

Abstract
Red yeast rice (RYR) is made by fermenting the yeast Monascus purpureus over rice. It is a source of natural red food colorants, a food garnish and a traditional medication. Results of the current study demonstrated that polar fractions of the RYR preparations contained herbal-drug interaction activity, which if left unremoved, enhanced P-glycoprotein activity and inhibited the major drug metabolizing cytochromes P450, i,e, CYP 1A2, 2C9 and 3A4. The data from Caco-2 cell absorption and animal model studies further demonstrated that the pharmacokinetic modulation effect by RYR preparations containing the polar fractions (“untreated” preparation) was greater than that from RYR preparations with the polar fractions removed (“treated” preparation). The data indicates a potential for herb-drug interactions to be present in RYR commonly sold as nutritional supplements when the polar fractions are not removed and this should be taken into consideration when RYR is consumed with medications, including verapamil.
Red yeast rice (RYR) is a traditional food spice consumed throughout Asia. Also known as “red koji”, its food and medicinal values date back to more than a thousand years, with the first recorded use being in 800A.D1,2. RYR is derived from rice that has been allowed to ferment with the mold Monascus purpureus (M. purpureus). RYR contains starch, sugar, sterols, isoflavones, pigments, fatty acids and yeast polyketides3,4. The classes of polyketides structures that arise from the fermentation process are called monacolins, and the major monacolin found in RYR is monacolin K, which is identical in structure to lovastatin. Other polyketides in RYR are structural analogs of monacolin K5.
Lovastatin is a reversible competitive inhibitor of 3-hydroxy-3-methyl-glutary CoA (HMG CoA) reductase, which is the key enzyme that controls cholesterol biosynthesis. It has been used as a cholesterol lowering drug6,7. 20–40 mg lovastatin significantly reduced levels of cholesterol and low density lipoprotein (LDL)8. In clinical studies of hypercholesterolemic patients, individuals receiving a dose of 2,400 mg/day of RYR had 18% decrease in total cholesterol, 23% decrease in LDL cholesterol and 15% decrease in triglycerol concentrations3. In the same clinical study, a daily dose of 2,400 mg of RYR powder containing about 5 mg of monacolin K reduced cholesterol levels in hypercholesterolemic subjects to a degree equivalent to what is typically observed with the use at 20 mg of lovastatin.
Whilst the non-polar fraction of RYR has been studied extensively9, there has been no studies to-date which investigates the pharmacokinetic activity of the polar fraction of RYR extract. In this study, we have used in vitro P-glycoprotein (P-gp) activity assay, P450 CYP inhibition assay and Caco-2 cell absorption study and in vivo (animal study) systems to compare the pharmacokinetic activities of extracts from raw RYR (untreated preparation) and the extract of RYR after removal of the polar fraction by soaking RYR in water overnight (treated preparation).
Results
HPLC analysis
The chemical profiles of “untreated” and “treated” RYR were revealed by HPLC analysis. 5 µl of “untreated” and “treated” RYR extracts at 50 mg/ml were injected and analyzed by HPLC system (Figure 1). The area under the curves was measured by the software, Class-VP (Shimadzu). In untreated and treated RYR extracts, the distribution of peak area at retention time between 20–40 min, were 95.6% and 98.2% respectively (Table 1). These indicated that majority of compounds presence in RYR extracts are organic in nature and these organic fractions profile remains with water extraction. Table 1 also revealed that the polar fractions (retention time between 0–20 min) were 60% removed by water extraction in treated RYR samples. In addition, washed fractions of the “treated RYR” extract was found to contain monascumic acid and citrinin (a nephotoxin)10 along with many other unknown compounds when analyzed with LCMS-IT-TOF. The monascumic acid was determined from one of the fractionated washed fraction which showed HRMS [M+H]+ = 216.1241 (calcd for C10H17NO4 + H, 216.1236) and characteristic MS fragments which are correspond to losses of water molecule ([M-18]+) followed by the elimination of COOH group ([M+H−45]+)11. Meanwhile the citrinin was determined by referring to retention time and mass of pure citrinin purchased from Fluka.
Figure 1. HPLC profiles of “untreated” and “treated” RYR extracts were measured at wavelength of 243nm, sample concentration both at 50 mg/ml.

Table 1. Distribution (%) of area under the curve at different range of retention times. Total peak area was the sum of all peaks area from 0 to 40 min. The peaks area within particular retention time (0–10 min, 10–20 min, 20–30 min and 30–40 min) were added up and expressed in percentage over total peaks area. Retention time 0 to 20 minutes and 20 to 40 minutes were classified as polar fractions and organic fractions respectively.
| Distribution of peak (%) | |||
|---|---|---|---|
| Retention time (min) | Treated RYR | Untreated RYR | |
| Polar Fractions | 0–10 | 0.8 | 1.2 |
| 10–20 | 1 | 3.2 | |
| Organic Fractions | 20–30 | 33 | 31 |
| 30–40 | 65.2 | 64.6 | |
P-gp activity assay
The influence of “untreated” and “treated” RYR extract on P-gp activity which is the key enzyme to determine bioavailability of drug administrated was investigated by in-vitro P-gp activity assay. In this experiment, verapamil (P-gp subtract, as positive control) and “treated” or “untreated” RYR extract were incubated independently with the P-gp membrane. It was observed to have a significant change in luminescence compared to the Na3VO4 (P-gp inhibitor) treated samples. However, the “untreated” RYR extract showed more than 2-fold in enhancement in P-gp ATPase activity when compared with the “treated” RYR extract.
CYP activity assay
The inhibition effect of “untreated” and “treated” RYR extract was determined by in-vitro CYP activity assay. The luminescence signals observed in the CYP1A2, CYP2C9 and CYP3A4 activity assays are directly proportional to the enzyme activity. The net signals from untreated (added with 1x PBS) CYP1A2, CYP2C9 and CYP3A4 reactions represent total CYP activity (without any inhibition). 100 µg/ml of naringenin (NG, a CYP inhibitor) was added as negative control in the experiments. Our results suggests that the “untreated” RYR extracts exhibited stronger inhibition on CYP1A2 (about 2- fold), CYP2C9 (about 4- fold) and CYP3A4 (about 2- fold) than the “treated” RYR extract at same concentration of RYR extracts (Figure 3).
Figure 3. The study of effects of “untreated” and “treated” RYR extract on (A) CYP1A2, (B) CYP2C9 and (C) CYP3A4 activity, final concentration of samples was 1.25 mg/ml.

Data are expressed as mean ± SD (n = 3). *P<0.05 and **P<0.01 vs the untreated group (UT) control value.
Caco-2 cell absorption assay
In vitro study using monolayers of Caco-2 cells was conducted to evaluate the changes in intestinal permeability of the drug tested (verapamil) with addition of “untreated” and “treated” RYR extracts. In the Caco-2 absorption study, the concentrations of the samples taken from the basolateral and apical compartments were calculated using a calibration curve (data not shown) which was constructed by measuring the peak area obtained from injections of verapamil standards (250 μg/ml, 125 μg/ml, 30 μg/ml, 15 μg/ml, 7.5 μg/ml and 3 μg/ml). The linear equation for regression analysis from this calibration curve was found to be y = 27043x - 11153 (r2 = 0.98). The intestinal absorption permeability co-efficienct (Peff (A–B)) of the control was 5.99 ± 0.41×10−6 and that of the treated and untreated RYR extract were 7.85 ± 0.83×10−6 and 17.0 ± 0.43×10−6 respectively (Figure 4).
Figure 4. Effects of “untreated” and “treated” RYR extract on the permeability coefficients (Peff (A–B)) of verapamil in the CaCO-2 absorption study, final concentration of samples was 0.5 mg/ml.
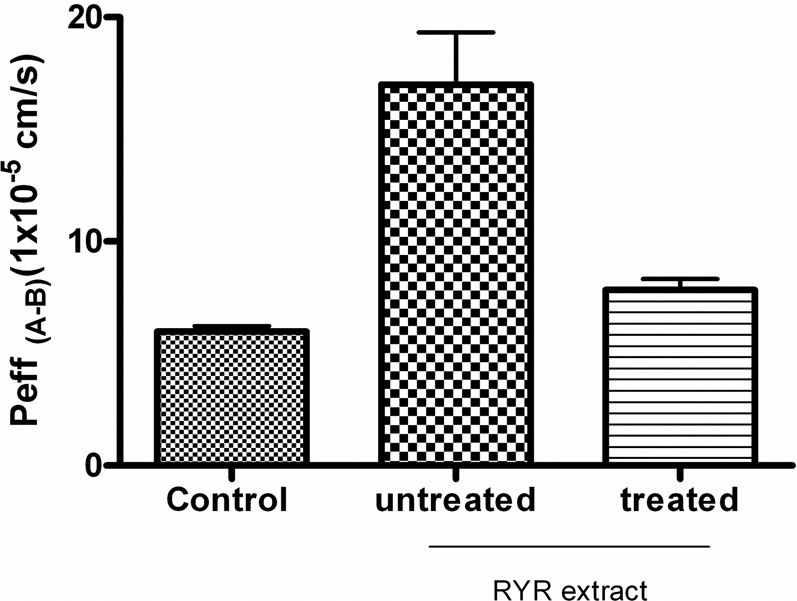
Data are expressed as mean ± SD (n = 4).
Animal study
The pharmacokinetic effect of “untreated” and “treated” RYR extract on co-administration of drug (verapamil) was investigated using an animal model. In the this study, the concentrations of verapamil present in the blood samples collected at different time points in the animal absorption study were calculated using the calibration curve (y = 17596x + 3604.2 [r2 = 0.998]). In this study, the rats were pre-treated with saline (control group) and either “treated” or “untreated” RYR extract followed with oral administration of verapamil at 10 mg/kg. After 30 minutes, the absorption of verapamil in the group that received “untreated” RYR was about 1.6-fold higher than the control group and the group that received” treated” RYR (Figure 5).
Figure 5. Plasma concentrations of verapamil in the SD rats of “untreated” and “treated” RYR treated group and the control treated group (saline).

Data are expressed as mean ± SD (n = 4–6).
Discussion
Foods, beverages and herbs are commonly consumed together with medicinal drugs. The chemicals from food and herbs may interact with medicinal drugs (e.g. grapefruit juice has been reported to interfere with the uptake of drugs12) resulting in either the reduction or increase of their bioavailability, which is an important pharmacokinetic parameter, and thus affecting treatment efficacy or tiggering toxicity. The seriousness of food-drug interactions is dependent on the therapeutic index of each drug. With modern drugs generally having lower therapeutic indices, there is a greater possibility of toxic effects and changed treatment efficacy13. P-gp is an ATP-dependent efflux pump with broad substrate specificity. The bioavailability of a drug is determined by its absorption, distribution, metabolism and excretion. These processes are mainly mediated by nuclear receptor-mediated detoxification system involving a variety of CYPs, conjugation enzymes and drug transporters (P-gp)14,15,16. Among these, the cytochrome CYP3A4 and P-gp are most important17,18. CYP3A4 accounts for about 60% of the breakdown of clinically used drugs19. P-gp transports many drugs from the intestine into the bloodstream and has been postulated to evolve as a defense mechanism against harmful substances, such as drugs and xenobiotics. Therefore, compounds that interact and inhibit the activity of P-gp and/or CYPs may change the bioavailability of drugs. For instance, naringenin (a CYP inhibitor) could enhance the bioavailability of verapamil in rabbits20, while verapamil (P-gp substrate) could enhance bioavailability of ivermectin in rats21.
Recently, RYR has been developed as a dietary supplement to lower blood lipids, including cholesterol and triglycerides. In addition to rice starch, protein, isoflavones, fiber, sterols, and fatty acids, RYR contains numerous biologically active constituents, including monacolin K, which has the ability to reduce blood cholesterol levels. Since isoflavones and sterols22,23 found in some herbs were reported to show inhibitory effect on P-gp, the question arose whether isoflavones, sterols and other compounds present in RYR caused similar effect. In order to show that the herb-drug interactions of RYR could be reduced by specific treatment of the raw material, we compared the pharmacokinetic activities of “treated” and “untreated” RYR by in vitro assay (P-gp, CYPs and Caco-2 cell assay) and in vivo assay (animal study).
Figure 1 suggested that the organic fractions (of retention time 20 to 40 minute), containing monacolin K and its analogs, of “untreated” and “treated” RYR were very similar. Therefore, the blood cholesterol lowering effect (contributed by monacolin K and its analogs) of “treated” RYR should be similar to the “untreated” RYR. The major difference in the chemical profile was in the polar fractions (of retention time 0 to 20 minute) of “treated” RYR extract, which had smaller peak areas than “untreated” RYR extract (Table 1). In conclusion, the polar compounds of RYR were effectively removed by water extraction. Nevertheless, the organic profile in the RYR remains unchanged by the extraction.
The effects of “treated” and “untreated” RYR extracts on P-gp activity were studied. The P-gp assay kit (Promega), provided the necessary reagents for performing luminescent P-gp ATPase assays. P-gp, also known as MDR1 and ABCB1, is a 170 kDa integral plasma membrane protein that functions as an ATP-dependent drug efflux pump and plays an important role in multi-drug resistance and certain adverse drug-drug interactions24. Compounds that are substrates for transport by P-gp typically stimulate its ATPase activity. P-gp dependent decreases in luminescence reflect ATP consumption by P-gp; thus the greater the decrease in signal, the higher the P-gp activity. The difference in luminescence signal between Na3VO4-treated samples and untreated samples (control) represented the basal P-gp ATPase activity, while the difference in luminescence signal between Na3VO4-treated samples and sample with the test compound (i.e. “treated” or “untreated” RYR extract) represented P-gp ATPase activity in the presence of the test compound. In Figure 2, the “untreated” RYR extract showed more than 2-fold decrease in P-gp ATPase activity as compared to the “treated” RYR extract. This suggested that compounds which interacted with P-gp ATPase in “untreated” RYR extract have been removed in the “treated” RYR samples and that these compounds activated P-gp.
Figure 2. The influences of “untreated” and “treated” RYR extract on P-gp activity, final concentration of samples was 1.25 mg/ml.
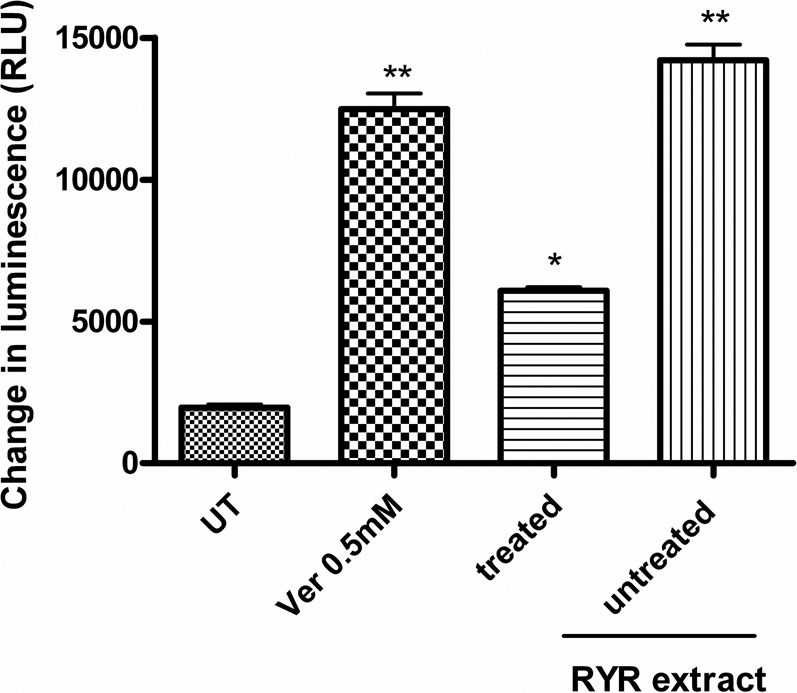
Data are expressed as mean ± SD (n = 3). *P<0.05 and **P<0.01 vs the untreated group (UT) control value.
In addition to P-gp, to determine whether the “treated” and “untreated” RYR extracts had effects on the cytochrome P450 (CYP) system, these were tested using the CYP1A2, CYP2C9 and CYP3A4 activity assays. These assay systems included membrane preparations containing a recombinant human CYP1A2, CYP2C9 or CYP3A4 enzyme, a luminogenic substrate specific to the CYP1A2, CYP2C9 or CYP3A4 enzyme, an NADPH Regeneration System and Luciferin Detection Reagent. In this assay, the modulation of CYP activity by a test compound was determined by comparing the changes from the average net signal of “untreated” CYP1A2, CYP2C9 and CYP3A4 reactions with the changes observed due to “treated” or “untreated” RYR extracts. The level of luminescence measured from the control or the test compound was directly proportional to the CYP enzyme activity. Our results revealed that addition of “untreated” RYR extract showed stronger inhibition of CYP1A2, CYP2C9 and CYP3A4 activity when compared to the addition of “treated” RYR extract (Figure 2). Since CYP1A2, CYP2C9 and CYP3A4 are known to represent over 70% of the human liver's ability to detoxify drugs, their inhibition by “untreated” RYR has implication on the safety and efficacy of the drug when consumed with RYR. Furthermore, the data also indicated the presence of as yet uncharacterized compound(s) in the polar fraction of RYR which inhibited the three CYPs. Since the P-gp enhancing activity of RYR also occurred in the polar fraction of RYR, it is unclear whether the same unknown compounds are responsible for the effects on P-gp and CYP or that these effects are due to different herb-drug interacting compounds.
To further study the pharmacokinetic effect of “treated” and “untreated” RYR extract with other drugs, a Caco-2 drug absorption study was performed. In vitro study with Caco-2 cell monolayers (i.e. a human colon carcinoma cell line) is a valuable tool for predicting human in vitro intestinal permeability. Caco-2 cells express a wide range of transporter proteins on their cell membranes similar to those of intestinal endothelium cells25. Thus, this cell line is ideal intestinal absorption simulations and studies. In the experiments, 50 µl of 1xPBS (control) or “treated” and “untreated” RYR extracts (sample concentration was 0.5 mg/ml) were added into the apical compartment of the tissue culture plates with verapamil at 100 µg/ml. Our data revealed that the addition of “untreated” RYR extract resulted in a 2-fold increase in the net absorption of verapamil (P-gp substrate) when compared with the addition of “treated” RYR extract in the Caco-2 study. Verapamil was chosen for the study on herbl-drug interaction of RYR water soluble extract in the Caco-2 absorption study because verapamil is not only a well-known P-gp substrate but also a P-gp inhibitor26. The absorption rate of the verapamil is determined by the P-gp activity. Therefore, the increase in absorption of verapamil by the addition of “treated” RYR extract revealed the potential P-gp inhibitory effect of RYR water soluble polar extract.
To determine whether the effects observed with Caco-2 cells when adding “treated” or “untreated” RYR extracts to verapamil absorption were similarly observed in an intact animal model system, the pharmacokinetic effects of co-administration of “untreated” and “treated” RYR extracts with drug (verapamil) were evaluated by an absorption study using the rat model system. The animals were pre-treated with either saline (control group) or “treated” or “untreated” RYR extract. Blood samples were collected from the jugular vein of the animal. Our data showed that at 30 minutes post-administration of verapamil, the absorption of verapamil in the group that received “untreated” RYR was significantly higher (about 2-fold) than that of the control group and the group that received “treated” RYR. In other words, co-administration of “untreated” RYR extract with verapamil caused over-dosage of verapamil. This finding implied that compared to “treated” RYR extract, “untreated” RYR extract affected the pharmacokinetics of verapamil in rats with respect to potential herb-drug interaction by inhibiting both P-gp and CYPs.
The results of this study provide the first evidence of the differences in pharmacokinetic effects of “treated” and “untreated” RYR extracts and suggest that the co-administration of “untreated” RYR with other CYP/P-gp substrate drugs (verapamil as an example) may result in changes in the pharmacokinetic profile (since the untreated RYR extract could enhance drug bioavailability).
This study implies that current use of RYR as a dietary supplement when taken together with drugs/medications can potentially cause undesirable herb-drug interactions. We have shown that CYP3A4, which accounts for 60% of the breakdown of clinically used drugs, was significantly inhibited by “untreated” RYR extract. We have also shown that P-gp activity was significantly inhibited by addition of “untreated” RYR extract (Figure 2 and Figure 3C). The study also points to the need for dietary supplement and Traditional Chinese Medicine (TCM) manufactures to develop better and safer RYR products for consumers by removing the polar fraction. In addition, the study also provided evidence of potential endogenous drug absorption modulators in M. purpureus which could explain why only about 5 mg of monacolin K (equivalent in chemical structure to lovastatin) in 2,400 mg of RYR powder can reduce cholesterol levels in hypercholesterolemic patients to a similar degree as 20 mg of lovastatin. Further studies are in progress to identify and characterize these endogenous drug absorption modulator(s) in RYR.
Methods
Preparation of RYR samples
Raw Red Yeast Rice (i.e. “untreated” RYR) was purchased from local supplier. The “treated” RYR was prepared by soaking 100 g of RYR overnight in 500 ml of distilled water at room temperature (25°C). After centrifugation at 4,000 rpm for 30 minutes, the supernatant was discarded and the RYR residue was collected and dried at 60°C for 24 hour (i.e. “treated” RYR). The “untreated” and “treated” RYR were subsequently soaked in 50% (v/v) ethanol overnight. The ethanol extracts were then collected and dried using a Speedvac concentrator. The extracts of “untreated” and “treated” RYR were analyzed by HPLC, LC-MS/MS, biochemical assays, and used in Caco-2 cell absorption study as well as rat pharmacokinetic studies.
Chemicals and reagents
Verapamil hydrochloride and lovastatin was purchased from Aldrich-Sigma (USA). Methanol and acetonitrile were obtained from Fisher Scientific (USA). All other reagents were standard laboratory reagents of analytical grade.
HPLC analysis
The HPLC system (Shimadzu Corp) used in the measurement of chemical profile and verapamil concentration in the cell absorption assay and animal experiment includes the following: LC-10AT liquid chromatograph, DGU-14A/ DGU-12A degasser, SCL-10A system controller, SIL-10AD auto injector, SPD-M10A diode array detector and CTO-10A column oven. For chemical profiling, a PFP HPLC column (150×4.60 mm i.d., 3 µm particle size, Phenomenex Inc., Torrance, CA) was used in the HPLC analysis. The flow rate was set at 0.7 ml/min with a mobile phase of 0.1% formic acid and 3% acetonitrile in distilled water. A gradient elution was set from 3-100% of acetonitrile at 40°C for 40 mins. The detector was set at 243 nm with an injection volume of 10 μl. For cell absorption assay and animal experiment, an Oryx Monolithic C18 HPLC column (100×3.0 mm i.d., 3 µm particle size, Phenomenex Inc., Torrance, CA) was used in the HPLC analysis. The flow rate was set at 0.7 ml/min with a mobile phase of 0.1% formic acid and 30% acetonitrile in distilled water. An isocratic elution was set at 30% of acetonitrile at 40°C. The detector was set at 318 nm with an injection volume of 10 μl.
LC-MS/MS analysis
The LC-MS-IT-TOF system was used to identify compounds in the polar extract. LC-MS analysis was performed using the Shimadzu Prominence Series LC coupled to the LCMS-IT-TOF. Prominence Series components included two LC-20AD pumps, DGU-20A3degasser, CBM-20A system controller, SIL-20AC auto injector and SPD-M20A PDA detector, CTO-20AC column oven, LCMS-IT-TOF MS detector and LCMS solution ver 3.50 SP2 software. The column used was a Shimadzu Shimpack VP-ODS packed with 4.6 µm particles (250×2.0 mm i.d). The flow rate was set at 0.2 ml/min with a mobile phase of 0.1% formic acid in acetonitrile and distilled water. A gradient elution was set 0 min 3% acetonitrile, 40 min 90% acetonitrile, 58 min 95% acetonitrile, 63 min 3% acetonitrile and stop time 70 min. A 5 µL injection volume was used. The LCMS-IT-TOF was operated under the following conditions: ESI in positive mode; drying gas: 1.5 L/min; CDL temperature: 200°C; interface temperature: 250°C; MS1 scan range: 100 – 900 m/z, MS2 scan range: 101 – 800 m/z.
P-Glycoprotein (P-gp) substrate assay
Pgp-Glo™ Assay System (Promega, USA) was used to analyze the effects of “treated” and “untreated” RYR extract on P-gp activity. Preparations of P-gp reaction mixtures and ATP detection reagent were done in accordance with the manufacturer's instructions with modifications as follows: 20 µl of RYR water soluble extract was prepared in different concentrations (1.25, 2.5 and 5 mg/ml); Verapamil (positive control as P-gp substrate, 0.5 mM) and Na3VO4 (P-gp inhibitor, 0.25 mM) were added into a white opaque 96-well plate (with clear flat bottom, Costar Inc., NY). P-gp was then added into the wells containing the test compounds and incubated at 37°C for 5 min. The reactions were initiated by the addition of MgATP solution and left to incubate at 37°C for 40 min. Then reactions were stopped and luminescence was initiated by adding ATP Detection Reagent to all wells. The luminescences in all samples were measured by an Infinite F200 plate reader (Tecan, Austria).
Cytochrome (CYP) 1A2, 2C9 and 3A4 P450 activity assay
CYP1A2A, CYP2C9 and CYP3A4 activity assay (CYP1A2, CYP2C9 and CYP3A4 P450-GloTM, Promega) employs luminogenic P450 probe substrates that are derivatives of beetle luciferin, a substrate for the luciferase enzyme with modification. In brief, the CYP1A2, CYP2C9 and CYP3A4 membranes were treated with 12.5 μl of luciferin-free water (as control), 100 μg/ml of naringenin (CYP inhibitor, as negative control) or the RYR water-soluble extract at different concentrations (1.25 mg/ml, 2.5 mg/ml and 5 mg/ml, drug treated) in a white opaque 96-well plate (Costar, USA). The plate was pre-incubated at 37°C for 10 min followed by addition of NADPH Regeneration solution to start the reaction. The mixture was further incubated at 37°C for 20 min before detection of luminescence by addition of ATP Detection Reagent to all wells to stop reaction. The NADPH Regeneration solution and ATP Detection Reagent were prepared in accordance with the manufacturer's instructions. The luminescences in all samples were measured by an Infinite F200 plate reader (Tecan).
Cell culture
Human colon adenocarcinoma (Caco-2) cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). Stocks of Caco-2 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM medium, Gibco, Grand Island, NY) containing high glucose (4.5 g/l) plus 20% fetal bovine serum (FBS, Gibco), 0.1 mM non-essential amino acids, 2 mM L-glutamine and penicillin and streptomycin (Gibco). Cells used in this study were between passages 20 and 40. Caco-2 cells were cultured in 75 cm2 T-flask (Corning, USA) at 37°C, 5% CO2, with constant humidity controlled environment. The medium was replaced twice a week. Cells were subcultured when they reached 80–90% confluence at a split ratio of 1∶10 using 0.05% trypsin-0.02% EDTA (Gibco)
Caco-2 cell absorption assay
Flasks with Caco-2 cells which were 90–100% confluent were harvested with trypsin/EDTA, neutralized with serum-containing medium, and the cells were harvested by centrifugation. The cell pellet was resuspended in a serum-free medium consisting of the Basal Seeding medium (BD Biosciences) and Mito+ Serum Extender (BD Biosciences) and seeded onto 6-well size Cell Culture Inserts (Corning, NY) at a concentration of 6×105 cells/cm2. The inserts contain a 0.4 µm polyester microporous membrane with collagen type I. The seeding medium was replaced with Enterocytes Differentiation medium (BD Biosciences) after 24 hours. The differentiation medium was replaced every 48 hours thereafter and the cells were maintained at 37°C, 95% relative humidity, and 5% CO2. After three days of incubation in the Enterocytes Differentiation medium, the Caco-2 cell monolayers were used for the permeability studies. Physiologically and morphologically well-developed Caco-2 cell monolayers with transepithelial electrical resistance (TEER) values greater than 300 Ω cm2 were used for the studies.
The transport medium used for these studies was modified Hank buffer containing 10 mM HEPES at pH 7.5. Prior to all experiments, each monolayer was washed twice with buffer and their TEER was measured to ensure the integrity of the monolayers. The apical to basolateral (A to B) transport of verapamil was measured in the absence and presence of the test compound. The concentration of verapamil used was 100 μg/ml. The studies were initiated by adding an appropriate volume of buffer containing verapamil to the apical (A to B transport) side of the monolayer. The volumes of the apical and basolateral compartments were 1.6 and 2.8 ml, respectively, and 50 µl of “untreated” and “treated” RYR extract were added to the apical compartment of the monolayer (final concentration at 0.5 mg/ml). The monolayers were then incubated for 3 hrs at 37°C. Samples were taken from the basolateral compartment at 0 min, 10 min, 30 min, 60 min, 90 min, 120 min and 180 min; and from the apical compartment at 0 min, during the incubation period and were analyzed for concentrations of verapamil by HPLC. The A to B permeability coefficient (Peff) of verapamil was calculated in the presence and absence of RYR water soluble extract.
Animal studies
Male Sprague-Dawley rats weighing 200–270 g were purchased from the Animal Holding Unit of The National University of Singapore (NUS, Singapore). All procedures were approved by the Institutional Animal Care and Use committee, Nanyang Polytechnic (Singapore) and performed in accordance with international standards. The animals were housed in conventional conditions under controlled cycles of light/darkness (12 hr/12 hr) with a regulated temperature (25°C). Twelve rats were allocated into two study groups with six animals in each group.
The groups of animal that received “treated” and “untreated” RYR extract were pre-treated with 1 ml of 100 mg/ml of ”untreated” and “treated” RYR extract 30 minutes prior to verapamil (10 mg/kg) administration. Rats in the control group were pre-treated with 1 ml saline. The rats were then administrated with 10 mg/kg verapamil. Blood samples were collected from the jugular vein at 0, 30, 60, 120, 240, 360 and 480 minutes after drug administration into a 1.5 ml eppendorf tube after they were anesthetized with ketamine & xylazine (both at 100 mg/ml). The samples collected were immediately centrifuged at 13,000 rpm for 2 minutes. The clear plasma layer was transferred to a clean tube and then equal volume of acentonitrile was added to denature the proteins in the samples. After a 10 sec mixing via vortex, the samples were centrifuged at 13,000 rpm for 2 minutes. The supernatants were transferred into HPLC vials and then analyzed for levels of verapamil by HPLC. The peak area ratio for an unknown sample was converted to concentration by reference to a calibration curve of verapamil constructed with drug-free pooled rat plasma.
Calculation of absorption permeability coefficient (Peff)
Absorption permeability coefficient (Peff (A–B)) in Caco-2 cell model was determined by the following equation:
Where dQ/dt is the rate of appearance of the drug in receiver chamber (B), C0 is the initial concentration of drug in the donor chamber (A), and A is the surface area across which transport occurred.
Statistical analysis
All in vitro experiments were conducted in triplicate sets. All results are expressed as mean ± SD (standard deviation). Statistical differences were evaluated using Student's unpaired t-test and one-way ANOVA when appropriate. A p<0.05 was considered to be significantly different for all tests.
Author Contributions
W. T. Fung, G. Subramaniam, J. Lee, H. M. Loh and P. H. H. Leung wrote the main manuscript. G. Subramaniam prepare figure 1, W. T. Fung prepare figures 2–4 and W. T. Fung and P. H. H. Leung prepared figure 5. All authors reviewed the manuscript.
Acknowledgments
This work was supported by SPRING- Innovation Development Scheme, Singapore. We also gratefully acknowledge Ms. He ZX for her laboratory assistance.
References
- Heber D., Lembertas A., Lu Q. Y., Bowerman S. & Liang V. An Analysis of Nine Proprietary Chinese Red Yeast Rice Dietary Supplements: Implications of Variability in Chemical Profile and Contents. J Altern Complement Med. 7, 231–236 (2001). [DOI] [PubMed] [Google Scholar]
- Ma J. et al. Constituents of red yeast rice, a traditional Chinese food and medicine. J Agric Food Chem. 48, 5220–5225 (2000). [DOI] [PubMed] [Google Scholar]
- Heber D. Dietary supplement or drug? The case for cholestin. Am J Clin Nutr. 70,106–108 (1999). [DOI] [PubMed] [Google Scholar]
- Lin Y. L., Wang T. H., Lee M. H. & Su N. W. Biologically active components and nutraceuticals in the Monascus-fermented rice: a review. Appl Micorbiol and Biotechnol. 77, 965–973 (2008). [DOI] [PubMed] [Google Scholar]
- Retterstøl K., Stugaard M., Gørbitz C., & Ose L. Results of intensive long-term treatment of familial hypercholesterolemia. Am J Cardio. 78, 1369–1374 (1996). [DOI] [PubMed] [Google Scholar]
- Demierre M. F., Higgins P. D. R., Gruber S. B., Hawk E. & Scott M. Statins and cancer prevention. Nat Rev Cancer. 5, 930–942 (2005). [DOI] [PubMed] [Google Scholar]
- Lewis B. N. J. & Ball M. J. Beneficial effect of simvastatin in patients with drug resistant familial hypercholesterolaemia. The New Zealand Medical Journal. 105, 284–286 (1992). [PubMed] [Google Scholar]
- David J. A. et al. Effects of a Dietary Portfolio of Cholesterol-Lowering Foods vs Lovastatin on Serum Lipids and C-Reactive Protein. JAMA. 290, 502–510 (2003). [DOI] [PubMed] [Google Scholar]
- Flajs D. & Peraica M. Toxicological properties of citrinin. Arh Hig Rada Toksikol. 60, 457–464 (2009). [DOI] [PubMed] [Google Scholar]
- Seden K., Dickinson L., Khoo S. & Back D. Grapefruit-drug interactions. Drugs. 70, 2373–2407 (2010). [DOI] [PubMed] [Google Scholar]
- Toshihiro A. et al. (+)- and (-)-syn-2-Isobutyl-4-methylazetidine-2,4-dicarboxylic Acids from the Extract of Monascus pilosus-Fermented Rice. J. Nat. Prod. 67, 479–480 (2004) [DOI] [PubMed] [Google Scholar]
- Chen J. & Raymond K. The role of CYP3A4 and p-glycoprotein in food-drug and herb-drug interactions. Australian Pharmacist. 25, 732–738 (2006). [Google Scholar]
- Goodwin B., Hodgson B. E., D'Costa D. J., Robertson G. R. & Liddle C. Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol Pharmacol. 62, 359–365 (2002). [DOI] [PubMed] [Google Scholar]
- Kliewer S. A. The nuclear pregnane X receptor regulates xenobiotic detoxification. J Nutr. 133, 2444S–2447S (2003). [DOI] [PubMed] [Google Scholar]
- Lau Y. Y., Okochi H., Huang Y. & Benet L. Z. Multiple transporters affect the disposition of atorvastatin and its two active hydroxy metabolites: application of in vitro and ex situ systems. J Pharmacol Exp Ther. 316, 762–771 (2006). [DOI] [PubMed] [Google Scholar]
- Fromm M. F. Importance of P-glycoprotein for drug disposition in humans. Eur J Clin Invest. 33 Suppl; 2, 6–9 (2003). [DOI] [PubMed] [Google Scholar]
- Christians U. Transport proteins and intestinal metabolism: P-glycoprotein and cytochrome P450 3A4. Ther Drug Monit. 26, 104–106 (2004). [DOI] [PubMed] [Google Scholar]
- Gibson G. G., Plant N. J., Swales K. E., Ayrton A. & El-Sankary W. Receptor-dependent transcriptional activation of cytochrome P4503A genes: induction mechanisms, species differences and interindividual variation in man. Xenobiotica. 32, 165–206 (2002). [DOI] [PubMed] [Google Scholar]
- Sharom F. J., Lugo M. R. & Eckford P. D. W. New insights into the drug binding, transport and lipid flippase activities of the p-glycoprotein multidrug transporter. J Bioenerg Biomembr. 37, 481–7 (2005). [DOI] [PubMed] [Google Scholar]
- Yeum C. H. & Choi J. S. Effect of naringin pretreatment on bioavailability of verapamil in rabbits. Arch Pharm Res. 29, 102–107 (2006). [DOI] [PubMed] [Google Scholar]
- Alvinerie M., Dupuy J., Eeckhoutte C. & Sutra J. F. Enhanced absorption of pour-on ivermectin formulation in rats by co-administration of the multidrug-resistant-reversing agent verapamil. Parasitol Res. 85, 920–922 (1999). [DOI] [PubMed] [Google Scholar]
- Ji B. S. & He L. CJY, an isoflavone, reverses P-glycoprotein-mediated multidrug-resistance in doxorubicin-resistant human myelogenous leukaemia (K562/DOX) cells. J Pharm Pharmacol. 59, 1011–1015 (2007). [DOI] [PubMed] [Google Scholar]
- Nabekura T., Yamaki T., Ueno K. & Kitagawa S. Effects of plant sterols on human multidrug transporters ABCB1 and ABCC1. Biochem Biophys Res Commun. 369, 363–378 (2008). [DOI] [PubMed] [Google Scholar]
- Aller S. G. et al. G. Structure of P-glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science. 27, 1718–1722 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siissalo S. et al. A.M. Effect of cell differentiation and passage number on the expression of efflux proteins in wild type and vinblastine-induced Caco-2 cell lines. Eur J Pharm Biopharm. 67, 548–554 (2007). [DOI] [PubMed] [Google Scholar]
- Summers M. A., Moore J. L. & McAuley J. W. Use of Verapamil as a Potential P-Glycoprotein Inhibitor in a Patient with Refractory Epilepsy. The Annals of Pharmacotherap. 38, 1631–1634 (2004). [DOI] [PubMed] [Google Scholar]