

Abstract
Packaging of proteins from the ER into COPII-vesicles is essential for secretion. In cells, most COPII-vesicles are ~60-80nm in diameter, yet some must increase their size to accommodate 300-400nm procollagen fibers or chylomicrons. Impaired COPII function results in collagen deposition defects, cranio-lenticulo-sutural dysplasia, or chylomicron retention disease, but mechanisms to enlarge COPII-coats have remained elusive. Here, we have identified the ubiquitin ligase Cul3Klhl12 as a regulator of COPII coat formation. Cul3Klhl12 catalyzes the monoubiquitination of the COPII-component Sec31 and drives the assembly of large COPII coats. As a result, ubiquitination by Cul3Klhl12 is essential for collagen export, yet less important for the transport of small cargo. We conclude that monoubiquitination controls the size and function of a vesicle coat.
Keywords: ubiquitin, Cul3, COPII, Sec31, collagen
The extracellular matrix (ECM) provides a scaffold for cell attachment and binding sites for membrane receptors, such as integrins, making it essential for the development of all metazoans 1,2. When engaged with the ECM, integrins trigger signaling cascades that regulate cell morphology and division, yet in the absence of a functional ECM, integrins are removed from the plasma membrane by endocytosis 3. The proper interplay between integrins and the ECM is particularly important during early development 4, as stem cells depend on integrin-dependent signaling for division and survival 5.
The establishment of the ECM requires secretion of several proteins, including its major constituent collagen. Following its synthesis in the ER, the export of collagen from cells depends on COPII-vesicles 6-9, and mutations in genes encoding COPII-proteins lead to collagen deposition defects, skeletal aberrations, and developmental diseases, such as cranio-lenticulo-sutural dysplasia 10,11.
COPII-vesicles are surrounded by a coat consisting of the Sar1 GTPase, Sec23-Sec24 adaptors, and an outer layer of Sec13-Sec31 heterotetramers 12. These coat proteins self-assemble into cuboctahedral structures with a diameter of ~60-80nm, which are too small to accommodate a procollagen fiber with a length of 300-400nm 13-15. Thus, collagen transport in cells must involve factors that are absent from in vitro self-assembly reactions. Indeed, TANGO1 and its partner cTAGE5 interact with collagen and Sec23/24, thereby recruiting collagen to nascent COPII coats 16,17. The deletion of TANGO1 in mice resulted in collagen deposition defects similar to those caused by loss of COPII 18, and mutations in human TANGO1 are associated with premature myocardial infarction 19. However, TANGO1 is not known to regulate the size of COPII-coats and mechanisms that permit the COPII coat to accommodate a large cargo remain poorly understood.
By analyzing mouse embryonic stem cell (mESC) division, we have identified Cul3Klhl12 as a regulator of COPII coat formation. Cul3Klhl12 monoubiquitinates Sec31 and drives assembly of large COPII-coats. As a result, ubiquitination by Cul3Klhl12 is essential for collagen export, a step that is required for integrin-dependent mESC division. We conclude that monoubiquitination determines the size and function of a vesicle coat.
Cul3 regulates mESC morphology
To provide insight into stem cell-specific division networks, we depleted ubiquitination enzymes from mESCs and scored for effects on proliferation and morphology. We found that loss of the ubiquitin ligase Cul3 caused mESCs to form tightly packed cell clusters with prominent actin cables and aberrant adhesions, as seen by confocal microscopy analysis of actin and vinculin localization (Fig. 1a). A similar phenotype was observed upon depletion of UBA3, a component of the Nedd8-pathway that activates Cul3 (Fig. S1a). Cul3-depleted mESCs were delayed in proliferation (Fig. S1b, d), yet retained their pluripotency, as seen by Oct4- and alkaline phosphatase-staining and the absence of differentiation markers in expression analyses (Fig. S1c, e, f; Fig. S2b). In contrast to mESCs, depletion of Cul3 had weaker consequences in fibroblasts (Fig. 1a), although a previously reported increase in multinucleation was observed (Fig. S1g; 20).
Figure 1. Cul3 regulates mESC morphology.

a. Left: D3 mESCs were plated on gelatin and transfected with siRNAs targeting Cul3, which resulted in cell clustering (phase microscopy; upper panel) and compaction (confocal microscopy: vinculin, green; actin, red; DNA, blue). Right: Depletion of Cul3 from mouse 3T3 fibroblasts did not cause cell compaction. b. Cul3 is required for integrin-localization to the mESC plasma membrane. D3 mESCs were plated on gelatin (top two rows), growth-factor depleted matrigel, or collagen-IV. Following Cul3-depletion, cell compaction and integrin-targeting to the plasma membrane were analyzed by confocal microscopy (actin, red; β1-integrin, green; DNA, blue).
Several observations show that the mESC-phenotypes were caused by specific depletion of Cul3. First, multiple siRNAs targeting distinct regions of the Cul3-mRNA had the same effects on mESCs, with a close correlation between knockdown efficiency and strength of phenotype (Fig. S2a). Second, microarray analysis showed a strong reduction in Cul3-mRNA upon siRNA-treatment, whereas no other gene was significantly and reproducibly affected (Fig. S2b). Third, siRNAs that target closely related proteins, such as other cullins, did not disturb the morphology of mESCs (Fig. S2c).
The aberrant morphology of Cul3-depleted mESCs was reminiscent of increased RhoA GTPase-activity, which triggers actin filament bundling 21. Accordingly, a reduction in RhoA-levels or inhibition of the RhoA-effector kinase ROCK1 rescued Cul3-depleted mESCs from compaction (Fig. S3a). Among several possibilities, higher RhoA-activity in the absence of Cul3 could result from RhoA stabilization or defective integrin-signaling. Stabilization of RhoA by co-depletion of all RhoA-specific Cul3-adaptors, the Bacurds 22, did not affect mESC morphology (data not shown). By contrast, depletion of components of integrin-signaling pathways phenocopied the loss of Cul3 in mESCs (Fig. S3b); partial reduction in Cul3-levels showed synthetic lethality with dasatinib, an inhibitor of the Src kinase that acts downstream of integrin-activation (Fig. S3c); and β1-integrin was absent from the plasma membrane of Cul3-depleted mESCs (Fig. 1b).
Cul3 could regulate integrin-synthesis and trafficking, or it could allow for efficient deposition of ECM proteins to prevent integrin internalization 3. To distinguish between these possibilities, we grew mESCs on growth factor-depleted matrigel to provide an exogenous ECM. Strikingly, under these conditions, β1-integrin was found at the plasma membrane of Cul3-depleted mESCs and no cell clustering was observed (Fig. 1b). Thus, Cul3 controls integrin-signaling in mESCs, most likely by supporting the establishment of a functional ECM.
Klhl12 is a key Cul3-adaptor in mESCs
Cul3 recruits substrates through adaptors with BTB-domains 23-26, yet siRNA-approaches did not yield roles for BTB-proteins in ESCs. As an alternative strategy to isolate Cul3-adaptors, we made use of the observation that stem cell regulators are highly expressed in ESCs, but downregulated upon differentiation 27. Using affinity purification and mass spectrometry, we identified 31 BTB-proteins that interact with Cul3 in mESCs (Fig. S4a; Table S1). When analyzed by qRT-PCR and immunoblot, we found that three adaptors, Klhl12, KBTBD8 and IBTK, were highly expressed in mESCs, but downregulated upon differentiation (Fig. 2a, b; Fig. S3d). Next, we depleted these adaptors from mESCs that were sensitized for changes in integrin-signaling by treatment with dasatinib. Importantly, depletion of Klhl12, but no other BTB-protein, resulted in mESC compaction, as seen with loss of Cul3 (Fig. 2c). Accordingly, endogenous Klhl12 effectively binds Cul3 in mESCs (Fig. S4b). These experiments, therefore, identify Klhl12 as a key substrate-adaptor for Cul3 in mESCs and the Cul3Klhl12 ubiquitin ligase as an important regulator of mESC morphology.
Figure 2. Klhl12 is a substrate adaptor for Cul3 in mESCs.
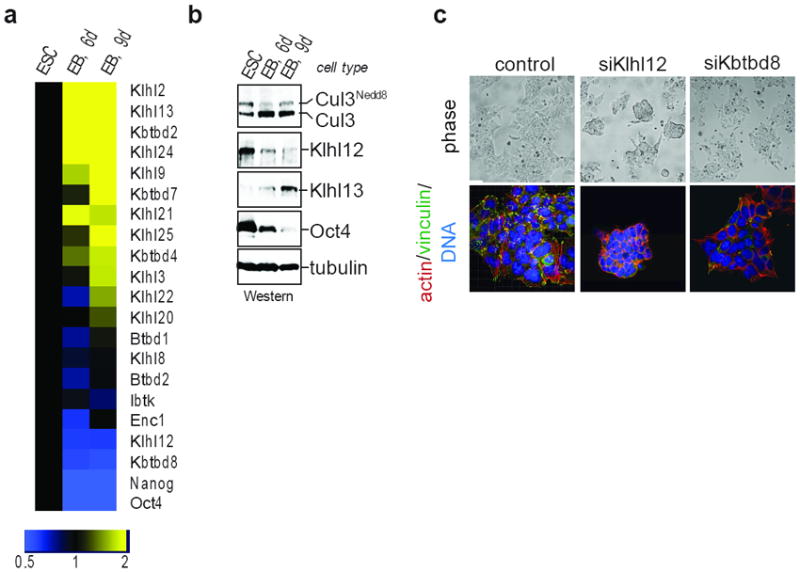
a. D3 mESCs were subjected to differentiation, and mRNA-levels of indicated proteins were measured by qRT-PCR. b. Klhl12 protein is downregulated upon differentiation, as observed by immunoblot of above samples. c. Klhl12 is a critical Cul3-adaptor in mESCs. D3 mESCs were sensitized towards altered integrin-signaling with dasatinib and monitored for compaction by phase (upper panel) or confocal microscopy (actin, red; vinculin, green; DNA, blue).
Cul3 monoubiquitinates Sec31
To isolate the substrates of Cul3Klhl12, we constructed 293T cell lines that allowed for the inducible expression of FLAGKlhl12. By affinity chromatography and mass spectrometry, we identified the COPII-proteins Sec13 and Sec31 as specific binding partners of Klhl12 (Fig. 3a; Table S2). Immunoblotting confirmed retention of endogenous Sec13 and Sec31 in Klhl12-purifications, but not in precipitates of other BTB-proteins (Fig. S5a). As seen in pulldown assays, Klhl12 directly bound Sec31, but not Sec13 (Fig. S5c, d), and this interaction was mediated the N-terminus of Sec31 (Fig. S6a) and the Kelch-domain of Klhl12 (Fig. S6b). In cells, ~30% of endogenous Klhl12 was associated with Sec13/31 (Fig. 3b; Fig. S5b). Consistent with such a prominent interaction, Sec13/31 and Klhl12 co-localized in punctae, which likely represent ER-exit sites of COPII-vesicles (Fig. 3c; 28). Importantly, siRNAs that compromise COPII resulted in mESC compaction (Fig. 3d), suggesting that Cul3Klhl12 and the COPII-coat act in the same pathway.
Figure 3. Cul3Klhl12 monoubiquitinates Sec31.

a. Immunoprecipitates of FLAGKlhl12 or FLAGKlhl9 were analyzed by Silver staining and mass spectrometry. Asterisk: non-specific band; double asterisk: breakdown product of Klhl12. b. Sec13 was immunoprecipitated from HeLa cell lysates, and Sec31 and Klhl12 were detected by immunoblot. c. Klhl12 co-localizes with COPII, as seen by confocal microscopy (Klhl12, green; Sec13, red; DNA, blue). d. D3 mESCs grown on gelatin and depleted of Sec13 were analyzed for compaction by phase (top) or confocal microscopy (actin, red; vinculin, green; DNA, blue). e. Cul3Klhl12 monoubiquitinates Sec31. Cul3Nedd8-Rbx1 was incubated with Klhl12, Sec13/31, and ubiquitin or Hisubiquitin. f. In vitro ubiquitination of Sec31 by Cul3Klhl12 or Cul3Klhl12-FG289AA was performed as above. g. Sec31 is monoubiquitinated in vivo. Upper panels: Ubiquitin conjugates were purified under denaturing conditions from MG132-treated 293T cells expressing Hisubiquitin, HASec31, Klhl12, Cul3, or dominant-negative Cul3, and analyzed by αSec31-Western. Lower panels: the same experiment was performed with lysine-free Hisubiquitin, which only allowed Sec31-monoubiquitination on at least two sites (Sec31ubi1 and Sec31ubi*). h. Ubiquitin conjugates were purified from 293T cells expressing Klhl12 or Sec31-binding deficient Klhl12-mutants. i. Cul3 is essential for Sec31-ubiquitination in vivo. 293T cells were transfected with Hisubiquitin and siRNAs, and ubiquitin conjugates were analyzed for Sec31 by Western.
In vitro, Cul3Klhl12 catalyzed the monoubiquitination of Sec31 (Fig. 3e), which was not observed if a Klhl12-mutant with a defective Sec31-binding interface was employed (Fig. 3f). Sec31 was also monoubiquitinated in cells, which was strongly increased upon expression of Klhl12 (Fig. 3g). Klhl12-mutants unable to bind Sec31 abolished its monoubiquitination (Fig. 3h), which is likely due to dimerization with and inactivation of endogenous Klhl12 (Fig. 3a, Fig. S6c). Sec31-monoubiquitination was also strongly diminished upon expression of dominant-negative Cul3 (Fig. 3g) or depletion of Cul3Klhl12 by siRNA (Fig. 3i). As seen upon expression of lysine-free ubiquitin, Sec31 was monoubiquitinated at one preferred and an alternative, less prominently used lysine (Fig. 3g), consistent with proteomic analyses that identified Lys647 and Lys1217 in Sec31A as ubiquitination sites 29,30. However, neither mutation of these residues nor any other of the 65 lysine residues of Sec31 blocked ubiquitination by Cul3Klhl12 (data not shown), revealing flexibility in the actual modification site.
Co-expression of Klhl12 and Cul3 triggered Sec31-multiubiquitination and degradation (Fig. 3g, 4e; Fig. S6d), which was not observed with lysine-free ubiquitin (Fig. 3g). However, whereas Sec31 was monoubiquitinated by endogenous Cul3Klhl12, its multiubiquitination was only seen when Cul3 and Klhl12 were overexpressed. Depletion of Cul3Klhl12 or proteasome inhibition did not change Sec31-levels in untransfected cells (Fig. 3i, Fig. S6e), and blockade of ubiquitin chain formation or proteasome inhibition did not impair Cul3Klhl12-function (see Fig. 5). Thus, multiubiquitination of Sec31 is unlikely a key outcome of Cul3Klhl12-activity in mESCs. Instead, it appears that Cul3Klhl12 acts by catalyzing monoubiquitination, with the COPII-protein Sec31 as a major substrate.
Figure 4. Cul3Klhl12-dependent monoubiquitination enlarges COPII-structures.

a. Localization of induced FLAGKlhl12 (green) and Sec31 (red) in 293T cells, monitored by confocal microscopy. Scale bar: 3μm. b. Klhl12-expressing HeLa cells were analyzed for Klhl12 (green) and Sec31, Sec13, or Sec24C (red) by confocal microscopy. Scale bar: 3μm. c. COPII-structures in HeLa cells transfected with FLAGKlhl12, lysine-free ubiquitin, or Cul3-siRNA, analyzed by confocal microscopy. Scale bar: 500nm. d. Upper panel: Thin-section EM of Klhl12-expressing or control HeLa cells (red arrow: Klhl12-dependent structures; blue arrow: small control vesicles). Scale bar: 500nm. Lower panel: Immunogold-EM of Klhl12 in transiently transfected HeLa (left) or stable 293T cells (right). Scale bar: 200nm. e. HeLa cells transfected with FLAGKlhl12, lysine-free ubiquitin, FLAGKlhl12FG289AA, FLAGCul31-250, or FLAGCul3, were analyzed for localization of Klhl12/Cul3 (green) and Sec31 (red) by confocal microscopy. Scale bar: 5μm.
Figure 5. Cul3Klhl12 promotes collagen export.
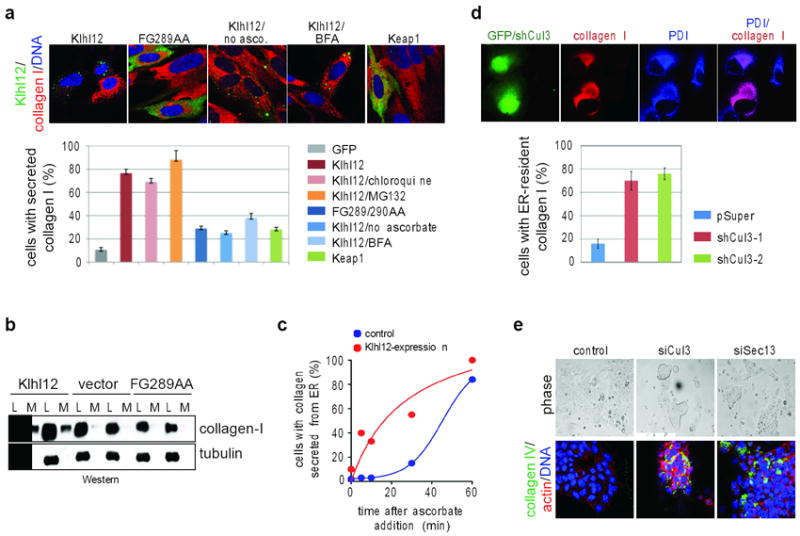
a. IMR90 cells transfected with FLAGKlhl12, FLAGKlhl12FG289AA, or FLAGKeap1 were analyzed by confocal microscopy (BTB, green; collagen-I, red; DNA, blue). When noted, cells were treated with chloroquine, MG132, brefeldin A (BFA), or dialyzed medium lacking ascorbate. Errors: standard deviation, n=3. b. Cell lysate (L) or culture medium (M) of IMR90 cells transfected with FLAGKlhl12, or FLAGKlhl12FG289AA was analyzed by immunoblotting. c. Collagen-I localization was analyzed IMR90 cells expressing Klhl12, after re-addition of ascorbate. d. HT1080 cells stably expressing collagen-I were transfected with shRNAs against Cul3 and analyzed by confocal microscopy (transfection control GFP, green; PDI, blue; collagen-I, red). Error bars: standard deviation, n=3. e. D3 mESCs were treated with control siRNAs or siRNAs targeting Cul3 or Sec13 and analyzed by confocal microscopy (collagen-IV, green; actin, red; DNA, blue).
Cul3 regulates the size of COPII coats
To identify a role for monoubiquitination by Cul3Klhl12, we induced Klhl12-expression in cells and followed the fate of Sec31 by microscopy. Shortly after Klhl12-induction, the majority of Klhl12 and Sec31 co-localized in small punctae (Fig. 4a). Over time, these punctae grew into much larger structures that contained most of Sec31, as well as other COPII-components, such as Sec13 or Sec24C (Fig. 4a, b). As seen by high-resolution confocal imaging, the large structures were hollow and spherical with a diameter of 200-500nm, and they were decorated with the proteins of the COPII-coat and with Klhl12 (Fig. 4c). Accordingly, thin-section electron microscopy revealed large, crescent-shaped tubules, possibly of ER origin, in cells transfected with Klhl12 (Fig. 4d). Immunogold-labeling EM showed comparable structures of 200-500nm that were decorated with Klhl12 (Fig. 4d). The Klhl12-dependent structures neither contained a cis-Golgi protein; Ergic-53, which is absent from procollagen transport vesicles 31; ER-membrane markers that do not accumulate at ER-exit sites 32; nor endosomal or autophagosomal markers (Fig. S7a-c). Importantly, Sec31-binding deficient mutants, including Klhl12FG289/290AA, neither co-localized with Sec31 nor induced formation of large structures (Fig. 4e; Fig. S7d), and depletion of Sec31 blocked formation of large structures by Klhl12 (Fig. 4b). Thus, binding of Klhl12 to Sec31 triggers formation of large COPII-containing structures.
When Klhl12 was expressed with a Cul3-mutant that blocks Sec31-ubiquitination (Cul31-250), COPII-structures were not enlarged (Fig. 4e). In addition, depletion of Cul3 by siRNAs, which also abolishes Sec31-monoubiquitination, prevented formation of large COPII-structures by Klhl12 (Fig. 4a, c). By contrast, if Klhl12 was expressed with lysine-free ubiquitin to allow mono-, but not multiubiquitination, large COPII-structures were readily detected (Fig. 4c, e), and these structures were enriched for ubiquitin, consistent with monoubiquitination being non-proteolytic (Fig. S7e). Thus, monoubiquitination by CulKlhl12 promotes formation of large COPII-structures, which likely represent a mixture of nascent coats at ER-exit sites and budded coats on large COPII-vesicles or tubules.
Cul3 is required for collagen export
Our screen linked Cul3Klhl12 to the establishment of the stem cell ECM, which requires collagen secretion. Thus, the Cul3Klhl12-dependent increase in COPII-size might function to promote collagen export from the ER. To test this hypothesis, we expressed Klhl12 in IMR90 cells, which at steady state accumulate collagen in the ER due to inefficient export. Strikingly, Klhl12, but not Klhl12FG289/290AA or unrelated BTB-proteins, triggered depletion of procollagen I from intracellular ER-pools (Fig. 5a). As a result, increased collagen levels were detected in the supernatant of cells expressing Klhl12, but not Klhl12FG289/290AA (Fig. 5b). When secretion was inhibited with brefeldin A, or if collagen folding in the ER was impaired by removal of ascorbate from the medium, procollagen remained within Klhl12-expressing cells (Fig. 5a). Time-resolved experiments showed that Klhl12 strongly accelerated collagen export from IMR90 cells (Fig. 5c). Shortly after inducing secretion, Klhl12 and collagen were detected at overlapping locations (Fig. S7f), all of which indicates that Cul3Klhl12 facilitates collagen traffic from the ER.
Blockade of Sec31-ubiquitination by dominant-negative Cul3 interfered with the Klhl12-dependent export of collagen from IMR90 cells (Fig. S8a). Similarly, depletion of Cul3Klhl12 from engineered HT1080 fibrosarcoma cells severely impaired collagen export, and most cells retained high levels of collagen in their ER (Fig. 5d; Fig. S8b). In contrast, smaller COPII-cargoes, such as fibronectin or EGF receptor, were properly localized in the absence of Cul3 (Fig. S8c, d). Similar observations were made in mESCs, where depletion of Cul3 led to a strong intracellular accumulation of collagen IV, comparable to the effects observed upon loss of Sec13 (Fig. 5e; Fig. S8e). Thus, Cul3Klhl12 is required for collagen-export, while it is less important for the trafficking of smaller COPII-cargo.
If promoting collagen export were the key role of Cul3 in mESCs, the phenotypes of Cul3-depletion might be mitigated by addition of collagen in trans. Indeed, this was the case: when mESCs were plated on purified collagen-IV, depletion of Cul3 did not cause cell clustering, and β1-integrin was detected at the plasma membrane (Fig. 1c). We conclude that promoting collagen secretion is a key a function of Cul3, in agreement with its role in driving the assembly of large COPII-coats.
Discussion
In this study, we have identified Cul3Klhl12 as an essential regulator of collagen export, which is required for mESC division. Deletion of Cul3 in mice results in early embryonic lethality with completely disorganized extraembryonic tissues 33, a phenotype that can in part be attributed to its role in collagen secretion. Moreover, Klhl12 has been identified as an autoantigen in the connective tissue disorder Sjogren’s syndrome 34, raising the possibility that aberrant function of Cul3Klhl12 might be related to disease.
Cul3Klhl12 monoubiquitinates Sec31 and promotes formation of large COPII-coats that can accommodate unusually shaped cargo. As a result, Cul3 is essential for the secretion of procollagen fibers, while it is not required for the transport of smaller or more flexible molecules, such as fibronectin, EGF receptor or β1-integrin. Thus, Cul3Klhl12 appears to be specifically required for the COPII-dependent transport of large cargo.
How ubiquitination affects COPII-size or structure coats is not known. None of the 65 lysine residues of Sec31 was essential for ubiquitination by Cul3Klhl12, showing that Cul3 can target alternative lysine residues if the primary site is blocked. Despite this flexibility, Cul3Klhl12 does not stoichiometrically ubiquitinate Sec31. Thus, if Sec31-ubiquitination performs a structural role, then few ubiquitinated molecules must suffice to produce large COPII-coats, and these vesicles must tolerate considerable variation in the modification site. Alternatively, as often seen with monoubiquitinated proteins, modified Sec31 might recruit an effector that delays COPII-budding or promotes coat polymerization. As Cul3Klhl12 ubiquitinates other proteins 35, Sec31 may not be its only substrate in the secretory pathway. Identification of the complete set of Cul3Klhl12-substrates and potential effector molecules should reveal the mechanism underlying the ubiquitin-dependent regulation of vesicle size.
Our findings have the potential to be translated into therapeutic strategies. We envision that agonists of Cul3Klhl12-function mitigate consequences of Sec23A-mutations in cranio-lenticulo-sutural dysplasia or Sar1-mutations in chylomicron retention disease 10,11. By contrast, interfering with Cul3-activity may counteract increased collagen deposition during fibrosis or keloid formation 36. Given the strong clustering phenotypes observed in Cul3-depleted mESCs, inhibition of Cul3Klhl12 might impair the proliferation of metastatic cells, which display features of undifferentiated cells 37,38. Thus, our identification of Cul3Klhl12 as a regulator of COPII size and function provides an exciting starting point to understand and therapeutically exploit key events in protein trafficking.
Methods summary
For stem cell culture, mouse D3 ESCs were maintained in GIBCO Dulbecco’s Modified Eagle ESC medium containing 15% FBS, 1x sodium pyruvate, 1x NEAA, 1mM β-ME and 1000u/ml LIF (Millipore), and grown on gelatin-coated culture plates. Doxycycline-inducible 293T Trex FLAGBTB stable cell lines were made with Flp-In™ T-REx™ 293 Cell Line system (Invitrogen) and maintained with blasticidin and hydromycin B.
For screening, two siRNA oligos were designed against 40 mouse ubiquitin ligases (Qiagen). 10pmol of siRNA oligo and Lipofectamine 2000 were pre-incubated in a gelatin-coated 96-well plate. D3 mESCs were seeded at 15000 cells/well on top of the siRNA mixture, and the morphology of ESC colonies was examined by bright-field microscopy 48h post transfection.
To identify Cul3Klhl12 substrates, doxycycline-inducible 239T cell lines expressing FLAGKlhl12 or FLAGKlhl9 were induced for 48h. Cleared lysate was subjected to anti-FLAG M2 affinity gel (Sigma), and precipitations were eluted with 3xFLAG peptide (Sigma). Concentrated eluates were analyzed by SDS-PAGE, and specific bands were identified by mass spectrometry analysis by the Vincent J. Coates Proteomics/Mass Spectrometry Laboratory.
For in vitro ubiquitination reactions, Cul3/Rbx1 purified from Sf9 cells was conjugated to NEDD8 using recombinant APPBP1-UBA3, Ubc12, and NEDD8. Klhl12 purified from E. coli and Sec31A/Sec13 complexes from Sf9 cells were added together with energy mix, E1, UbcH5c, and ubiquitin and incubated at 30°C for 1hr.
For confocal microscopy, cells fixed in paraformaldehyde and permeabilized with TritonX-100 were incubated with primary antibodies for 2h and Alexa-labeled secondary antibodies (Invitrogen) for 1h. Pictures were taken on Zeiss LSM 510 and 710 confocal microscopes and analyzed with LSM image browser and Imaris 3D imaging processing software. Images were processed for contrast enhancement to remove noise.
Supplementary Methods
Plasmids, protein, antibodies
Human Cul3 and Klhl12 were cloned into pcDNA4 and pcDNA5 vectors for expression in mammalian cells. Cul3, Sec31A and Sec13 were also cloned into pCS2 vector for IVT/T and expression in mammalian cells. pcDNA4-Cul3N250 contains the first cullin repeat of the N-terminal Cul3 (1-250aa) which is sufficient for binding BTB proteins, but not Rbx1 and serves as a dominant negative for Cul3/BTB-mediated ubiquitination. The Klhl12 mutants FG289AA, RL342AA, RGL369AAA, RE416AA, YDG434AAA and RCY510AAA were made by site-directed mutagenesis.
Cul3 and Rbx1 were cloned into pFastBac, co-expressed in Sf9 ES insect cells using the Bac-to-Bac baculovirus expression system (Invitrogen) and purified as a complex by Ni-NTA agarose (Qiagen). Similarly, the Sec31A/Sec13 heterodimer and UBA1 were purified from Sf9 ES insect cells. UbcH5c and Ubc12 were cloned into pQE vector and purified from BL21(DE3) bacterial cells. Ubiquitin was cloned into pET and pCS2 vector with a N-terminal 6xHis tag. The pET-His-ubiquitin was used for bacterial purification whereas pCS2-His-ubiquitin was expressed in mammalian cells. Wildtype ubiquitin, APPBP1-UBA3 and NEDD8 were purchased from Boston Biochem.
To purify recombinant Klhl12 for ubiquitination assays, we expressed pMAL-TEV-Klhl12-his and pMAL-TEV-Klhl12FG289AA-his in BL21(DE3) cells, purified the proteins on amylose resin, cleaved them by TEV protease, and re-purified them on Ni-NTA agarose. WT-Klhl12 and mutants were also cloned into pMAL vector and purified as MBP-tagged proteins for in-vitro protein binding assays.
All shRNAs were cloned in pSuper-GFP neo vector (from Oligoengine) into BglII and XhoI sites. The GFP-Bcl2-Cb5 construct, a fusion between Bcl2 and cytochrome b5, was purchased from Clontech.
We raised mouse monoclonal antibodies against human Klhl12 and human Klhl13. Both antibodies are available at Promab Biotechnologies (cat. # 30058 and # 30067). We also raised antibodies against Sec13, Sec24C, and Sec24D. Other antibodies used in this study are: Cul3 (Bethyl Laboratories, cat. # A301-109A), Sec31A (BD Biosciences, cat. # 612350), Collagen IV (Abcam, cat. # ab19808), anti-FLAG (Sigma, cat. #F3165, #F7425), Ubiquitin (Santa Cruz, cat. # sc-8017, P4D1), Rhodomine phalloidin (Invitrogen, cat. # R415), PDI (1D3) (Assay Designs, cat. #SPA-891), anti LC-3 (Sigma, Cat # L-7543), anti-alpha tubulin (DM1A, Abcam, Cat # ab7291), anti-fibronectin (Abcam, ab2413), anti-GM130 (BD Biosciences, cat. # 610822), and anti-EGFR (Ab12, Neomarkers, MS-400P1). LF-67 (Anti sera for Type I procollagen) was obtained as a generous gift from Dr. Larry Fisher.
Cell culture
The D3 mouse embryonic stem cells (mESC) were maintained in ESC medium containing 15% FBS, 1x sodium pyruvate, 1x NEAA, 1mM β-ME and 1000u/ml LIF (Millipore, cat. # ESG1107) in GIBCO Dulbecco’s Modified Eagle Medium, and grown on 0.1% gelatin-coated tissue culture plates. Hela cells, 293T cells, 3T3 cells and IMR90 cells were maintained in DMEM plus 10% FBS. Dialyzed FBS was bought from Hyclone. The doxycycline-inducible 293T Trex Klhl12-3xFLAG stable cell line was made with Flp-In™ T-REx™ 293 Cell Line system from Invitrogen. Stable cell lines expressing other BTB-proteins were generated accordingly. These cell lines were maintained with 10% TET(-) FBS, blasticidin and hydromycin B as instructed and expression was induced by 1ug/ml doxycycline.
Human lung fibroblasts IMR-90 cells were obtained from the Corielle Institute: NIA (National Institute on Aging) Aging Cell Repository. For generating procollagen stable HT-1080cell lines, we cloned proalpha(1) into a pRMc/CMV-vector and selected for neomycin resistance 39. This vector was provided as a generous gift by Neil Bulleid. Cells were kept in a 37°C incubator at 5%CO2.
siRNA screen in mouse ES cells
siRNA oligos against 40 mouse ubiquitin E3 enzymes were pre-designed by Qiagen and handled as instructed. Two different siRNA oligos against each gene were included in the initial screen. 10pmol of siRNA oligos and 0.25ul of Lipofectamine2000 were pre-incubated in a 0.1% gelatin-coated 96-well plate in 20ul of OPTIMEM for 15min at room temperature. The D3 mESCs were trypsinized and seeded at 15000 cells/well in 80ul of ESC medium on top of the siRNA mixture. Fresh medium was added to the cells the next day and the morphology of ES cell colonies were examined using bright-field microscopy at 48h post transfection. Hit validation was performed with additional siRNAs that were purchased from two distinct vendors (Qiagen, Dharmacon) and that target different sites of the Cul3 mRNA. Knockdown efficiency was tested by qRT-PCR and immunoblot.
Rescue of Cul3-siRNA phenotype in mESCs by matrigel and collagen-IV
D3 mESCs were cultured on tissue culture dishes coated with gelatin (negative control), growth-factor depleted matrigel (BD Biosciences, cat# 356231), or purified collagen-IV (BD Biosciences, cat# 354233). Matrigel and collagen-IV were applied at 10μg/cm2. Cul3 was depleted 24h later using our standard siRNA transfection protocol, and mESC morphology was analyzed by confocal microscopy against β1-integrin, actin, and DNA.
Drug treatments of Cul3-depleted cells
To study the synthetic lethal effect of Src-inhibition with Cul3 knockdown, we treated wildtype and Cul3-depleted D3 mESCs with 0, 25, 50, 100nM of dasatinib for 18h before the phenotypes were analyzed by light microscopy.
To study the effect of Rho-inhibition on Cul3 knockdown, Cul3-depleted D3 mESCs were treated with ROCK inhibitor Y27632 at 10uM for 24h before phenotype analysis. Alternatively, RhoA was co-depleted using specific siRNAs.
Cell cycle analysis
To assess the division rate of Cul3-depleted mESCs, we treated cells with control, Cul3-, or Ube2C/Ube2S-siRNA and seeded at 3×105 cells/well in gelatin-coated 6-well plates. The specificity of Ube2S- and Ube2C-siRNAs was tested before 40. The cells were trypsinized at 2, 3 and 4d post transfection and counted by hemocytometer.
ES cell differentiation analysis
To differentiate mouse ES cells into embryoid bodies (EBs), we trypsinized undifferentiated D3 mouse ES cells, washed once with LIF-free ESC media, and seeded the cells at 2×106 cells/dish onto 10-cm Corning Ultra-Low-Attachment Dishes (Corning cat. # 3262) containing 10 ml of ESC medium without LIF. After 24h, the cells were dissociated from the plate by gentle pipetting of the medium and collected in a 15ml Falcon tube by centrifugation. The supernatant was aspirated off and the cells were re-seeded onto 10-cm Corning Ultra-Low-Attachment Dishes containing fresh ESC medium without LIF. Medium was changed every other day for a total of 6 or 9d. Total RNA of ESCs and EB samples was extracted using TRIzol (Invitrogen, cat. # 15596-026) and chloroform. The expression of pluripotent markers and BTB genes at various time points during differentiation was analyzed using quantitative real-time PCR.
As a complementary experiment, D3 mESCs were treated with control or Oct4 siRNA. 48h after transfection, cells were collected and total RNA was extracted using TRIzol as above. The expression of pluripotent markers, tissue specific genes and BTB genes in control and Oct4-depleted cells were analyzed using qRT-PCR.
Quantitative real-time PCR analysis
We used TRIzol (Invitrogen, cat. # 15596-026) and chloroform to extract total RNA from cells. The first-strand cDNAs were synthesized by using Revertaid first strand cDNA synthesis kit (Fermentas, cat. # K1621). Gene-specific primers for qRT-PCR were designed by using NCBI Primer-Blast. The quantitative RT-PCR reaction was done with the Maxima SYBR Green/Rox qPCR system (Fermentas, cat. # K0221).
Identification of Cul3Klhl12-substrates
To identify Cul3Klhl12 substrates, we generated a doxycycline-inducible hKlhl12-3xFLAG stable cell line using the Flp-In™ T-REx™ 293 Cell Line system (Invitrogen). As controls, we generated stable cell lines expressing other BTB proteins including Klhl9. Klhl12-3xFLAG and Klhl9-3xFLAG expression was induced in 30×15cm-plates by 1ug/ml of doxycycline for 48h, and cells were collected by centrifugation and lysed by douncing 40 times in PBS+0.1%NP40. The cell lysate was cleared by centrifugation and then subjected to anti-FLAG M2 affinity gel (Sigma, cat. # A2220-5mL) at 4C for 4h on a rotator. Immunoprecipitations were eluted by 300ul of 200ug/ml 3xFLAG peptide (Sigma, cat. # F4799-4MG) in PBS. The elution was repeated three times for 1h at room temperature. Eluates were pooled, concentrated to 100ul using Amicon Ultra-0.5, Ultracel-10 Membrane (Millipore, cat. # UFC501008) and run on a SDS-PAGE gel. The gel was stained by SimplyBlue™ SafeStain (Invitrogen, cat. # LC6060), and specific gel bands were cut out and sent for mass spectrometry analysis by the Vincent J. Coates Proteomics/Mass Spectrometry Laboratory at UC Berkeley.
Immunoprecipitation of endogenous protein complexes
To confirm the interaction of endogenous proteins, we lysed Hela cells or D3 mESCs by freeze-thaw twice in 20mM HEPES buffer pH7.5, 5mM KCl, 1.5mM MgCl2, 1x protease inhibitor cocktail (Roche). Specific antibodies against Cul3, Sec13 or Sec31 conjugated to protein G agarose beads were added to the cleared cell lysate and incubated at 4C for 4h. Protein complexes were eluted with gel-loading buffer at 95°C. Endogenous proteins in complexes were detected by immunoblot using specific antibodies against Cul3, Sec13, Sec31, or Klhl12.
To detect ubiquitination of endogenous COPII components, we incubated Hela cell extract with pre-immune serum or antibody against Sec13 conjugated to protein G agarose beads at 4C for 4h. Protein complexes were eluted with SDS gel-loading buffer at 95°C. Ubiquitinated proteins in the complex were detected by immunoblot against ubiquitin.
In-vitro protein interaction assays
To dissect the Klhl12 and Sec31A interaction, we coupled 20ug recombinant MBPKlhl12, various mutants or MBP as a control to 15ul amylose resin by incubating at 4C for 1h. Cul3, Sec31A and mutants were expressed from pCS2 and labeled with 35S-Met using TnT Sp6 Quick Coupled Trsnc/trans Syst (Promega, cat. # L2080). The labeled Cul3 or Sec31A were incubated with MBP-Klhl12 or mutants at 4C for 3h. Beads were washed 4x with TBST and 2x with TBS, and incubated in SDS loading buffer at 95°C. Samples were run on SDS-PAGE and results were visualized by autoradiography.
In vitro ubiquitination assays with Cul3Klhl12
Cul3/Rbx1 was conjugated to NEDD8 at 30C for 1h with the following conditions: 2.5 mM Tris/HCl pH 7.5, 5 mM NaCl, 1 mM MgCl2, 1 mM DTT, 1x energy mix 40, 1uM APPBP1-UBA3, 1.2 uM Ubc12, 4 uM Cul3/Rbx1, and 60 uM NEDD8. For in-vitro ubiquitination of Sec31A, we set up a 10ul reaction as follows: 2.5 mM Tris/HCl pH 7.5, 5 mM NaCl, 1 mM MgCl2, 1 mM DTT, 1x energy mix, 100nM UBA1, 1uM UbcH5c, 1uM Cul3~Nedd8/Rbx1, 1uM Klhl12, 150uM ubiquitin, 0.05ug Sec13/31A. The reaction was carried out at 30C for 1hr and stopped by adding SDS gel loading buffer.
In vivo ubiquitination assays with Cul3Klhl12
293T cells grown in 10cm dishes were transfected with pCS2-HA-Sec13/31A, pCS2-His-ubiquitin, pcDNA5-Klhl12-FLAG, pcDNA4-Cul3-FLAG, or pcDNA4-Cul3N250-FLAG, as indicated, using calcium phosphate. 24h later, 1μM MG132 was added and cells were incubated overnight. Cells were harvested with gentle scraping and resuspended in 1ml buffer A (6M guanidine chloride, 0.1 M Na2HPO4/NaH2PO4 and 10mM immidazole,pH 8.0). Cells were lysed by sonication for 10s and incubated with 25ul Ni-NTA agarose at room temperature for 3h. The beads were washed 2x with buffer A, 2x with buffer A/TI (1 volume buffer A and 3 volumes buffer TI), 1x with buffer TI (25mM Tris-Cl, 20mM imidazole, pH6.8), and incubated in 60ul SDS gel loading buffer containing 300mM imidazole and 50mM βME at 95°C. Samples were separated by SDS-PAGE and ubiquitinated Sec31A was detected by immunoblot using antibody against Sec31A.
To detect Sec31A ubiquitination upon Cul3/Klhl12 depletion, we co-transfected 100nM siRNAs against Cul3 or Klhl12 with pCS2-HA-Sec13/31A and pCS2-His-ubiquitin using calcium phosphate. The Ni-NTA purification was performed 48h post transfection and Sec31A ubiquitination was detected as described above.
Confocal microscopy
Cells were fixed in 4% paraformaldehyde and permeabilized with 0.5% TritonX-100 in 1X TBS, 2% BSA. Cells were incubated with primary antibodies against Sec31A, Sec13, Sec24C, ERGIC53, CD63, BiP, or ubiquitin for 2h and secondary antibodies (Invitrogen, Alexa Fluor® 546 goat anti-rabbit IgG (H+L); Alexa Fluor 488 goat anti-mouse IgG (H+L); HOECHST 33342,) for 1h at room temperature followed by extensive washing. Pictures were taken on Zeiss LSM 510 and 710 Confocal Microscope systems and analyzed with LSM image browser and Imaris 3D imaging processing software.
Transmission Electron Microscopy
Mock- and Klhl12-transfected HeLa cells were grown to 70% confluence as a monolayer on an Aclar ® sheet (Electron Microscopy Sciences, Hartfield, PA). The cells were fixed for 30min in 0.1M cacodylate buffer, pH 7.2, containing 2% glutaraldehyde, and subsequently washed with buffer prior to post-fixation with 1% Osmium tetroxide on ice. This was followed by staining with 1 % aqueous Uranyl Acetate for 30 min at room temperature. For dehydration with progressive lowering of temperature, each incubation period was 10 min, with exposure to 35% ethanol at 4°C, to 50% ethanol and 70% ethanol at −20°C, and 95%, and 100% ethanol at −35°C. Cells were restored to room temperature in 100% ethanol before flat embedding in an Epon resin. Thin (70-100nm) sections were collected on Formvar-coated 200-mesh copper grids and post-stained with 2% aqueous uranyl acetate and 2% tannic acid. The sections were imaged at 120 kV using a Tecnai 12 Transmission Electron Microscope (FEI, Eindhoven, Netherlands).
For the purpose of immunolabeling, HeLa cells expressing FLAGKlhl12 or doxycycline-inducible 293T Trex FLAGKlhl12 stable cell lines were fixed in 2% paraformaldehyde and 0.5% glutaraldehyde and embedded in LR white resin. Fixation and infiltration were performed in a microwave oven (Pelco model 3450, Ted Pella, Inc., Redding, CA). 70 nm thick sections were picked on 100-mesh nickel grids coated with Formvar film and carbon, incubated in blocking buffer (5% BSA, 0.1% fish gelatin, 0.05% Tween20 in PBS) for 30 min, and followed by incubation with αFLAG antibody at a dilution of 1/40 for 1h. Goat anti-mouse IgG conjugated with 10 nm gold (BD Biosciences) was used as the secondary antibody at a diltuion of 1/40 for 1 h. Sections were poststained in 2% uranyl acetate for 5 min.
Gene expression analysis by microarray
To compare gene expression profiles of WT-mESCs versus Cul3-depleted mESCs, we transfected D3 mESCs with control or Cul3-siRNA, followed by growth on gelatin-coated 6-well plates. 48h later, total RNA was extracted by TRIzol and chloroform, and further purified using RNeasy Mini Kit (Qiagen, cat. # 74104). Microarray analysis was performed by the Functional Genomics Laboratory (UC Berkeley) using Affymetrix Mouse 430A 2.0 chip.
Analysis of collagen export from cells
IMR-90 human lung fibroblasts grown on 100mm dishes in DMEM/10% FBS were transfected with FLAGKlhl12, FLAGKlhl12FG289AA, FLAGKeap1 and pcDNA5-flag using nucleofection kit R (bought from Lonza) as described in the manufacturer’s protocol and plated on 6 well plate with 25mm coverslips. When indicated, co-transfections with 2μg each of FLAGKlhl12 and dominant-negative Cul3 were performed. Dialyzed 10% FBS media was used for ascorbate free transfections. Brefeldin A (Sigma) was used at a concentration of 2.5mg/ml and cells were incubated for 30min. MG132 was used at 20μM for 2h, choloroquine was used at 200μM for 1h. Media was collected the next day and cells on coverslips were fixed with 3% paraformaldehyde for 30min and remaining cells on a plate were used to prepare lysates. Cells on coverslips were permebealized with 0.1% Triton for 15min at room temperature followed by blocking with 1%BSA for 30min. Primary antibodies used were polyclonal anti Procollagen (LF-67,diluted 1:1000) and anti-flag (diluted 1:200). Secondary antibodies were Alexa fluor 546 donkey anti-rabbit IgG and Alexa Fluor 488 goat ant-rabbit IgG (diluted 1:200). After staining cells with appropriate primary and secondary antibodies, we fixed coverslips on slides using mounting reagent containing DAPI. Images were analyzed with a Zeiss LSM710 confocal microscope and captured with Zen10 software. Merges of images were performed with ImageJ and LSM image Browser. Media collected from 6-well plates was normalized with respect to lysate protein concentration estimated using BCA method. Media and lysates of each reaction were checked by immunoblot analysis. Tubulin was used as loading control for lysates. Ascorbate chase experiments were done by adding ascorbate (0.25mM ascorbic acid and 1mM asc-2-phosphate) to Klhl12-transfected cells, followed by incubation for 5, 10, 30 and 60min.
A human fibrosarcoma cell line (HT1080) stably transfected with proalpha1(1) was used for Cul3 knockdowns. Cul3- and Klh12-shRNAs targeting two different regions in both genes were cloned into pSuperGFP and transfected using Lipofectamine 2000. pSuper GFP was used as negative control. Cells were grown on 25 mm coverslips in 6-well plates and fixed 2d post transfection. Collagen staining was done using LF-67 (1:1000) and ER was stained with anti-PDI (1:1000) antibody. Fibronectin and EGFR were stained in parallel experiments. Fibronectin expression was induced in HT1080 using 1uM dexamethasone before Cul3 knockdowns. ER retention or secretion was scored in cells expressing GFP shRNAs. Cells without GFP shRNAs and transfected with pSUPER GFP were quantified as well. Images were taken on a Zeiss LSM 710 confocal microscope and visualized with LSM image browser. Lysates were prepared from remaining cells on 6 well and checked for knockdown efficiency.
siRNA oligos used in this study
| RNAi oligos | Targeted sequence (5’ – 3’) |
| mCul3 #1 | GAAGGAATGTTTAGGGATA |
| mCul3 #2 | GGAAGAAGATGCAGCACAA |
| mCul3 #3 | GGTGATGATTAGAGACATA |
| mCul3 #4 | CAACTTTCTTCAAACACTA |
| mCul3 #5 | CATTATTTATTGATGATAA |
| mUBA3 | CGTTTGAAGCAGAGAGAAA |
| mKlhl12 | CCTTGAGAGTGGAGCAGAA |
| hKlhl12 | CCAAAGACATAATGACAAA |
| mKBTBD8 | GAACATGAGCAGAGTGAAA |
| mOct4 | AGGCAAGGGAGGTAGACAA |
| hSec31 | CCTGAAGTATTCTGATAAA |
| mSec13 (pool of 4 oligos) | CCATGTGTTTAGTAATTTA |
| GGCAATATGTGGTCACCTA | |
| GCTGAAAGTATTCATGTAA | |
| GGAACAAATGACTATTATT | |
| mCdc42 (pool of 4 oligos) | GATCTAATTTGAAATATTA |
| GGATTGAGTTCCTAATTAA | |
| AGAGGATTATGACAGACTA | |
| AAATCAAACTAAAGATTAA | |
| mBcar1/CAS (pool of 4 oligos) | GACTAATAGTCTACATTTA |
| GGAGGTGTCTCGTCCAATA | |
| CTATGACAATGTTGCTGAA | |
| GGGCGTCCATGCTCCGGTA | |
| mSrc (pool of 4 oligos) | CCCTTGTGTCCATATTTAA |
| CCACGAGGGTTGCCATCAA | |
| CAGACTTGTTGTACATATT | |
| GCAACAAGAGCAAGCCCAA | |
| mRhoG (pool of 4 oligos) | GGTTTACCTAAGAGGCCAA |
| GCTGTGCCTTAAGGACTAA | |
| GCACAATGCAGAGCATCAA | |
| GGCGCACCGTGAACCTAAA | |
| mRhoA (pool of 4 oligos) | GGATTTCCTAATACTGATA |
| GAAAGTGTATTTGGAAATA | |
| AGCCCTATATATCATTCTA | |
| CGTCTGCCATGATTGGTTA | |
| mRac1 (pool of 4 oligos) | GGTTAATTTCTGTCAAACA |
| GCGTTGAGTCCATATTTAA | |
| GCTTGATCTTAGGGATGAT | |
| GGAGTAATTCAACTGAATA | |
| mCdh1/E-cadherin (pool of 4 oligos) | GGAGGAGAACGGTGGTCAA |
| CGCGGATAACCAGAACAAA | |
| CCATGTTTGCTGTATTCTA | |
| GGGACAATGTGTATTACTA | |
| mIqgap1 (pool of 4 oligos) | ACATGATGATGATAAACAA |
| GGTTGATTTCACAGAAGAA | |
| GTATAAATTTATTTCTTAA | |
| GGTGGATCAGATTCAAGAA | |
| mCul1 (pool of 2 oligos) | GCATGATCTCCAAGTTAAA |
| CGTGTAATCTGCTATGAAA | |
| mCul2 (pool of 2 oligos) | GCGCTGATTTGAACAATAA |
| CCAGAGTATTTATATCTAA | |
| mCul4a (pool of 2 oligos) | GTGTGATTACCATAATAAA |
| CCAGGAAGCTGGTCATCAA | |
| mCul5 (pool of 2 oligos) | CCCTCATATTTACAGCAAA |
| ACATGAAGTTTATAATGAA | |
| mCul7 (pool of 2 oligos) | GCATCAAGTCCGTTAATAA |
| GGATGTGATTGATATTGAA |
Supplementary Material
Acknowledgments
We thank Brenda Schulman for advice and gifts of cDNAs and proteins. We are grateful to Julia Schaletzky for critically reading the manuscript and many discussions. We thank the members of the Rape and Schekman labs for advice and suggestions, Lillian Lim for providing Cul3-shRNAs, Christina Glazier for contributions on BTB-protein cloning, and Ann Fischer and Michelle Richner for tissue culture support. This work was funded by grants from the Pew Foundation (MR), the NIH (NIGMS-RO1, MR; NIH Director’s New Innovator Award, MR), and the Howard Hughes Medical Institute (RS). LJ was funded by a CIRM predoctoral fellowship; she is Tang fellow. KBP is HFSP long term post-doctoral fellow.
Footnotes
Author Contributions Experiments were designed by LJ, KRB, RS, and MR. LJ performed the mESC screen, identified Klhl12 and Sec31, and analyzed the role of Cul3 in COPII formation in cells and in collagen export in mESCs; KRB analyzed collagen export in fibroblasts; KEW analyzed COPII formation in cells; CB identified inactive Klhl12; AG performed EM; LJ, KRB, and MR prepared the manuscript.
The authors declare no competing financial interests.
References
- 1.Leitinger B. Transmembrane Collagen Receptors. Annu Rev Cell Dev Biol. 2011 doi: 10.1146/annurev-cellbio-092910-154013. [DOI] [PubMed] [Google Scholar]
- 2.Wickstrom SA, Radovanac K, Fassler R. Genetic analyses of integrin signaling. Cold Spring Harb Perspect Biol. 2011;3 doi: 10.1101/cshperspect.a005116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol. 2009;10:843–853. doi: 10.1038/nrm2799. [DOI] [PubMed] [Google Scholar]
- 4.Stephens LE, et al. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995;9:1883–1895. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
- 5.Chen SS, Fitzgerald W, Zimmerberg J, Kleinman HK, Margolis L. Cell-cell and cell-extracellular matrix interactions regulate embryonic stem cell differentiation. Stem Cells. 2007;25:553–561. doi: 10.1634/stemcells.2006-0419. [DOI] [PubMed] [Google Scholar]
- 6.Lang MR, Lapierre LA, Frotscher M, Goldenring JR, Knapik EW. Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation. Nat Genet. 2006;38:1198–1203. doi: 10.1038/ng1880. [DOI] [PubMed] [Google Scholar]
- 7.Townley AK, et al. Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J Cell Sci. 2008;121:3025–3034. doi: 10.1242/jcs.031070. [DOI] [PubMed] [Google Scholar]
- 8.Sarmah S, et al. Sec24D-dependent transport of extracellular matrix proteins is required for zebrafish skeletal morphogenesis. PLoS One. 2010;5:e10367. doi: 10.1371/journal.pone.0010367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohisa S, Inohaya K, Takano Y, Kudo A. sec24d encoding a component of COPII is essential for vertebra formation, revealed by the analysis of the medaka mutant, vbi. Dev Biol. 2010;342:85–95. doi: 10.1016/j.ydbio.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 10.Boyadjiev SA, et al. Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet. 2006;38:1192–1197. doi: 10.1038/ng1876. [DOI] [PubMed] [Google Scholar]
- 11.Fromme JC, et al. The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Dev Cell. 2007;13:623–634. doi: 10.1016/j.devcel.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jensen D, Schekman R. COPII-mediated vesicle formation at a glance. J Cell Sci. 2011;124:1–4. doi: 10.1242/jcs.069773. [DOI] [PubMed] [Google Scholar]
- 13.Stagg SM, et al. Structural basis for cargo regulation of COPII coat assembly. Cell. 2008;134:474–484. doi: 10.1016/j.cell.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fath S, Mancias JD, Bi X, Goldberg J. Structure and organization of coat proteins in the COPII cage. Cell. 2007;129:1325–1336. doi: 10.1016/j.cell.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 15.Fromme JC, Schekman R. COPII-coated vesicles: flexible enough for large cargo? Curr Opin Cell Biol. 2005;17:345–352. doi: 10.1016/j.ceb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 16.Saito K, et al. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell. 2009;136:891–902. doi: 10.1016/j.cell.2008.12.025. [DOI] [PubMed] [Google Scholar]
- 17.Saito K, et al. cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Mol Biol Cell. 2011 doi: 10.1091/mbc.E11-02-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilson DG, et al. Global defects in collagen secretion in a Mia3/TANGO1 knockout mouse. J Cell Biol. 2011;193:935–951. doi: 10.1083/jcb.201007162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kathiresan S, et al. Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet. 2009;41:334–341. doi: 10.1038/ng.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sumara I, et al. A Cul3-based E3 ligase removes Aurora B from mitotic chromosomes, regulating mitotic progression and completion of cytokinesis in human cells. Dev Cell. 2007;12:887–900. doi: 10.1016/j.devcel.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Shaw LM, Rabinovitz I, Wang HH, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the alpha6beta4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, et al. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol Cell. 2009;35:841–855. doi: 10.1016/j.molcel.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 23.Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–171. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geyer R, Wee S, Anderson S, Yates J, Wolf DA. BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol Cell. 2003;12:783–790. doi: 10.1016/s1097-2765(03)00341-1. [DOI] [PubMed] [Google Scholar]
- 25.Pintard L, et al. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003;425:311–316. doi: 10.1038/nature01959. [DOI] [PubMed] [Google Scholar]
- 26.Xu L, et al. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–321. doi: 10.1038/nature01985. [DOI] [PubMed] [Google Scholar]
- 27.Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hughes H, et al. Organisation of human ER-exit sites: requirements for the localisation of Sec16 to transitional ER. J Cell Sci. 2009;122:2924–2934. doi: 10.1242/jcs.044032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim W, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–340. doi: 10.1016/j.molcel.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emanuele MJ, et al. Global Identification of Modular Cullin-RING Ligase Substrates. Cell. 2011;147:459–474. doi: 10.1016/j.cell.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stephens DJ, Pepperkok R. Imaging of procollagen transport reveals COPI-dependent cargo sorting during ER-to-Golgi transport in mammalian cells. J Cell Sci. 2002;115:1149–1160. doi: 10.1242/jcs.115.6.1149. [DOI] [PubMed] [Google Scholar]
- 32.Zhu W, et al. Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J. 1996;15:4130–4141. [PMC free article] [PubMed] [Google Scholar]
- 33.Singer JD, Gurian-West M, Clurman B, Roberts JM. Cullin-3 targets cyclin E for ubiquitination and controls S phase in mammalian cells. Genes Dev. 1999;13:2375–2387. doi: 10.1101/gad.13.18.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uchida K, et al. Identification of specific autoantigens in Sjogren’s syndrome by SEREX. Immunology. 2005;116:53–63. doi: 10.1111/j.1365-2567.2005.02197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Angers S, et al. The KLHL12-Cullin-3 ubiquitin ligase negatively regulates the Wnt-beta-catenin pathway by targeting Dishevelled for degradation. Nat Cell Biol. 2006;8:348–357. doi: 10.1038/ncb1381. [DOI] [PubMed] [Google Scholar]
- 36.Schafer M, Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–638. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 38.Zhang XH, et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell. 2009;16:67–78. doi: 10.1016/j.ccr.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geddis AE, Prockop DJ. Expression of human COL1A1 gene in stably transfected HT1080 cells: the production of a thermostable homotrimer of type I collagen in a recombinant system. Matrix (Stuttgart, Germany) 1993;13:399–405. doi: 10.1016/s0934-8832(11)80045-4. [DOI] [PubMed] [Google Scholar]
- 40.Williamson A, et al. Identification of a physiological E2 module for the human anaphase-promoting complex. Proc Natl Acad Sci U S A. 2009;106:18213–18218. doi: 10.1073/pnas.0907887106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.