int. J. Biochem. 24, 71–77 [DOI] [PubMed] [Google Scholar]
- Carling D., Hardie D. G. (1989). The substrate and sequence specificity of the AMP-activated protein kinase. Phosphorylation of glycogen synthase and phosphorylase kinase. Biochim. Biophys. Acta 1012, 81–86 [DOI] [PubMed] [Google Scholar]
- Carnegie G. K., Means C. K., Scott J. D. (2009). A-kinase anchoring proteins: from protein complexes to physiology and disease. IUBMB Life 61, 394–406 10.1002/iub.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A. M., Broadwell R. D. (1986). Cytochemical identification of cerebral glycogen and glucose-6-phosphatase activity under normal and experimental conditions. II. Choroid plexus and ependymal epithelia, endothelia and pericytes. J. Neurocytol. 15, 511–524 [DOI] [PubMed] [Google Scholar]
- Chee N. P., Geddes R., Wills P. R. (1983). Metabolic heterogeneity in rabbit brain glycogen. Biochim. Biophys. Acta 756, 9–12 [DOI] [PubMed] [Google Scholar]
- Chen Y., Hertz L. (1999). Noradrenaline effects on pyruvate decarboxylation: correlation with calcium signaling. J. Neurosci. Res. 58, 599–606 [DOI] [PubMed] [Google Scholar]
- Cohen P. (1989). The structure and regulation of protein phosphatases. Annu. Rev. Biochem. 58, 453–508 10.1146/annurev.bi.58.070189.002321 [DOI] [PubMed] [Google Scholar]
- Cohen P., Burchell A., Foulkes J. G., Cohen P. T. (1978). Identification of the Ca2+-dependent modulator protein as the fourth subunit of rabbit skeletal muscle phosphorylase kinase. FEBS Lett. 92, 287–293 [DOI] [PubMed] [Google Scholar]
- Crerar M. M., Karlsson O., Fletterick R. J., Hwang P. K. (1995). Chimeric muscle and brain glycogen phosphorylases define protein domains governing isozyme-specific responses to allosteric activation. J. Biol. Chem. 270, 13748–13756 10.1074/jbc.270.23.13748 [DOI] [PubMed] [Google Scholar]
- Devos P., Hers H. G. (1979). A molecular order in the synthesis and degradation of glycogen in the liver. Eur. J. Biochem. 99, 161–167 10.1111/j.1432-1033.1979.tb13242.x [DOI] [PubMed] [Google Scholar]
- Devos P., Hers H. G. (1980a). Glycogen in rat adipose tissue: sequential synthesis and random degradation. Biochem. Biophys. Res. Commun. 95, 1031–1036 10.1016/0006-291X(80)91576-4 [DOI] [PubMed] [Google Scholar]
- Devos P., Hers H. G. (1980b). Random, presumably hydrolytic, and lysosomal glycogenolysis in the livers of rats treated with phlorizin and of newborn rats. Biochem. J. 192, 177–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel G. A. (2011). Brain lactate metabolism: the discoveries and the controversies. J. Cereb. Blood Flow Metab. [Epub ahead of print]. 10.1038/jcbfm.2011.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel G. A., Ball K. K., Cruz N. F. (2007). A glycogen phosphorylase inhibitor selectively enhances local rates of glucose utilization in brain during sensory stimulation of conscious rats: implications for glycogen turnover. J. Neurochem. 102, 466–478 10.1111/j.1471-4159.2007.04595.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M., Mangia S., Maraviglia B., Giove F. (2010). Glycogenolysis in astrocytes supports blood-borne glucose channeling not glycogen-derived lactate shuttling to neurons: evidence from mathematical modeling. J. Cereb. Blood Flow Metab. 30, 1895–1904 10.1038/jcbfm.2010.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNuzzo M., Maraviglia B., Giove F. (2011). Why does the brain (not) have glycogen? Bioessays 33, 319–326 10.1002/bies.201000151 [DOI] [PubMed] [Google Scholar]
- Doherty M. J., Young P. R., Cohen P. T. (1996). Amino acid sequence of a novel protein phosphatase 1 binding protein (R5) which is related to the liver- and muscle-specific glycogen binding subunits of protein phosphatase 1. FEBS Lett. 399, 339–343 10.1016/S0014-5793(96)01357-9 [DOI] [PubMed] [Google Scholar]
- Dringen R., Gebhardt R., Hamprecht B. (1993). Glycogen in astrocytes: possible function as lactate supply for neighboring cells. Brain Res. 623, 208–214 10.1016/0006-8993(93)91429-V [DOI] [PubMed] [Google Scholar]
- Elsner P., Quistorff B., Hansen G. H., Grunnet N. (2002). Partly ordered synthesis and degradation of glycogen in cultured rat myotubes. J. Biol. Chem. 277, 4831–4838 10.1074/jbc.M108226200 [DOI] [PubMed] [Google Scholar]
- Ferrer J. C., Favre C., Gomis R. R., Fernandez-Novell J. M., Garcia-Rocha M., de la Iglesia N., Cid E., Guinovart J. J. (2003). Control of glycogen deposition. FEBS Lett. 546, 127–132 [DOI] [PubMed] [Google Scholar]
- Ganesh S., Puri R., Singh S., Mittal S., Dubey D. (2006). Recent advances in the molecular basis of Lafora's progressive myoclonus epilepsy. J. Hum. Genet. 51, 1–8 10.1007/s10038-005-0321-1 [DOI] [PubMed] [Google Scholar]
- Garcia-Rocha M., Roca A., de la Iglesia N., Baba O., Fernandez-Novell J. M., Ferrer J. C., Guinovart J. J. (2001). Intracellular distribution of glycogen synthase and glycogen in primary cultured rat hepatocytes. Biochem. J. 357, 17–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs M. E., Anderson D. G., Hertz L. (2006). Inhibition of glycogenolysis in astrocytes interrupts memory consolidation in young chickens. Glia 54, 214–222 10.1002/glia.20377 [DOI] [PubMed] [Google Scholar]
- Gibbs M. E., Lloyd H. G., Santa T., Hertz L. (2007). Glycogen is a preferred glutamate precursor during learning in 1-day-old chick: biochemical and behavioral evidence. J. Neurosci. Res. 85, 3326–3333 10.1002/jnr.21307 [DOI] [PubMed] [Google Scholar]
- Goldsmith E., Sprang S., Fletterick R. (1982). Structure of maltoheptaose by difference Fourier methods and a model for glycogen. J. Mol. Biol. 156, 411–427 10.1016/0022-2836(82)90336-9 [DOI] [PubMed] [Google Scholar]
- Gorman J., Greene E. C. (2008). Visualizing one-dimensional diffusion of proteins along DNA. Nat. Struct. Mol. Biol. 15, 768–774 10.1038/nsmb.1441 [DOI] [PubMed] [Google Scholar]
- Graham T. E., Yuan Z., Hill A. K., Wilson R. J. (2010). The regulation of muscle glycogen: the granule and its proteins. Acta Physiol. (Oxf) 199, 489–498 10.1111/j.1748-1716.2010.02131.x [DOI] [PubMed] [Google Scholar]
- Greenberg C. C., Jurczak M. J., Danos A. M., Brady M. J. (2006). Glycogen branches out: new perspectives on the role of glycogen metabolism in the integration of metabolic pathways. Am. J. Physiol. Endocrinol. Metab. 291, E1–E8 10.1152/ajpendo.00652.2005 [DOI] [PubMed] [Google Scholar]
- Gruetter R. (2003). Glycogen: the forgotten cerebral energy store. J. Neurosci. Res. 74, 179–183 10.1002/jnr.10785 [DOI] [PubMed] [Google Scholar]
- Gunja-Smith Z., Marshall J. J., Mercier C., Smith E. E., Whelan W. J. (1970). A revision of the Meyer-Bernfeld model of glycogen and amylopectin. FEBS Lett. 12, 101–104 10.1016/0014-5793(70)80573-7 [DOI] [PubMed] [Google Scholar]
- Harik S. I., Busto R., Martinez E. (1982). Norepinephrine regulation of cerebral glycogen utilization during seizures and ischemia. J. Neurosci. 2, 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmann B., Zander N. F., Kilimann M. W. (1991). Isoform diversity of phosphorylase kinase alpha and beta subunits generated by alternative RNA splicing. J. Biol. Chem. 266, 15631–15637 [PubMed] [Google Scholar]
- Hertz L., Lovatt D., Goldman S. A., Nedergaard M. (2010). Adrenoceptors in brain: cellular gene expression and effects on astrocytic metabolism and Ca(2+)i. Neurochem. Int. 57, 411–420 10.1016/j.neuint.2010.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertz L., Peng L., Dienel G. A. (2007). Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 27, 219–249 10.1038/sj.jcbfm.9600343 [DOI] [PubMed] [Google Scholar]
- Howarth C., Peppiatt-Wildman C. M., Attwell D. (2010). The energy use associated with neural computation in the cerebellum. J. Cereb. Blood Flow Metab. 30, 403–414 10.1038/jcbfm.2009.231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson D. S., Summers R. J., Gibbs M. E. (2007). Beta2- and beta3-adrenoceptors activate glucose uptake in chick astrocytes by distinct mechanisms: a mechanism for memory enhancement? J. Neurochem. 103, 997–1008 10.1111/j.1471-4159.2007.04789.x [DOI] [PubMed] [Google Scholar]
- Ikemoto A., Bole D. G., Ueda T. (2003). Glycolysis and glutamate accumulation into synaptic vesicles. Role of glyceraldehyde phosphate dehydrogenase and 3-phosphoglycerate kinase. J. Biol. Chem. 278, 5929–5940 10.1074/jbc.M211617200 [DOI] [PubMed] [Google Scholar]
- Ishida A., Noda Y., Ueda T. (2009). Synaptic vesicle-bound pyruvate kinase can support vesicular glutamate uptake. Neurochem. Res. 34, 807–818 10.1007/s11064-008-9833-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limon-Boulez I., Bouet-Alard R., Gettys T. W., Lanier S. M., Maltier J. P., Legrand C. (2001). Partial agonist clonidine mediates alpha(2)-AR subtypes specific regulation of cAMP accumulation in adenylyl cyclase II transfected DDT1-MF2 cells. Mol. Pharmacol. 59, 331–338 [DOI] [PubMed] [Google Scholar]
- Lomako J., Lomako W. M., Whelan W. J. (1991). Proglycogen: a low-molecular-weight form of muscle glycogen. FEBS Lett. 279, 223–228 10.1016/0014-5793(91)80154-U [DOI] [PubMed] [Google Scholar]
- Lomako J., Lomako W. M., Whelan W. J. (2004). Glycogenin: the primer for mammalian and yeast glycogen synthesis. Biochim. Biophys. Acta 1673, 45–55 10.1016/j.bbagen.2004.03.017 [DOI] [PubMed] [Google Scholar]
- Lowry O. H., Passonneau J. V., Hasselberger F. X., Schulz D. W. (1964). Effect of ischemia on known substrates and cofactors of the glycolytic pathway in brain. J. Biol. Chem. 239, 18–30 [PubMed] [Google Scholar]
- Lucia A., Nogales-Gadea G., Perez M., Martin M. A., Andreu A. L., Arenas J. (2008). McArdle disease: what do neurologists need to know? Nat. Clin. Pract. Neurol. 4, 568–577 10.1038/ncpneuro0913 [DOI] [PubMed] [Google Scholar]
- Lundgren K., Rassov A., Bols M. (1996). NN 42–1007 is a novel, potent inhibitor of hepatic glycogen phosphorylase, and of hepatocyte glycogenolysis. Diabetes 45, 521 [Google Scholar]
- Marchand I., Chorneyko K., Tarnopolsky M., Hamilton S., Shearer J., Potvin J., Graham T. E. (2002). Quantification of subcellular glycogen in resting human muscle: granule size, number, and location. J. Appl. Physiol. 93, 1598–1607 10.1152/japplphysiol.00585.2001 [DOI] [PubMed] [Google Scholar]
- Marchand I., Tarnopolsky M., Adamo K. B., Bourgeois J. M., Chorneyko K., Graham T. E. (2007). Quantitative assessment of human muscle glycogen granules size and number in subcellular locations during recovery from prolonged exercise. J. Physiol. 580, 617–628 10.1113/jphysiol.2006.122457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melendez-Hevia E., Waddell T. G., Shelton E. D. (1993). Optimization of molecular design in the evolution of metabolism: the glycogen molecule. Biochem. J. 295(Pt 2), 477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgard C. B., Littman D. R., van Genderen C., Smith M., Fletterick R. J. (1988). Human brain glycogen phosphorylase. Cloning, sequence analysis, chromosomal mapping, tissue expression, and comparison with the human liver and muscle isozymes. J. Biol. Chem. 263, 3850–3857 [PubMed] [Google Scholar]
- Nichols D. (1994). Proteins, Transmitters and Synapses. Edinburgh, UK: Blackwell Scientific Publications [Google Scholar]
- Nielsen J. N., Derave W., Kristiansen S., Ralston E., Ploug T., Richter E. A. (2001). Glycogen synthase localization and activity in rat skeletal muscle is strongly dependent on glycogen content. J. Physiol. 531, 757–769 10.1111/j.1469-7793.2001.0757h.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen J., Suetta C., Hvid L. G., Schroder H. D., Aagaard P., Ortenblad N. (2010). Subcellular localization-dependent decrements in skeletal muscle glycogen and mitochondria content following short-term disuse in young and old men. Am. J. Physiol. Endocrinol. Metab. 299, E1053–E1060 10.1152/ajpendo.00324.2010 [DOI] [PubMed] [Google Scholar]
- Obel L. F., Andersen K. M. H., Bak L. K., Schousboe A., Waagepetersen H. S. (2012). Effects of adrenergic agents on intracellular Ca2+ homeostasis and metabolism of glucose in astrocytes with an emphasis on pyruvate carboxylation, oxidative decarboxylation and recycling: implications for glutamate neurotransmission and excitotoxicity. Neurotox. Res. [Epub ahead of print]. 10.1007/s12640-011-9296-1 [DOI] [PubMed] [Google Scholar]
- O'Dowd B. S., Barrington J., Ng K. T., Hertz E., Hertz L. (1995). Glycogenolytic response of primary chick and mouse cultures of astrocytes to noradrenaline across development. Brain Res. Dev. Brain Res. 88, 220–223 10.1016/0165-3806(95)00084-Q [DOI] [PubMed] [Google Scholar]
- Ortenblad N., Nielsen J., Saltin B., Holmberg H. C. (2011). Role of glycogen availability in sarcoplasmic reticulum Ca2+ kinetics in human skeletal muscle. J. Physiol. 589, 711–725 10.1113/jphysiol.2010.195982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegri G., Rossier C., Magistretti P. J., Martin J. L. (1996). Cloning, localization and induction of mouse brain glycogen synthase. Brain Res. Mol. Brain Res. 38, 191–199 10.1016/0169-328X(95)00305-C [DOI] [PubMed] [Google Scholar]
- Pellerin L., Magistretti P. J. (2011). Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. [Epub ahead of print]. 10.1038/jcbfm.2011.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prats C., Helge J. W., Nordby P., Qvortrup K., Ploug T., Dela F., Wojtaszewski J. F. (2009). Dual regulation of muscle glycogen synthase during exercise by activation and compartmentalization. J. Biol. Chem. 284, 15692–15700 10.1074/jbc.M900845200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proux D., Alexandre Y., Delain D., Dreyfus J. C. (1974). The isozymes of phosphorylase kinase in various mammalian tissues. Biochimie 56, 1559–1564 [DOI] [PubMed] [Google Scholar]
- Psarra A. M., Sotiroudis T. G. (1996). Subcellular distribution of phosphorylase kinase in rat brain. Association of the enzyme with mitochondria and membranes. Int. J. Biochem. Cell Biol. 28, 29–42 10.1016/1357-2725(95)00117-4 [DOI] [PubMed] [Google Scholar]
- Ransom B. R., Fern R. (1997). Does astrocytic glycogen benefit axon function and survival in CNS white matter during glucose deprivation? Glia 21, 134–141 [PubMed] [Google Scholar]
- Richter F., Bohme H. J., Hofmann E. (1983). Developmental changes of glycogen phosphorylase b isozymes in rat tissues. Biomed. Biochim. Acta 42, 1229–1235 [PubMed] [Google Scholar]
- Richter P. H., Eigen M. (1974). Diffusion controlled reaction rates in spheroidal geometry. Application to repressor–operator association and membrane bound enzymes. Biophys. Chem. 2, 255–263 10.1016/0301-4622(74)80050-5 [DOI] [PubMed] [Google Scholar]
- Riggs A. D., Bourgeois S., Cohn M. (1970). The lac repressor-operator interaction. 3. Kinetic studies. J. Mol. Biol. 53, 401–417 [DOI] [PubMed] [Google Scholar]
- Roach P. J. (2002). Glycogen and its metabolism. Curr. Mol. Med. 2, 101–120 [DOI] [PubMed] [Google Scholar]
- Sato K., Satoh K., Sato T., Imai F., Morris H. P. (1976). Isozyme patterns of glycogen phosphorylase in rat tissues and transplantable hepatomas. Cancer Res. 36, 487–495 [PubMed] [Google Scholar]
- Schmid H., Pfeiffer-Guglielmi B., Dolderer B., Thiess U., Verleysdonk S., Hamprecht B. (2009). Expression of the brain and muscle isoforms of glycogen phosphorylase in rat heart. Neurochem. Res. 34, 581–586 10.1007/s11064-008-9825-3 [DOI] [PubMed] [Google Scholar]
- Schousboe A., Bak L. K., Sickmann H. M., Sonnewald U., Waagepetersen H. S. (2007). Energy substrates to support glutamatergic and GABAergic synaptic function: role of glycogen, glucose and lactate. Neurotox. Res. 12, 263–268 [DOI] [PubMed] [Google Scholar]
- Schousboe A., Sickmann H. M., Walls A. B., Bak L. K., Waagepetersen H. S. (2010). Functional importance of the astrocytic glycogen-shunt and glycolysis for maintenance of an intact intra/extracellular glutamate gradient. Neurotox. Res. 18, 94–99 10.1007/s12640-010-9171-5 [DOI] [PubMed] [Google Scholar]
- Schousboe A., Sickmann H. M., Bak L. K., Schousboe I., Jajo F. S., Faek S. A., Waagepetersen H. (2011). Neuron-glia interactions in glutamatergic neurotransmission: Roles of oxidative and glycolytic ATP as energy source. J. Neurosci. Res. 89, 1926–1934 10.1002/jnr.22746 [DOI] [PubMed] [Google Scholar]
- Schurr A. (2006). Lactate: the ultimate cerebral oxidative energy substrate? J. Cereb. Blood Flow Metab. 26, 142–152 10.1038/sj.jcbfm.9600174 [DOI] [PubMed] [Google Scholar]
- Shulman R. G., Hyder F., Rothman D. L. (2001). Cerebral energetics and the glycogen shunt: neurochemical basis of functional imaging. Proc. Natl. Acad. Sci. U.S.A. 98, 6417–6422 10.1073/pnas.101129298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann H. M., Waagepetersen H. S., Schousboe A., Benie A. J., Bouman S. D. (2010). Obesity and type 2 diabetes in rats are associated with altered brain glycogen and amino-acid homeostasis. J. Cereb. Blood Flow Metab. 30, 1527–1537 10.1038/jcbfm.2010.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmann H. M., Waagepetersen H. S., Schousboe A., Benie A. J., Bouman S. D. (2012). Brain glycogen and its role in supporting glutamate and GABA homeostasis in a type 2 diabetes rat model. Neurochem. Int. 60, 267–275 10.1016/j.neuint.2011.12.019 [DOI] [PubMed] [Google Scholar]
- Sickmann H. M., Walls A. B., Schousboe A., Bouman S. D., Waagepetersen H. S. (2009). Functional significance of brain glycogen in sustaining glutamatergic neurotransmission. J. Neurochem. 109(Suppl. 1), 80–86 10.1111/j.1471-4159.2009.05915.x [DOI] [PubMed] [Google Scholar]
- Sorg O., Magistretti P. J. (1991). Characterization of the glycogenolysis elicited by vasoactive intestinal peptide, noradrenaline and adenosine in primary cultures of mouse cerebral cortical astrocytes. Brain Res. 563, 227–233 10.1016/0006-8993(91)91538-C [DOI] [PubMed] [Google Scholar]
- Subbarao K. V., Hertz L. (1990). Effect of adrenergic agonists on glycogenolysis in primary cultures of astrocytes. Brain Res. 536, 220–226 10.1016/0006-8993(90)90028-A [DOI] [PubMed] [Google Scholar]
- Subbarao K. V., Hertz L. (1991). Stimulation of energy metabolism by alpha-adrenergic agonists in primary cultures of astrocytes. J. Neurosci. Res. 28, 399–405 10.1002/jnr.490280312 [DOI] [PubMed] [Google Scholar]
- Suda S., Shinohara M., Miyaoka M., Lucignani G., Kennedy C., Sokoloff L. (1990). The Lumped Constant of the deoxyglucose method in hypoglycemia - effects of moderate hypoglycemia on local cerebral glucose-utilization in the rat. J. Cereb. Blood Flow Metab. 10, 499–509 10.1038/jcbfm.1990.92 [DOI] [PubMed] [Google Scholar]
- Suh S. W., Bergher J. P., Anderson C. M., Treadway J. L., Fosgerau K., Swanson R. A. (2007). Astrocyte glycogen sustains neuronal activity during hypoglycemia: studies with the glycogen phosphorylase inhibitor CP-316,819 (R-R*,S*-5-chloro-N-2-hydroxy-3-(methoxymethylamino)-3-oxo-1-(phenylmet hyl)propyl-1H-indole-2-carboxamide). J. Pharmacol. Exp. Ther. 321, 45–50 [DOI] [PubMed] [Google Scholar]
- Suzuki A., Stern S. A., Bozdagi O., Huntley G. W., Walker R. H., Magistretti P. J., Alberini C. M. (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144, 810–823 10.1016/j.cell.2011.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson R. A. (1992). Physiologic coupling of glial glycogen metabolism to neuronal activity in brain. Can. J. Physiol. Pharmacol. 70(Suppl.) S138–S144 [DOI] [PubMed] [Google Scholar]
- Swanson R. A., Choi D. W. (1993). Glial glycogen stores affect neuronal survival during glucose deprivation in vitro. J. Cereb. Blood Flow Metab. 13, 162–169 10.1038/jcbfm.1993.19 [DOI] [PubMed] [Google Scholar]
- Swanson R. A., Sagar S. M., Sharp F. R. (1989). Regional brain glycogen stores and metabolism during complete global ischaemia. Neurol. Res. 11, 24–28 [DOI] [PubMed] [Google Scholar]
- Vernia S., Rubio T., Heredia M., Rodriguez de C. S., Sanz P. (2009a). Increased endoplasmic reticulum stress and decreased proteasomal function in Lafora disease models lacking the phosphatase laforin. PLoS One 4:e5907 10.1371/journal.pone.0005907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernia S., Solaz-Fuster M. C., Gimeno-Alcaniz J. V., Rubio T., Garcia-Haro L., Foretz M., de Cordoba SR., Sanz P. (2009b). AMP-activated protein kinase phosphorylates R5/PTG, the glycogen targeting subunit of the R5/PTG-protein phosphatase 1 holoenzyme, and accelerates its down-regulation by the laforin-malin complex. J. Biol. Chem. 284, 8247–8255 10.1074/jbc.M808492200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ververken D., Van Veldhoven P., Proost C., Carton H., De Wulf H. (1982). On the role of calcium ions in the regulation of glycogenolysis in mouse brain cortical slices. J. Neurochem. 38, 1286–1295 [DOI] [PubMed] [Google Scholar]
- Vilchez D., Ros S., Cifuentes D., Pujadas L., Vallès J., García-Fojeda B., Criado-García O., Fernández-Sánchez E., Medraño-Fernández I., Domínguez J., García-Rocha M., Soriano E., Rodríguez de Córdoba S., Guinovart J. J. (2007). Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat. Neurosci. 11, 1407–1413 10.1038/nn1998 [DOI] [PubMed] [Google Scholar]
- Waagepetersen H. S., Westergaard N., Schousboe A. (2000). The effects of isofagomine, a potent glycogen phosphorylase inhibitor, on glycogen metabolism in cultured mouse cortical astrocytes. Neurochem. Int. 36, 435–440 10.1016/S0197-0186(99)00146-1 [DOI] [PubMed] [Google Scholar]
- Walls A. B., Heimburger C. M., Bouman S. D., Schousboe A., Waagepetersen H. S. (2009). Robust glycogen shunt activity in astrocytes: effects of glutamatergic and adrenergic agents. Neuroscience 158, 284–292 10.1016/j.neuroscience.2008.09.058 [DOI] [PubMed] [Google Scholar]
- Walls A. B., Sickmann H. M., Brown A., Bouman S. D., Ransom B., Schousboe A., Waagepetersen H. S. (2008). Characterization of 1,4-dideoxy-1,4-imino-d-arabinitol (DAB) as an inhibitor of brain glycogen shunt activity. J. Neurochem. 105, 1462–1470 10.1111/j.1471-4159.2008.05250.x [DOI] [PubMed] [Google Scholar]
- Wender R., Brown A. M., Fern R., Swanson R. A., Farrell K., Ransom B. R. (2000). Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J. Neurosci. 20, 6804–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby D., Wachten S., Masada N., Cooper D. M. (2010). Direct demonstration of discrete Ca2+ microdomains associated with different isoforms of adenylyl cyclase. J. Cell Sci. 123, 107–117 10.1242/jcs.062067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby C. A., Gentry M. S., Dixon J. E. (2008). Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J. Biol. Chem. 283, 4069–4076 10.1074/jbc.M708712200 [DOI] [PMC free article] [PubMed] [Google Scholar]