

Abstract
DNA methylation dynamics influence brain function and are altered in neurological disorders. 5-hydroxymethylcytosine (5-hmC), a DNA base that is derived from 5-methylcytosine, accounts for ~40% of modified cytosine in the brain and has been implicated in DNA methylation–related plasticity. We mapped 5-hmC genome-wide in mouse hippocampus and cerebellum at three different ages, which allowed us to assess its stability and dynamic regulation during postnatal neurodevelopment through adulthood. We found developmentally programmed acquisition of 5-hmC in neuronal cells. Epigenomic localization of 5-hmC–regulated regions revealed stable and dynamically modified loci during neurodevelopment and aging. By profiling 5-hmC in human cerebellum, we found conserved genomic features of 5-hmC. Finally, we found that 5-hmC levels were inversely correlated with methyl-CpG–binding protein 2 dosage, a protein encoded by a gene in which mutations cause Rett syndrome. These data suggest that 5-hmC–mediated epigenetic modification is critical in neurodevelopment and diseases.
Cytosine methylation serves as a critical epigenetic mark by modifying DNA-protein interactions that influence transcriptional states and cellular identity. Such methylation modulates core epigenomic processes, including gene expression, X-chromosome inactivation, silencing of repetitive elements, imprinting, and regulation of centro-meric and telomeric heterochromatin1. 5-methylcytosine (5-mC) has generally been viewed as a stable covalent modification to DNA; however, the fact that 5-mC can be enzymatically modified to 5-hmC by Tet family proteins through Fe(II) α-KG–dependent hydroxylation yields a new perspective on the previously observed plasticity in 5-mC–dependent regulatory processes2-4. Indeed, Tet1 and Tet2 are critical for embryonic stem cell (ES cell) self-renewal, normal myelopoiesis, myeloid leukemia and gliomas2,5-8. However, despite the emerging importance of 5-hmC in regulating pluripotency and myelopoeisis, very little is known about the function of 5-hmC during neurodevelopmental processes because of a lack of detailed genomic maps of 5-hmC in such contexts. Given the distinct genomic distributions that were recently reported for 5-hmC in ES cells and adult brain9-12, establishment of comprehensive 5-hmC maps in the context of neurodevelopment and aging is critical toward understanding the role of this base modification in these processes.
Epigenetic plasticity in DNA methylation–related regulatory processes influences activity-dependent gene regulation13-15, learning and memory16,17, and repeat-associated transcript expression18,19 in the CNS. Hydroxylation of 5-mC to 5-hmC presents a particularly intriguing epigenetic regulatory mechanism in the mammalian brain, where its dynamic regulation is critical20. In the CNS, 5-hmC is substantially enriched relative to many other tissues and cell types; for instance, 5-hmC levels in the brain are approximately tenfold greater than those seen in ES cells3,10,21. In Purkinje cells of the cerebellum, 5-hmC is approximately 40% as abundant as 5-mC and is enriched relative to granule cells22, further suggesting cell type–specific functions for 5-hmC. In addition, in certain neurodevelopmental contexts, neuronal stimulation dictates activity-dependent region-specific active 5-mC demethylation13-15, highlighting the importance of DNA methylation dynamics in neuronal function. Disruption of DNA methylation–related processes leads to diverse neurological disorders, often characterized by postnatal manifestation23. Mutations in genes encoding proteins that directly bind to methylated DNA (methyl-CpG–binding proteins such as methyl-CpG–binding protein 2, MeCP2) or place 5-mC marks (DNA methyltransferase, DNMT3B) result in distinct neurodevelopmental disorders involving dysregulation of both coding genes and repetitive elements24-27. MeCP2 also modulates activity-dependent gene expression in which neuronal stimulation results in site-specific phosphorylation, release from chromatin and localized loss of DNA methylation13. Despite the known interaction between MeCP2 and dynamic DNA methylation, the mechanisms by which such regulation occurs remain obscure. Insight into 5-hmC states in the presence and absence of MeCP2 therefore provides a new perspective through which to consider epigenomic regulation in the context of neurological disease.
To unravel the biology of 5-hmC, we recently developed a selective chemical-labeling method for 5-hmC that uses T4 bacteriophage β-glucosyltransferase to transfer an engineered glucose moiety containing an azide group onto the hydroxyl group of 5-hmC, which can then chemically incorporate a biotin group for detection, affinity enrichment and sequencing10. Using this technology, we generated, to the best of our knowledge, the first genome-wide maps of 5-hmC in mouse cerebellum and hippocampus during development and aging, which provide a detailed epigenomic view of regulated 5-hmC in CNS. We found that 5-hmC was depleted on the X chromosome in both males and females. Our analyses suggest that there is dynamic regulation of 5-hmC during neurodevelopment and aging. In addition to the association of 5-hmC with the bodies of developmentally activated genes, we also observed developmentally regulated dynamics of 5-hmC at repetitive loci during neurodevelopment. Comprehensive analysis of 5-hmC distributions in human cerebellum further revealed conserved characteristics of 5-hmC in mammals. Finally, we found dosage-dependent genomic 5-hmC content in mouse models of Rett syndrome, a neurodevelopmental disorder that is caused by de novo mutations in the MECP2 gene, which encodes the 5-mC–specific binding protein MECP2. Together, these data suggest that 5-hmC–mediated epigenetic regulation is critical in neurodevelopment, aging and human diseases.
RESULTS
Acquisition of 5-hmC during neurodevelopment and aging
DNA methylation is critical for proper postnatal neurodevelopment and undergoes age-dependent changes in the adult brain28,29. However, the neurodevelopmental and age-dependent dynamics associated with 5-hmC remain poorly understood. To address these issues, we assessed the abundance of 5-hmC in mouse cerebellum and hippocampus during neurodevelopment and aging by immunohistochemistry with a 5-hmC–specific antibody2,10. We examined 5-hmC in mice at postnatal day 7 (P7), 6 weeks and 1 year of age. Immunohistochemistry of 5-hmC in developing (P7) and adult cerebellum (6 weeks) indicated that there was substantial overlap between 5-hmC–positive and NeuN-positive neuronal cells in the granule cell layer (Fig. 1a,b). Compared to P7, the intensity of 5-hmC in granule cells was substantially increased in cerebellum at 6 weeks (Fig. 1a,b). Enrichment of 5-hmC in calbindin-positive Purkinje cells, relative to cells in the granule cell layer, occurred in the cerebellum at both P7 and 6 weeks (Fig. 1c,d); however, highly proliferative progenitor cells in the external granule layer of P7 cerebellum generally lacked 5-hmC (Fig. 1a,c and Supplementary Fig. 1a). In hippocampus, we found enrichment of 5-hmC in NeuN-positive cells in the dentate gyrus granule cell layer of both P7 and adult mice (Fig. 1e,f and Supplementary Fig. 1b). At P7, however, immature neurons (NeuN negative) have very low levels or a complete absence of 5-hmC. Similarly, there was a substantial reduction in 5-hmC staining in subgranular Nestin-positive neural progenitor cells (Supplementary Fig. 1c). We also observed enrichment of 5-hmC in NeuN-positive mature neurons in the granular cell layer of the olfactory bulb relative to cells in the anterior of the subventricular zone, a region of high neurogenic capacity that is driven by neural stem cells (Supplementary Fig. 2). Colocalization of 5-hmC with mature neuronal cells (calbindin and NeuN positive), but not immature neurons, suggests that 5-hmC is involved with neuronal development.
Figure 1. Immunostaining, quantification, and genomic mapping of 5-hmC across three ages and two brain regions in mouse.
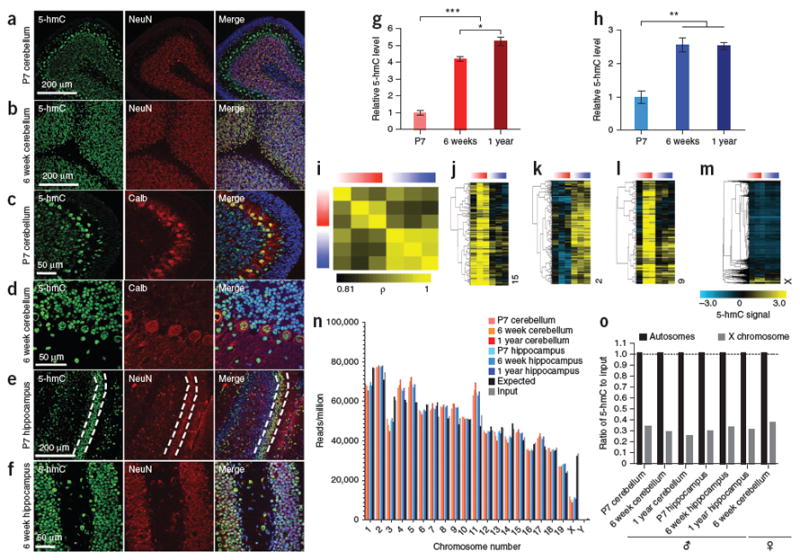
(a–f) 5-hmC–specific immunostaining in P7 (a,c) and 6 week (b,d) cerebellum, P7 (e) and 6 week (f) hippocampus. Dentate gyrus granular neurons in P7 are indicated by the dashed lines. (g,h) Quantification of 5-hmC–specific immunoblot in cerebellum and hippocampus at P7, 6 weeks and 1 year (n = 3, mean ± s.e.m.; *P < 0.05, **P < 0.01, ***P < 0.001, ANOVA). (i) Heat map of genome-wide Pearson correlations between input normalized 5-hmC binned data (10-kb bins) in hippocampus and cerebellum. In all heat maps, red indicates cerebellum and blue indicates hippocampus. (j,k) Example of tissue-specific 5-hmC loci in cerebellum and hippocampus based on genome-wide binned data (10-kb bins) identified by hierarchical clustering (uncentered complete linkage). (l) Example of developmentally programmed 5-hmC loci in cerebellum based on genome-wide binned data (10-kb bins) identified by hierarchical clustering (uncentered complete linkage). (m) Heat map of normalized 5-hmC binned data (10-kb bins) on the X chromosome (hierarchical clustering, uncentered complete linkage). For all heat maps, increasingly darker color represents increased age. In j–m, the chromosome number is indicated on the right. (n) Chromosomal read densities in hippocampus and cerebellum at each age for 5-hmC–enriched DNA, input DNA and values expected by chance. (o) Depletion of 5-hmC on the X chromosome relative to autosomes in male and female mouse brain. For the female sample, normalized 5-hmC densities were divided by input densities from the same female (GSE25398). Dashed line corresponds to 1, or no enrichment.
Furthermore, 5-hmC–specific immunoblot analyses revealed a significant increase from P7 to adult in both cerebellum and hippocampus, with a 4.22 ± 0.14–fold increase in cerebellum (P < 0.001) and a 2.57 ± 0.2–fold increase in hippocampus (P < 0.01) (Fig. 1g,h and Supplementary Fig. 1c,d), consistent with the developmental enrichment of 5-hmC observed by immunocytochemistry. We also noted a significant increase in 5-hmC during aging from 6 weeks to 1 year (21.1% increase, P < 0.05; Fig. 1g) in cerebellum, but not in hippocampus. Together, these data suggest that both developmental programming and age-dependent alterations of 5-hmC occur in the mammalian brain.
Features of neurodevelopmentally regulated 5-hmC
To assess genome-wide 5-hmC distributions during neurodevelopment and aging, we employed a previously established method for specific chemical labeling, affinity enrichment and deep sequencing of 5-hmC–containing DNA10. We generated 5-hmC–enriched sequences, as well as sequences from unenriched input genomic DNA, from multiple biological replicates per age and brain region, resulting in ~25 million nonduplicate reads per biological condition (Supplementary Table 1).
Genome-scale patterns of 5-hmC were first assessed using binned data. Overall, input-normalized binned data were highly correlated among all samples, with Pearson coefficients ranging from 0.81 to 0.96 (10-kb bins), indicating that the genomic distributions of 5-hmC were both reproducible and highly similar in mouse cerebellum and hippocampus (Fig. 1i and Supplementary Fig. 3). However, hierarchical clustering clearly distinguished patterns of cerebellum- and hippocampus-specific 5-hmC enrichment (Fig. 1j,k and Supplementary Fig. 3). Similarly, developmentally dependent loci, or those differentially marked in P7 and adult (both 6 weeks and 1 year) mice, were clear (Fig. 1l). Although 5-hmC signals were highly correlated genome wide, specific brain regions tended to be more similar to one another, with the largest same-tissue differences being observed between P7 and adult (6 weeks and 1 year) cerebellum. This difference is likely a result of the marked developmental changes between these tissues, as compared with those found in hippocampus (Fig. 1i and Supplementary Fig. 1). Together, these data strongly indicate that tissue-specific and developmentally regulated dynamics associated with 5-hmC occur in the brain.
Previously, we found that 5-hmC associates with the bodies of more highly expressed genes in adult cerebellum. We further assessed 5-hmC read density in and around genes for all of the ages and regions that we tested. We found that 5-hmC associated with gene bodies and defined gene proximal regions (~875 bp upstream of transcription termination site (TSS), ~160–200 bp downstream of TTS) in both hippocampus and cerebellum (Supplementary Fig. 4a,b). In cerebellum, metagene analysis indicated that genic 5-hmC reflected age-dependent changes observed in bulk genomic 5-hmC during development and aging, whereas less pronounced changes are detectable in hippocampus (Supplementary Fig. 4a,b).
To further explore the relationship between 5-hmC and gene expression in brain, we determined 5-hmC read densities in and around genes ranked by the expression level in P7 and adult cerebellum (Supplementary Fig. 5a and Supplementary Data Set 1). Notably, we found that 5-hmC displayed a TSS-associated bias at genes with lower expression only in P7 cerebellum, where a substantial number of progenitor cells remain. Such a distribution is reminiscent of that observed in mouse and human ES cells, where 5-hmC can be enriched at the TSS of repressed, but developmentally poised, genes12,30,31. On the other hand, in adult cerebellum, genes with low expression did not exhibit distinct TSS-associated 5-hmC and intragenic 5-hmC scaled more closely with gene expression levels (Supplementary Fig. 5a). To further assess the gene expression–dependent dynamics of genic 5-hmC, we measured 5-hmC densities at developmentally activated genes and compared them with 5-hmC densities at repressed or unaltered genes. We found an increase in 5-hmC signals at genes exhibiting activation from P7 to adult compared with unaltered genes, whereas genes that were repressed exhibited only a slight decrease in 5-hmC (Supplementary Fig. 5b–d). These results are consistent with the notion that 5-hmC is associated with actively transcribed genes in adult cerebellum and demonstrate that 5-hmC is acquired in developmentally activated genes.
The observation that 5-hmC is acquired in developmentally activated genes, but is not significantly reduced at repressed genes, suggests that a primary function of 5-hmC in brain may be to offset 5-mC–related repressive effects by reducing 5-mC levels. We therefore tested 5-mC levels using 5-mC–specific DNA immunoprecipitation (MeDIP) at ten intragenic loci that displayed increased mRNA and 5-hmC expression in cerebellum from 6-week-old mice relative to P7 mice. On average, the ten loci tested displayed a 32% reduction in 5-mC levels, whereas there was no substantial reduction in 5-mC in multiple control loci (with similar 5-hmC enrichment at P7 and 6 weeks and unaltered mRNA expression or no 5-hmC enrichment at P7 and 6 weeks; Supplementary Fig. 6a,b). Notably, however, a number of developmentally activated genes with increased 5-hmC did not display a concomitant decrease in 5-mC by MeDIP. Given the overall depletion of CpG dinucleotides in the 5-hmC–enriched regions that we tested (ratio of observed to expected CpG dinucleotide frequency = 0.28; Supplementary Fig. 6a), these data suggest the presence of non-CpG 5-hmC in these developmentally activated genes. In fact, non-CpG DNA methylation has previously been detected in 5-hmC–enriched regions in mouse ES cells9, and positively correlates with gene expression in human ES cells32.
The genomic distributions of 5-hmC in both hippocampus and cerebellum are notably distinct from those observed in ES cells, with some overlapping features9-12. Specifically, 5-hmC was enriched throughout gene bodies in the brain, whereas 5-hmC was also present in ES cells in the bodies of active genes, although to a lesser degree than that found in the brain. At the TSSs of repressed genes in ES cells, 5-hmC could co-occur with repressor complexes, whereas 5-hmC appeared to be largely depleted from TSSs in the brain. Notably, even at developmentally repressed genes, we found little to no change in TSS-associated 5-hmC. These data suggest distinct regulation of 5-hmC in brain as compared with ES cells, and may point toward diverse mechanisms regulating 5-hmC at TSSs and gene bodies.
Depletion of 5-hmC on the X chromosome
Chromosomal 5-hmC densities indicated that the distribution of 5-hmC in the genome is nonrandom, whereas non-enriched (input) genomic DNA read densities closely followed the values that would be expected by chance if reads were randomly distributed among chromosomes. We observed substantial depletion of 5-hmC on the X chromosome (Fig. 1m–o). Depletion on the X chromosome occurred in hippocampus and cerebellum at all ages, as well as in both males and females (Fig. 1o). Independent sequencing of 5-hmC in the rat brain also revealed a depletion of 5-hmC on the X chromosome relative to the autosomes (Supplementary Fig. 7a). The depletion of 5-hmC on the X chromosome may reflect the unique dynamics of DNA methylation on this particular chromosome required for dosage compensation. In fact, X inactivation in human ES cells has been reported to be uniquely sensitive to oxygen levels, which may influence Tet-mediated hydroxylation of 5-mC33.
Although 5-hmC was depleted at the chromosomal level, we found localized enrichment of 5-hmC at genes escaping X inactivation, including Xist and Utx (Supplementary Fig. 7c), specifically in females. Such enrichment was not a result of an increased number of the X chromosome reads in females, as more distal regions exhibited similar depletion in males and females (Supplementary Fig. 7c). These results may implicate 5-hmC–associated regulatory pathways in escape from X inactivation and may implicate 5-hmC in regulating dosage compensation.
Tissue-specific differentially hydroxymethylated regions
Immunostaining and bulk quantification of 5-hmC suggested that developmental programming of 5-hmC occurred in the brain. To comprehensively localize brain region–specific and neurodevelopmentally regulated 5-hmC–enriched regions (differential 5-hydroxymethylated regions, DhMRs) at the genome-wide level, we tested each of the 36 possible pairwise comparisons between different ages and brain regions using a Poisson-based method34. We identified a total of 555,195 DhMRs among all samples (P ≤ 10−5; Table 1 and Supplementary Data Sets 2–7). Overall, 60% of DhMRs were intragenic (1.79-fold > expected) and were most enriched in exons (5.7-fold greater than expected; Fig. 2a). Notably, we also observed a 2.0–2.7-fold enrichment of DhMRs at non–TSS-associated CpG islands (CGIs) and within ±2 kb of CGIs (Fig. 2a). Although we found a slight enrichment of DhMRs within ±500 bp of TSSs (1.87-fold greater than expected), DhMRs were depleted at CGI TSSs (1.53-fold less than expected) (Fig. 2a). Notably, DhMRs were also less frequent than would be expected intergenically (1.64-fold less than expected; Fig. 2a). Consistent with this, we found that, as a group, DhMRs tended to be slightly enriched for both GC content and CpG dinucleotide frequency, but differed substantially in each compared with CGIs (Supplementary Fig. 8a). These data demonstrate that, in mouse hippocampus and cerebellum, 5-hmC–regulated regions are most enriched in intragenic regions and intragenic CGIs of intermediate CpG and GC content.
Table 1.
DhMRs among each of 36 pairwise comparisons between each age and tissue
| Control | Treatment
|
|||||
|---|---|---|---|---|---|---|
| P7 cerebellum | 6 week cerebellum | 1 year cerebellum | P7 hippocampus | 6 week hippocampus | 1 year hippocampus | |
| P7 cerebellum | 0 | 25,224 | 24,073 | 13,339 | 19,559 | 12,386 |
| 6 week cerebellum | 17,058 | 0 | 4,411 | 22,282 | 23,952 | 13,363 |
| 1 year cerebellum | 30,010 | 7,539 | 0 | 19,892 | 25,269 | 8,549 |
| P7 hippocampus | 26,250 | 32,148 | 14,433 | 0 | 2,641 | 1,220 |
| 6 week hippocampus | 37,654 | 34,328 | 20,484 | 2,829 | 0 | 915 |
| 1 year hippocampus | 47,010 | 44,819 | 14,870 | 4,460 | 4,228 | 0 |
Figure 2. Identification and characterization of DhMRs.
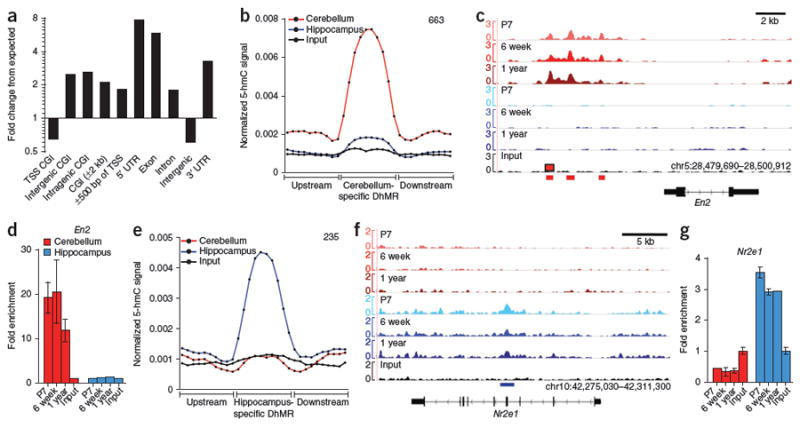
(a) Association of all identified DhMRs with various genomic features. (b) Cerebellum-specific DhMRs. DhMRs identified in each age-matched comparison between cerebellum (treatment) and hippocampus (control) were overlapped to identify the set of DhMRs specific to cerebellum at all ages. Total number of cerebellum-specific DhMRs (663) is indicated in the upper right-hand corner. (c) Genomic view of cerebellum-specific DhMRs upstream of the cerebellum-specific gene En2. Note that the cerebellum-specific peak most upstream of En2 exhibited developmentally regulated acquisition of 5-hmC from P7 to 6 week, remaining high at 1 year. (d) qPCR verification of 5-hmC enrichment in a cerebellum-specific DhMR upstream of En2 (n = 2 per age, mean ± s.e.m.). (e) Hippocampus-specific DhMRs. DhMRs identified in each age-matched comparison between hippocampus (treatment) and cerebellum (control) were overlapped to identify the set of DhMRs specific to hippocampus at all ages. Total number of hippocampus-specific DhMRs (235) is indicated in the upper right-hand corner. (f) Genomic view of a hippocampus-specific DhMR in Nr2e1. (g) qPCR verification of 5-hmC enrichment in the Nr2e1 hippocampus-specific DhMR (n = 2 per age, mean ± s.e.m.). For b and e, normalized 5-hmC signals (reads per million divided by bin size) are expressed in ten equally sized DhMR portions upstream, within and downstream of each tissue-specific DhMR. Normalized 5-hmC signals were determined at each age and then averaged across each specific brain region.
DNA methylation can direct cell- and tissue-specific transcriptional programs in brain. Whether or not 5-hmC is similarly involved in these processes is unclear. To examine the tissue specificity exhibited by 5-hmC, we identified hippocampus- and cerebellum-specific DhMRs unique to each tissue at all of the ages that we tested (Fig. 2). We identified 633 cerebellum-specific DhMRs (P < 10−5, 3.75-fold average increase in 5-hmC signal; Fig. 2b and Supplementary Data Set 8) and 235 hippocampus-specific DhMRs (P < 10−5, 4.5-fold average increase in 5-hmC signal; Fig. 2e and Supplementary Data Set 8). Tissue-specific DhMRs marked tissue-specific genes. For instance, among 633 cerebellum-specific DhMRs, we identified three regions (P = 1.774 × 10−44, 2.72 × 10−23 and 1.10 × 10−17; Fig. 2c) directly upstream of the Engrailed homeobox gene En2. En2 is a crucial regulator of cerebellum pattern formation and is highly enriched in cerebellum35. Notably, these peaks directly overlapped a previously identified paired-box group 2 (Pax2/5/8)-regulated enhancer element36, consistent with recent observations in mouse and human ES cells, suggesting that 5-hmC is involved in distinct gene regulatory elements including enhancers9,12,31. In hippocampus, a large intragenic peak (P = 9.79 × 10−44; Fig. 2f) in Nr2e1 (nuclear receptor subfamily 2, group E, member 1, also known as TLX) was also identified as being highly specific. Enrichment of 5-hmC specifically in cerebellum or hippocampus at En2 and Nr2e1, respectively, at all of the ages tested was verified in independent samples by quantitative PCR (qPCR; Fig. 2d,g), demonstrating the reproducibility of DhMR identification through direct comparison of 5-hmC–enriched sequence sets. Thus, 5-hmC likely influences brain region–specific transcriptional programs during early brain development (before P7), facilitating tissue-specific gene expression.
DNA sequences in tissue-specific DhMRs were analyzed for motifs overrepresented at 5-hmC–regulated regions. In each tissue, a highly similar 21-nucleotide motif was identified as being significantly overrepresented (E = 1.7 × 10−9 in cerebellum, E = 1.7 × 10−18 in hippocampus; Supplementary Fig. 8c,d). Notably, a similar motif was independently reported at differentially methylated regions in induced pluripotent stem cells that was detected using bisulfite sequencing, which does not distinguish 5-mC from 5-hmC37. The similarity among these three independent sets of differentially methylated regions might represent a common sequence motif associated with dynamically methylated regions in mammalian genomes.
Identification of stable and dynamic DhMRs
5-hmC has been proposed as an intermediate to 5-mC demethylation, a process that would suggest 5-hmC might be relatively transient. Alternatively, given that oxidation of 5-mC reduces the affinity of 5-mC–specific binding proteins (and thus histone deacetylase–associated repressive complexes), 5-hmC may thereby maintain chromatin states that are more permissive toward transcription. The latter possibility suggests that 5-hmC would be stable at specified loci, counteracting 5-mC–mediated chromatin repression. Direct comparison of 5-hydroxymethylation states across three distinct ages allowed us to address whether or not 5-hmC may be maintained or lost at specific genomic locations in brain, distinguishing stable from dynamically regulated 5-hmC loci.
The strong genome-wide correlations detected between different ages and brain regions are consistent with 5-hmC being relatively stable, but still exhibiting localized changes. We further examined developmentally regulated and age-dependent sets of DhMRs in detail to determine the relative stability of 5-hmC (Fig. 3). In adult mice, we identified 6,782 regions in cerebellum (P < 10−5, sixfold average increase in 5-hmC signal; Fig. 3c, Supplementary Data Set 8 and Supplementary Fig. 9c) and 90 regions in hippocampus (P < 10−5, 4.5-fold average increase in 5-hmC signal; Fig. 3g, Supplementary Data Set 8 and Supplementary Fig. 9d) that acquired 5-hmC between P7 and 6 weeks of age and then maintained the mark through 1 year. We classified such loci as stable in adult, as each subsequently maintained the mark through 1 year of age, demonstrating that 5-hmC can in fact be maintained in both cerebellum and hippocampus. Notably, we detect a slight increase in 5-hmC at 1 year relative to 6 weeks at such loci in both cerebellum and hippocampus (Fig. 3c,g). This could indicate that, at specified loci, 5-hmC may not only be maintained, but may be further acquired with age.
Figure 3. Dynamic and stable 5-hmC DhMRs in cerebellum and hippocampus.
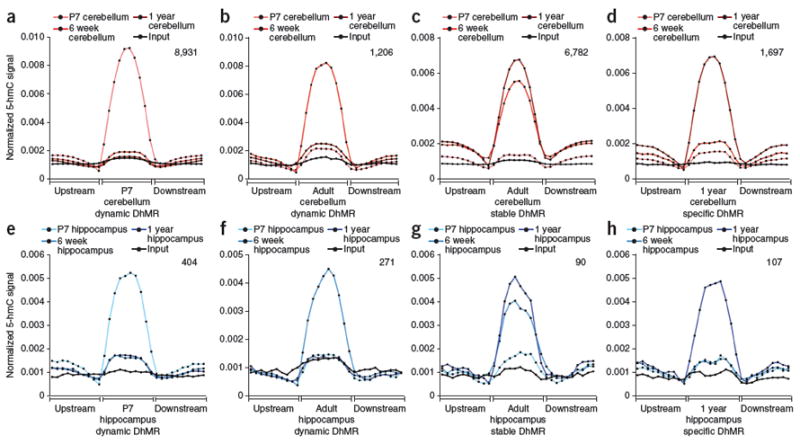
(a,e) P7 dynamic DhMRs, defined as DhMRs present at P7 relative to both 6 weeks and 1 year. Average normalized 5-hmC signals (reads per million divided by portion size) in ten equally sized DhMR portions upstream, in and downstream of DhMRs are plotted for each age. The total number of P7 cerebellum dynamic DhMRs (8,931) is indicated in the upper right-hand corner. Below is the identical analysis in hippocampus (404 total DhMRs). (b,f) 6 week dynamic DhMRs in cerebellum (1,206) and hippocampus (271) defined as DhMRs identified at 6 weeks relative to both P7 and 1 year. (c,g) Adult stable DhMRs in cerebellum (6,782) and hippocampus (90), defined as DhMRs identified at both 6 weeks and 1 year relative to P7. (d,h) 1 year–specific DhMRs in cerebellum (1,697) and hippocampus (107) defined as DhMRs identified at 1 year relative to both P7 and 6 weeks.
Other loci exhibited more dynamic regulation during neurodevelopment and/or aging; 8,931 and 404 regions in cerebellum and hippocampus, respectively, were marked in P7 samples, but lost 5-hmC at 6 weeks of age, without reacquisition at 1 year (Fig. 3a,e, Supplementary Data Set 8 and Supplementary Fig. 9a,b). Conversely, 1,967 regions in cerebellum and 107 regions in hippocampus were specific to samples taken at 1 year of age (Fig. 3d,h, Supplementary Data Set 8 and Supplementary Fig. 9f,g), indicating that acquisition of 5-hmC at specified loci was age dependent. Unexpectedly, we found 1,206 cerebellum loci and 271 hippocampal loci that exhibit acquisition of 5-hmC between P7 and 6 weeks of age, but loss of the mark at 1 year of age (Fig. 3b,f, Supplementary Data Set 8 and Supplementary Fig. 9g,h). Ontology analyses of genes harboring stable and dynamic DhMRs further indicated enrichment of genes involved in neuronal function, including synaptic transmission and neurodevelopment/neuronal differentiation, whereas genes associated with DhMRs expressed specifically at 1 year of age tended to be enriched for more general cellular processes (Supplementary Data Set 9). Together, these analyses suggest that, although some DhMRs are maintained in cerebellum and hippocampus, a substantial number of others display dynamic regulation at specified loci during both neurodevelopment and aging.
5-hmC at repeat elements during neurodevelopment and aging
DNA methylation is critical for regulating the ~45% of the mammalian genome that is classified as repetitive and/or derived from transposon sequences, thereby serving a defensive role against repeat-mediated genomic instability. Retrotransposition can occur during neuronal differentiation; however, the mechanisms by which retrotransposition may occur under normal conditions remain unclear38,39. Given that we found 5-hmC associated with the gene bodies of more highly expressed and developmentally activated genes, we sought to assess whether 5-hmC associates with repetitive and/or transposon-derived loci in mammalian brain, and whether distinct brain regions and ages exhibited changes in repeat-associated 5-hmC. We aligned sequences to the RepeatMasker annotated portion of the genome directly. Relative 5-hmC content was measured as the proportion of reads corresponding to each repeat class in 5-hmC–enriched DNA relative to unenriched input DNA. In adult cerebellum, we found enrichment of 5-hmC on short interspersed nuclear element (SINE) and long tandem repeat (LTR) class repeats relative to unenriched genomic DNA (Fig. 4a). Notably, 5-hmC marking of SINE and LTR class repeats increased both developmentally (from P7 to 6 weeks, 8.9% increase for SINE, 6.0% increase for LTR) and with age (from 6 weeks to 1 year, 3.1% for SINE, 1% at LTR). Such dynamic changes were further confirmed by qPCR at multiple genomic loci of each repeat subtype (Supplementary Fig. 10a). In hippocampus, marking of repeats was more stable across ages, but also exhibited enrichment in SINE and LTR class repeats, and depletion in long interspersed nuclear element (LINE) and satellite class repeats, similar to cerebellum (Fig. 4b and Supplementary Fig. 10b).
Figure 4. Analysis of repeat-associated 5-hmC.
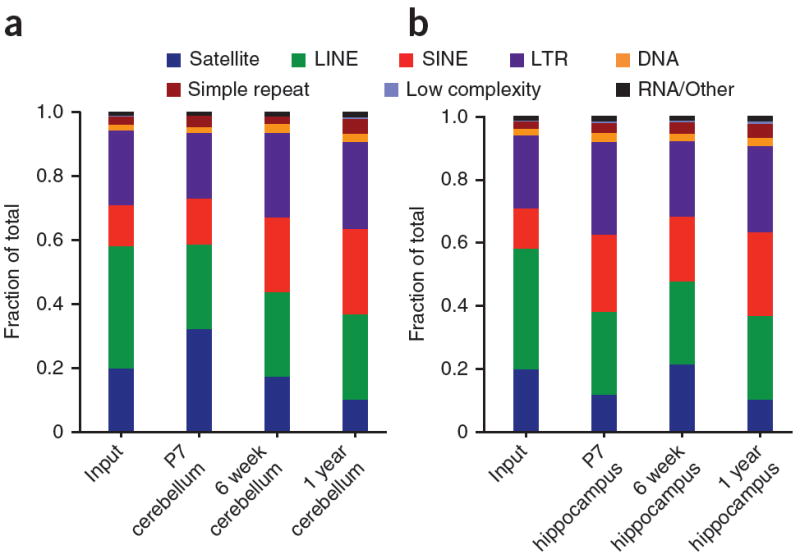
(a,b) Displayed are the fractions of total reads aligned to each class of repetitive elements annotated by RepeatMasker in P7, 6 week and 1 year cerebellum (a), and P7, 6 week and 1 year hippocampus (b). Other/RNA consists of all classes listed in RepeatMasker not otherwise specified in the legend.
Conserved characteristics of 5-hmC in human cerebellum
To address 5-hmC states in human cerebellum, we sequenced 5-hmC–enriched and input DNA derived from the cerebella of two normal individuals (34-year-old male and 40-year-old female, Fig. 5). Genomic distributions of 5-hmC in human samples indicated conserved characteristics when compared to mouse. In total, we identified 43,276 regions consistently enriched for 5-hmC in human cerebellum (P < 10−5; Supplementary Data Set 10). Interestingly, we find more dramatic alterations in chromosome-wide 5-hmC densities relative to both input DNA and values expected by chance, compared to what was observed in mouse (Fig. 5b,c). Consistent with mouse cerebellum, we found that 5-hmC was substantially depleted from the X chromosome in both male and female human samples (Supplementary Fig. 7b). As in mouse, most (71%) 5-hmC–enriched loci are intragenic and highly enriched within exons (11.98-fold > expected) (Fig. 5d). However, 5-hmC–enriched loci do tend to be more common amongst CGIs and TSSs (Fig. 5d). Similar to mouse, 5-hmC-enriched regions are depleted in intergenic regions and less frequent in introns (Fig. 5d). GC content and CpG frequencies are higher than randomly chosen genomic locations, but substantially lower than that of CGIs, as in mouse (Supplementary Fig. 8b). At repetitive loci, 5-hmC was highly enriched at SINE class elements relative to input DNA in both samples tested (Fig. 5e). There was also strong depletion in satellite repeats and substantial, albeit less, depletion in LINEs. In contrast to mouse cerebellum, we saw no substantial enrichment at LTR class repeats.
Figure 5. Conserved features of 5-hmC in human cerebellum.
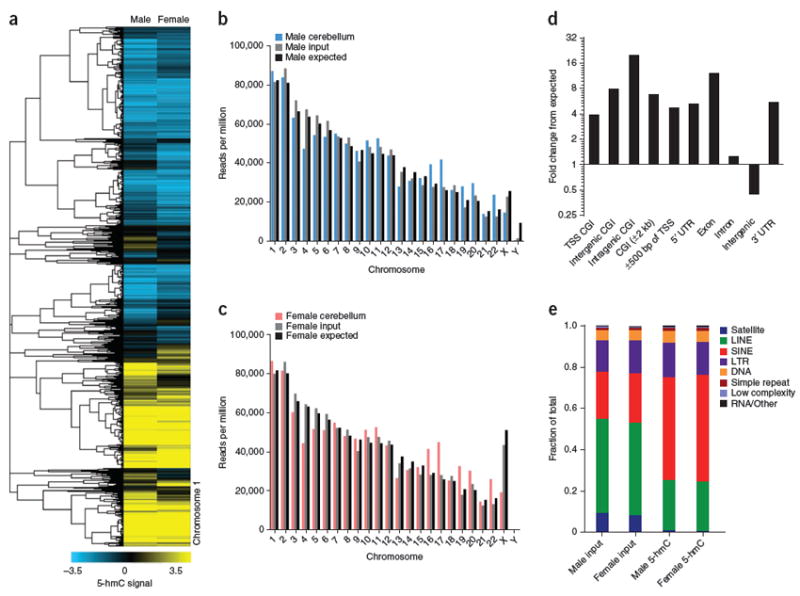
(a) Genome-wide input-normalized 5-hmC signals (10-kb bins) in two adult human cerebella (34-year-old male and 40-year-old female) at chromosome 1. (b,c) Chromosome-wide densities of 5-hmC in male and female cerebellum. Values are normalized to the total number of unique nonduplicate reads, in millions, for 5-hmC and input. Expected values are determined as 106 reads per hg18 length multiplied by chromosome length. In males, expected values for the X and Y chromosomes are divided by 2. (d) Association of 5-hmC–enriched regions with genomic features. The percentage of 5-hmC–enriched regions overlapping a defined genomic feature was compared with the percentage expected to fall in that particular region by chance based on the genomic coverage of that region. Values are expressed as fold change from expected. (e) Analysis of repeat-associated 5-hmC. Displayed are the fraction of total reads aligned to each class of repetitive elements annotated by RepeatMasker in unenriched input genomic DNA and 5-hmC–enriched DNA from each sample.
Altered 5-hmC in mouse models of Rett syndrome
DNA methylation–related processes are critical for a number of neurological disorders. Rett syndrome is one such disorder that is caused by de novo mutations in the MECP2 gene. Both loss-of-function and increased MeCP2 dosage result in overlapping, but distinct, neuropsychiatric disorders25. These observations demonstrate the importance of MeCP2 dosage in proper neuronal function. However, the state of DNA methylation–related dynamics in the presence or absence of MeCP2 on a genomic scale has gone unexplored. Given that 5-hmC is derived from 5-mC, a simple model predicts that, through its affinity for methyl-CpG, MeCP2 may protect conversion of 5-mC to 5-hmC. As such, we quantified genomic 5-hmC levels in mouse models of Rett syndrome, including multiple independent knockout strains (Mecp2–/ytm1.1Jae and Mecp2–/ytm1.1Bird) and a transgenic strain overexpressing MeCP2 at twice the levels seen in wild type (FVB-Tg(MECP2)1Hzo/J). Genomic 5-hmC was measured at 6–8 weeks of age, a time period relevant to the phenotypes arising in Mecp2 mutants26,27. We found that the overall abundance of 5-hmC in cerebellum was negatively correlated with Mecp2 dosage, exhibiting a ~20% increase in the absence of Mecp2 and a ~25% decrease in MeCP2-overexpressing animals (Fig. 6a). These data indicate that binding of Mecp2 to 5-mC may indeed serve to protect the conversion of 5-mC to 5-hmC. To further test this, we asked whether the methyl-CpG–binding domain (MBD) of Mecp2 could block hydroxylation of 5-mC DNA by the catalytic domain of Tet1 (Tet1-CD) in vitro. Consistent with bulk quantification of 5-hmC in mouse cerebellum, we found that Mecp2-MBD was capable of preventing Tet1-CD–mediated hydroxylation of ~90% of 5-mCs at a molecular ratio of 1:1 (Mecp2-MBD to 5-mC; Supplementary Fig. 11).
Figure 6. Altered 5-hmC states in mouse models of Rett syndrome.
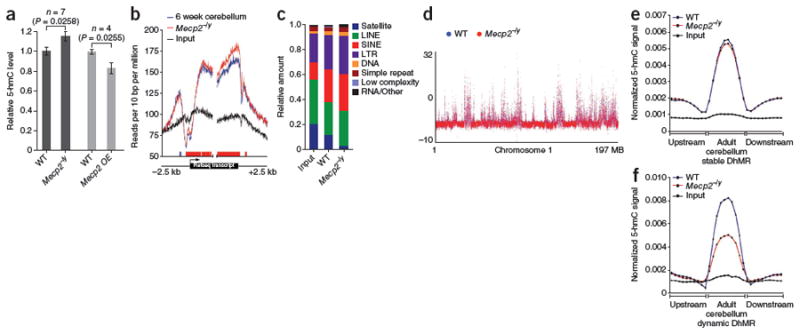
(a) Quantification of 5-hmC–specific immunoblot from 6–8-week-old wild-type (WT), Mecp2–/y cerebellum (n = 7, 5 Mecp2–/ytm1.1Jae and 2 Mecp2–/ytm1.1Bird; P = 0.0258, unpaired t test), and MECP2 transgenic (OE; FVB-Tg(MECP2)1Hzo/J) mice (n = 4, P = 0.0228, unpaired t test). (b) Metagene analysis read densities around all RefSeq transcripts centered on either TSS or TTS in wild-type and Mecp2–/ytm1.1Bird cerebellum. Blocks of significantly increased 5-hmC, color-coded based on the genotype in which the increase was observed. Red indicates increased 5-hmC in Mecp2–/y and blue indicates decreased 5-hmC in wild type (P < 0.05, unpaired t test, 50-bp windows, 10-bp increments). (c) Repeat-associated 5-hmC in wild-type and Mecp2–/ytm1.1Bird cerebellum. Shown are the fractions of total reads aligned to each class of repetitive elements annotated by RepeatMasker. (d) Genomic view of input normalized binned (10 kb) data across chromosome 1. Data from each genotype are superimposed. (e) Average normalized 5-hmC signals in wild type and Mecp2–/ytm1.1Bird at cerebellum DhMRs exhibiting stability in adult (6 weeks and 1 year) relative to P7. (f) Average normalized 5-hmC signals in wild type and Mecp2–/ytm1.1Bird at cerebellum DhMRs exhibiting dynamic regulation at 6 weeks (enriched in 5-hmC at 6 weeks relative to both P7 and 1 year). Normalized 5-hmC signal is the reads per million divided by DhMR portion size, for ten equally sized DhMR portions upstream, in and downstream of DhMRs, averaged across all DhMRs analyzed.
We subsequently captured and sequenced 5-hmC–containing DNA from multiple Mecp2–/ytm1.1Bird cerebella, such that genomic 5-hmC profiles could be compared with those described above (Fig. 6b–f). Consistent with the overall increase in 5-hmC, we found that intragenic 5-hmC was significantly increased in Mecp2–/y tissue (P < 0.05; Fig. 6c). Notably, the increase was restricted to the gene body, and we observed no increase in 5-hmC at the characteristic peak upstream of TSSs (Fig. 6c). This suggests a potential gene body–specific function of Mecp2 at regions marked with 5-hmC. At repetitive loci, we also found the enrichment of 5-hmC at each major repeat class (LTR, SINE, LINE), with the exception of satellites, relative to input genomic DNA (Fig. 6c). This is consistent with previous reports of Mecp2-mediated regulation of L1 (LINE)- and IAP (LTR)-type repeat expression in the brain18,19. Given the association of 5-hmC with more highly expressed genes, these data hint at a mechanism by which expression of retrotransposon transcripts may be activated in the absence of MeCP2, which has been observed in both mice and humans.
In adult (6 week) cerebellum, we identified DhMRs that obtained 5-hmC from P7 to 6 weeks of age, but then either retained or lost 5-hmC by 1 year of age. As these regions represent stable and dynamically modified 5-hmC loci, we sought to assess the fate of 5-hmC at these regions in the absence of Mecp2, such that any potential role for Mecp2 in regulating DNA methylation dynamics could be deciphered. To do so, we measured 5-hmC signals in each independent set of DhMRs. We found that, at regions stably modified in 6 week and 1 year cerebellum, 5-hmC signals were nearly identical between normal and Mecp2–/y mice (Fig. 6d); however, at dynamically modified loci, we found an average decrease of 39% in 5-hmC signals in the absence of Mecp2 (Fig. 6f). These findings together suggest that dynamic DhMRs could be important during neurodevelopment and that loss of Mecp2 could influence DNA methylation dynamics at DhMRs during adulthood.
DISCUSSION
To elucidate the influence of 5-hmC on epigenomic dynamics in mammalian brain, we generated detailed epigenomic maps of 5-hmC expression in mouse cerebellum and hippocampus during postnatal neurodevelopment and aging. We observed a marked depletion of 5-hmC on the X chromosome and identified both stable and dynamic DhMRs during postnatal neurodevelopment and aging. In addition to the association of 5-hmC with the bodies of developmentally activated genes, we also observed developmentally regulated dynamics of 5-hmC at repetitive loci, including acquisition of 5-hmC in SINEs and LTRs during neurodevelopment. Genome-wide analyses of 5-hmC distributions in human cerebella further revealed conserved characteristics of 5-hmC in mammals. Finally, we found that the overall abundance of 5-hmC was negatively correlated with MeCP2 dosage. Notably, the loss of Mecp2 led to the specific reduction of 5-hmC signals at dynamic DhMRs. Together, these data suggest that 5-hmC is important during postnatal neurodevelopment and aging, as well as in human neurological disorders.
We found that 5-hmC markedly increased from the early postnatal stage to adulthood, suggesting a strong correlation between 5-hmC and neurodevelopment. Tet proteins (Tet1, Tet2 and Tet3) are known to oxidize 5-mC to 5-hmC2,3. Notably, using microarray analyses, we found no significant increase in Tet family gene expression during neurodevelopment (P > 0.05; Supplementary Fig. 12), suggesting that the changes in genomic 5-hmC cannot be explained simply by changes in Tet expression. This indicates that, if Tet family proteins are responsible for 5-mC hydroxylation in brain, Tet activity might be modified post-transcriptionally or through additional co-factors.
Among all of the samples that we tested, analyses of DhMRs indicated that they most frequently associated with genes bodies, becoming enriched in developmentally activated genes during neurodevelopment. Gene ontology analyses on the set genes that both acquired 5-hmC and were activated in cerebellum from P7 to 6 weeks confirmed substantial enrichment of genes involved in cerebellum development and various neuronal functions (Supplementary Data Set 9), consistent with 5-hmC having a role in neurodevelopment. Furthermore, assessment of 5-mC levels at such genes demonstrated that a substantial fraction also displayed reduced 5-mC from P7 to 6 weeks, consistent with the notion that conversion of 5-mC to 5-hmC may serve to offset the repressive effects of 5-mC. However, we also found that some such genes did not show substantial differences in 5-mC levels as measured by MeDIP. Given that dynamically regulated 5-hmC loci tended to be depleted in CpG dinucleotides (Supplementary Figs. 6d and 8b), these data may indicate that non-CpG 5-hmC is involved in gene activation. Notably, a recent report found strand-specific non-CpG DNA methylation at 5-hmC–enriched loci in mouse ES cells, and strand-specific non-CpG methylation also correlates positively with gene expression in human ES cells9. Together, these data could suggest the involvement of intragenic non-CpG 5-hmC in gene activation in the CNS, although further studies resolving 5-mC and 5-hmC at single base resolution will be required to fully resolve the roles of each.
Intragenically, 5-hmC associates with CGIs, but is much less frequent at CGIs associated with TSSs. Previous observations have suggested that TSS-associated 5-mC hydroxylation, via Tet family proteins, is involved in both maintaining gene expression of pluripotent associated genes and poising expression of developmentally regulated genes in ES cells2,5,12,30,40. In ES cells, intragenic 5-hmC tends to be highest in moderately expressed genes11,12,31. Our data suggest a more substantial enrichment of 5-hmC in gene bodies in brain relative to ES cells. In the context of current genome-wide 5-hmC datasets9-12, the comprehensive 5-hmC maps that we generated suggest distinct regulation of 5-hmC in brain as compared with ES cells, and highlight the potentially diverse mechanisms regulating 5-hmC at TSSs and gene bodies.
The depletion of 5-hmC specifically on X chromosome in both males and females was surprising. In females, depletion of 5-hmC on the X chromosome may be expected from previous reports that found less DNA methylation on the inactive X chromosome41. However, depletion in males is rather unexpected, as all cells must harbor an active X chromosome. These results may suggest that the maintenance of an active X chromosome, in both males and females, is generally associated with lower 5-hmC levels. Mechanistically, it is possible that the X chromosome is subjected to unique epigenetic dynamics or signature sequences that would prevent conversion of 5-mC to 5-hmC or, perhaps equally likely, promote the further modification of 5-hmC to unmethylated cytosine. Whether this phenomenon functions in cis or requires in trans factor(s) would need further investigation.
Our data suggest that, although 5-hmC is maintained at many specified loci, it could also be dynamically regulated during neurodevelopment and aging. It has been proposed that hydroxylation of 5-mC serves either as an intermediate to 5-mC demethylation or as a stable epigenetic modification to DNA that is distinct from 5-mC, perhaps depending on its relative stability across the genome. Our data suggest that 5-hmC can be stable across three distinct ages in two mouse brain regions: cerebellum and hippocampus. Consistent with this, we identified sets of 5-hmC–regulated loci that clearly maintained 5-hmC from 6 weeks to 1 year of age in each region tested. However, more dynamically regulated loci were also present in both cerebellum and hippocampus. Clearly, the refinement of genome-scale 5-hmC maps in specific subtypes of neuronal cells will be important in the future to give us a full understanding of the influence of 5-hmC on DNA methylation dynamics in the CNS.
Transposable elements make up a substantial portion of most eukaryotic genomes and could affect genome diversity and human disease either by insertional mutagenesis or by contributing recombination substrates, both during and long after their integration42. Recent work suggests transposable elements may upregulate the expression of host genes and function as part of genome-wide regulatory networks43. Somatic retrotransposition of L1-type LINEs has been seen in the human brain and may influence neurogenesis and/or affect neuronal function39. In addition, epigenetic regulatory mechanisms have been directly linked to the regulation of transposon mobilization. Our analyses revealed that 5-hmC was also enriched in transposable elements, particularly SINE- and LTR-type repeats in mammalian brain. The enrichment of 5-hmC was dynamically regulated during neurodevelopment and aging in mouse cerebellum. At this time, however, whether 5-hmC–mediated epigenetic regulation is involved in transposon mobilization requires further investigation.
We also examined genomic 5-hmC distributions in the context of human neurological disorders. We focused on Rett syndrome, which is caused by de novo mutations in the MECP2 gene24. Mecp2 deficiency results in global changes in neuronal chromatin structure, and Mecp2 can dampen transcriptional noise genome-wide in a DNA methylation–dependent manner19. Mecp2 has also been implicated in neuronal activity–dependent gene regulation44,45. These observations indicate that Mecp2, through its affinity for 5-mC, could serve to temper transcription by modulating the conversion of 5-mC to 5-hmC. Our results indicate that the overall abundance of 5-hmC in cerebellum is negatively correlated with Mecp2 dosage and that the Mecp2-MBD may directly inhibit Tet1-CD–mediated hydroxylation of 5-mC. Consistent with this, our sequencing analyses suggest that, overall, both intragenic and transposable element–associated 5-hmC is increased in the absence of Mecp2. However, the loss of Mecp2 led to a 39% reduction of 5-hmC specifically at the dynamic, but not stable, DhMRs that we identified. This unexpected paradox might be explained by different modes of action of Mecp2 at epigenetically stable and dynamic regions. Globally, Mecp2 could dampen transcriptional noise by maintaining 5-mC and preventing conversion to 5-hmC, explaining the negative correlation between Mecp2 dosage and overall 5-hmC. Indeed, Mecp2 is expressed at near histone octamer levels and is bound genome-wide in cerebellum neurons19. However, redundancy exists in MBDs and may function to impart robustness at a subset of loci. At epigenetically stable regions, such as those maintaining 5-hmC from 6 weeks to 1 year, redundant MBD-associated mechanisms may maintain the equilibrium between 5-mC and 5-hmC in the absence of Mecp2. However, at more dynamically modified loci, such as those that acquire 5-hmC between P7 and 6 weeks, but subsequently lose the mark at 1 year, there may be a less redundant mechanism. At such loci, Mecp2 may be the predominant MBD, and the loss of Mecp2 would lead to a change in the equilibrium between 5-mC and 5-hmC.
An intriguing possibility raised by such a Mecp2-predominant mechanism would be its involvement at developmentally activated or neuronal activity–dependent loci. On stimulation, Mecp2 may disassociate, allowing specific transcription factor(s) to activate gene expression, which could involve recruitment of Tet proteins to hydroxylate 5-mC. In the absence of Mecp2, this dynamic would be compromised, leading to a reduction in 5-hmC. Notably, such a mechanism could explain transcriptional activating affects observed at a subset of genes in MeCP2 transgenic animals. More recently, in addition to its role during neurodevelopment, MeCP2 has been shown to be required for adult neurological function46. It is possible that these dynamic loci could be important in these processes as well; however, it remains to be determined whether the loss of MeCP2 in adulthood could alter 5-hmC modification at these loci.
To summarize, we generated detailed genome-wide maps of 5-hmC in mouse cerebellum and hippocampus during postnatal development and aging and characterized dynamic regulation of 5-hmC during neurodevelopment and aging, pointing to critical role(s) for 5-hmC in neurodevelopment. Our results could lay the groundwork for further exploration into the influence of 5-hmC on epigenomic dynamics in normal brain function and human diseases.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/natureneuroscience/.
ONLINE METHODS
Animal and human tissues
All of the mice that we used were on the C57BL/6 genetic background, except for the MeCP2 overexpression mice. The MeCP2 knockout mice, Mecp2–/ytm1.1Jae and Mecp2–/ytm1.1Bird, were described previously26,27. The MeCP2 overexpression line (FVB-Tg(MECP2)1Hzo/J) was described previously and obtained from Jackson Laboratory (stock number 008679)47. All animal procedures were performed according to protocols approved by Emory University Institutional Animal Care and Use Committee. Frozen human cerebella from both a normal male and female were obtained through the Emory Neuropathology Core Facility.
Genomic DNA preparation and dot blot
Genomic DNA was isolated from brain samples with proteinase K digestion. Tissues were homogenized on ice and then treated with proteinase K (0.667 μg μl−1) in digestion buffer (100 mM Tris-HCl (pH 8.5), 5 mM EDTA, 0.2% SDS (vol/vol), 200 mM NaCl) overnight at 55 °C. The second day, an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1, saturated with 10 mM Tris (pH 8.0) and 1 mM EDTA; P-3803, Sigma) was added, mixed completely, centrifuged for 5 min at 20,817g, and precipitated with an equal volume of isopropanol. Genomic DNA was recovered in 10 mM Tris-HCl (pH 8.0) and sonicated to ~500 bp by Misonix 3000 (microtip, 4 pulses of 27 s each, 1-min rest on ice, output 2).
Dot blot was performed on a Bio-Dot Apparatus (#170-6545, BIO-RAD) as described previously10 using rabbit antibody to 5-hmC (1:10,000, #39769, Active Motif) as the primary antibody, incubated overnight at 4 °C. Horseradish peroxidase–conjugated antibody to rabbit (1:5,000, #A-0545, Sigma) was used as a secondary antibody, and incubated for 30 min at 20–25 °C. Standard DNA templates were loaded (D5405, ZYMO) for the quantification and to verify the specificity of antibodies.
5-hmC–specific enrichment
5-hmC enrichment was as previously described with an improved selective chemical labeling method10. 5-hmC labeling reactions were performed in a 100-μl solution containing 50 mM HEPES buffer (pH 7.9), 25 mM MgCl2, 300 ng μl−1 sonicated genomic DNA (100–500 bp), 250 μM UDP-6-N3-Glu, and 2.25 μM wild-type β-glucosyltransferase. Reactions were incubated for 1 h at 37 °C. DNA substrates were purified via Qiagen DNA purification kit or by phenol-chloroform precipitation and reconstituted in H2O. Click chemistry was performed with the addition of 150 μM dibenzocyclooctyne modified biotin into the DNA solution, and incubated for 2 h at 37 °C. Samples were then purified by Pierce Monomeric Avidin Kit (Thermo) following the manufacturer’s recommendations. After elution, biotin-5-N3-gmC–containing DNA was concentrated by 10 K Amicon Ultra 0.5-ml Centrifugal Filters (Millipore) and purified using a Qiagen DNA purification kit.
Immunohistochemistry
Animals were anesthetized with chloral hydrate (50 mg per kg) and perfused with phosphate-buffered saline, followed by perfusion with 4% paraformaldehyde (vol/vol). Brains were dehydrated in 30% sucrose (vol/vol). We cut 40-μm brain sections with a cryostat (Leica). Immunostaining was performed as described previously48. For 5-hmC, cells and sections were treated with 1 M HCl for 30 min at 37 °C. For primary antibodies, we used rabbit antibody to 5-hmC (1:10,000, #39769, active motif), mouse antibody to neuronal nuclei (1:500, #MAB377, Millipore), mouse antibody to nestin (1:1,000, #556309, BD Bioscience) and mouse antibody to calbindin (1:200, #C9848, Sigma). For secondary antibodies, we used goat antibody to rabbit Alexa488 (1:500, #A11008, Invitrogen) and goat antibody to mouse Alexa568 (1:500, #A11031, Invitrogen). Samples were counter-stained with the fluorescent nuclear dye 4′,6-diamidino-2-phenylindole (#B2261, Sigma) and visualized with a Zeiss confocal microscope and ZEN 2009 software. For photomontages of hippocampus and cerebellum, all images were taken using a Zeiss AX10 microscope at the same exposure time, and images were processed with Photoshop software.
Sequencing of 5-hmC–enriched and input DNA
Libraries were generated following the Illumina protocol for “Preparing Samples for ChIP Sequencing of DNA” (Part# 111257047 Rev. A). We used 25 ng of input genomic DNA or 5-hmC–captured DNA to initiate the protocol. DNA fragments of ~150–300 bp were gel-purified after the adaptor ligation step. PCR-amplified DNA libraries were quantified on an Agilent 2100 Bioanalyzer and diluted to 6–8 pM for cluster generation and sequencing. We performed 38-cycle single-end sequencing using Version 4 Cluster Generation and Sequencing Kits (Part #15002739 and #15005236 respectively) and Version 7.0 recipes. Image processing and sequence extraction were done using the standard Illumina Pipeline.
Sequence alignment, binning and peak identification
FASTQ sequence files from biological replicates were concatenated and aligned to mouse NCBI37v1/mm9, human (NCBI36, hg18) or rat (Baylor 3.4/rn4) references using Bowtie 0.12.6, retaining only unique non-duplicate genomic matches with no more than 2 mismatches in the first 25 bp.
Unique, non-duplicate reads from input genomic DNA and each 5-hmC–enriched sequence set were counted in 100-, 1,000- and 10,000-bp bins genome wide and normalized to the total number of non-duplicate reads in millions. Input normalized values were subtracted from 5-hmC–enriched values per bin to generate normalized 5-hmC signals. Bins from each sample were clustered using uncentered correlation with pairwise complete linkage (Cluster3.0) to broadly asses patterns of 5-hmC–regulated loci. Overall, clustering of different sized bins generated similar clustering profiles. All data presented are those binned at 10,000 bp.
Chromosome-wide densities were determined as reads per chromosome divided by the total number of reads in millions. Expected values were determined by dividing 106 by the total genome length and multiplying by chromosome length. Expected values were divided by 2 for chromosomes X in male samples. For comparison between autosomes and the X chromosome, chromosome-wide 5-hmC read densities were divided by input read densities to assess 5-hmC enrichment. Chromosome densities from rat were compared expected values assuming a random distribution of reads amongst chromosomes. A summary of sequence output is provided in Supplementary Table 1.
DhMR identification and annotation
To identify DhMRs between samples in mouse brain, we employed a Poisson-based peak identification algorithm (MACS) using unique non-duplicate reads. DhMRs were determined among all pairs of 5-hmC–enriched samples by directly comparing all samples to one another in each direction, rather than comparing each to input before pairwise comparisons (effective genome size = 1.87 × 109, tag size = 38, bandwidth = 200, P value cutoff = 1.00 × 10−5; ranges for calculating regional lambda: peak region = 200 and 1,000; mfold values for modeling peaks were adjusted per pairwise comparison, ensuring ≥1,000 peaks to build the model).
In human cerebellum, 5-hmC–enriched regions were those regions consistently detected in both of the individuals that we tested. Peak analysis was performed using MACS with the identical parameters described above, comparing each to unenriched input genomic DNA from the same individual to prevent variation in read mapping that may exist between individuals. Effective genome length was set to 2.7 × 109. Regions overlapping ≥1 bp are defined as enriched for 5-hmC in human cerebellum.
Association of DhMRs with genomic features was performed by overlapping defined sets of DhMRs with known genomic features obtained from UCSC Tables for NCBI37v1/mm9 or NCBI36/hg18: RefSeq Whole Gene, 5′ UTR, Exon, Intron, 3′ UTR, ±500 bp of TSS, RefSeq Intergenic (complement of Whole Gene), CpG Islands (±2 kb of CGI, Intergenic/Intragenic based on RefSeq Whole Gene). DhMRs were assigned to a given genomic feature if overlapping ≥1 bp. The percentage of total DhMRs in a defined set was divided by the percentage expected to overlap each genomic feature by chance, based on the percentage of genomic space occupied by that genomic feature, to determine the fold change from expected values.
DhMR-associated sequence motif identification
The top 50 tissue-specific DhMRs, based on MACS P value, from hippocampus and cerebellum were analyzed independently using MEME v4.6.0 (http://meme.sdsc.edu/meme/intro.html). Motifs with an E value < 10−3 were considered to be significant.
Generation of metagene profiles
Metagene RefSeq transcript profiles were generated by determining the distance between each read and the closest TSS or TTS, summing the number of 5′ ends in 10-bp bins centered on either TSS or TTS. We examined 10-bp bins 2.5 kb upstream and 2.5 kb downstream of TSSs and TTSs (RefSeq reference downloaded from UCSC 05/20/2010). Read densities (reads per 10 bp) were normalized per million reads of unique non-duplicate reads.
Read coverage and visualization
Genomic views of read coverage were generated using Integrated Genomics Viewer tools and browser (IGV 1.4.2, http://www.broadinstitute.org/igv/) with a window size of 25 and a read extend of 200.
Repeat-associated 5-hmC
Reads were aligned to the RepeatMasker track of NCBI37v1/mm9 or NCBI36/hg18 using Bowtie–q –best parameters, allowing no more than two mismatches across the entire 38-bp read. Aligned reads were assigned to repeat class annotations defined by RepeatMasker. Relative enrichment of 5-hmC between different samples at different repeat classes was assessed by determining the fraction of total aligned reads corresponding to each class compared with the fraction of total aligned reads detected for each class in input genomic DNA.
MeDIP
MeDIP was performed as previously described using 4 μg of heat-denatured sonicated DNA and 10 μg of 5-methylcytidine antibody (Eurogentec/Anaspec #BI-MECY-0500)48.
qPCR validation of 5-hmC–enriched regions and MeDIP
For 5-hmC, 250 pg of input or 5-hmC–enriched DNA was used in triplicate 20-μl qPCR reactions, each with 1X PowerSYBR Green PCR Master Mix (ABI), 0.5 mM forward and reverse primers, and water. For MeDIP, reactions were identical except that 1 ng of template DNA was used. Reactions were run on an SDS 7500 Fast Instrument using standard cycling conditions. Fold enrichment was calculated as 2−dCt, where dCt = Ct (5-hmC enriched) − Ct (input). MeDIP data was normalized to one P7 cerebellum sample and fold change in 5-mC level was determined in other samples based on the difference in Ct. Primer sequences are provided in Supplementary Table 2.
Microarray analyses
Affymetrix GeneChip Mouse Exon 1.0 ST arrays were used following the instructions from Affymetrix (P/N 701880 Rev.5). Total RNA from mouse brain tissues was isolated with TRIzol. Quantity and quality were determined by Agilent 2100 bioanalyzer RNA Nano assay. We spiked 1 μg total RNA with Affymetrix Poly-A RNA controls. Prior to cDNA synthesis, ribosomal RNAs were removed using the RiboMinus Transcriptome Isolation Kit (Invitrogen, cat. no. K1550-01) and ribosomal RNA–reduced total RNA was cleaned with the IVT cRNA Clean Up Kit (Affymetrix, P/N900547). Biotinylated sense-strand cDNA was generated with the GeneChip WT Terminal Labeling Kit and subsequently hybridized to the arrays. CEL intensity files were processed by robust multichip averaging using Affymetrix Power Tools, distributed through AltAnalyze49, using core probes with DABG P values < 0.05, and a raw expression threshold of 70. Differentially expressed genes in mouse brain tissues were established by ANOVA, Benjamini-Hochberg–adjusted P value threshold of ≤ 0.05. Microarray data are available through NCBI GEO (accession number GSE32187).
Gene ontology analyses
Gene ontology analyses were performed on sets of unique RefSeq identifiers using DAVID Bioinformatics Resources 6.7 Functional Annotation Tool50. Gene sets were identified by joining subsets of 5-hmC–enriched regions with RefSeq Tables obtained from the UCSC Tables. The ‘Adult_activated_5hmC_GO’ table includes genes that exhibited activated gene expression in 6 week cerebellum relative to P7 cerebellum with ≥1 region in which 5-hmC was enriched in 6 week cerebellum relative to P7 cerebellum (Supplementary Data Set 2). The ‘Adult_dynamic_DhMR_GO’ table includes genes with ≥1 region overlapping adult dynamic DhMRs from both cerebellum and hippocampus (Supplementary Data Sets 2, 3, 5 and 6). The ‘Adult_stable_DhMR_GO’ table includes genes with ≥1 region overlapping adult stable DhMRs from both cerebellum and hippocampus (Supplementary Table 2). The ‘1yr_specific_DhMR_GO’ table includes genes with ≥1 region overlapping 1 year–specific DhMRs from both cerebellum and hippocampus (Supplementary Table 2).
In vitro 5-mC hydroxylation assay
Mouse Tet1-CD (residues 1,367–2,039 of GenBank GU079948; pXC823) and human MeCP2 MBD domain (residues 77–167; pXC732) were expressed in BL21-Gold (DE3) E. coli cells (Stratagene) as a 6xHis-SUMO fusion construct harbored in a modified pET28b vector (Novagen). Protein expression was induced with 0.4mM isopropyl-β-d-thiogalactopyrano-side in Luria-Bertan broth. Fusion proteins were purified via three-column chromatography (Ni affinity, ion exchange and size exclusion). 6xHis-SUMO tag was cleaved off by Ulp1 protease, leaving two extraneous N-terminal amino acids (His-Met) fused to the target protein. The 6xHis-SUMO tag and the Ulp1 protease were removed by passing the mixture through the nickel column again Purified proteins were stored in 20 mM Tris (pH 8.0), 300 mM NaCl, 1 mM DTT and 5% glycerol (vol/vol).
For in vitro 5-mC hydroxylation assay, MeCP2-MBD was pre-incubated with full 5-mC DNA (Zymo Research) for 30 min at 37 °C for 30 min in reaction buffer (75 μM of Fe (II), 50 mM HEPES (pH 8.0), 2 mM of ascobate, 1 mM α-KG). Full 5-mC DNA (corresponding to 68 pmol 5-mC) was held constant and MeCP2-MBD was varied to produce molecular ratios (MeCP2-MBD to 5mC) of 1:4, 1:2, 1:1, 2.5:1, 5:1 and no MeCP2-MBD) while keeping total protein constant by supplementing decreasing amounts of MeCP2-MBD with bovine serum albumin TET1-CD was then added at 0.78 μM and the reaction was allowed to proceed for 3 h at 37 °C. We then added 20 μg proteinase K to digest protein at 50 °C for 1 h DNA was purified with a Qiagen PCR purification Kit and 5-hmC was measured by dot blot as described above.
Supplementary Material
Acknowledgments
We would like to thank the members of the Jin laboratory for their assistance, and S.T. Warren and C. Strauss for critical reading of the manuscript. H.I. is supported by the Training Program in Human Disease Genetics funded by the US National Institutes of Health (NIH, T32MH087977). L.A.G. is supported by NIH grants CA129831 and CA129831-03S1. C.H. was partially supported by NIH grant GM071440. X.C. was supported by NIH grant GM049245 and is a Georgia Research Alliance Eminent Scholar. M.G. and A.I.L. are partially supported by Emory Alzheimer’s Disease Center (P50AG025688). P.J. is supported by NIH grants (NS051630, MH076090 and P50AG025688) and Simons Foundation Autism Research Initiative. P.J. is also the recipient of a Beckman Young Investigator Award, Basil O’Connor Scholar Research Award and Alfred P. Sloan Research Fellow in Neuroscience. This work was supported, in part, by the Emory Genetics Discovery Fund.
Footnotes
AUTHOR CONTRIBUTIONS K.E.S., X.L. and P.J. designed the experiments. The experiments were performed by K.E.S., X.L., Y.L., C.-X.S., Q.D., H.I., A.V., L.A.G. and C.H. Data analyses were performed by K.E.S., X.L., H.W., H.I., and P.J. M.G., A.I.L. and Q.C. contributed reagents and tissues. A.K.U. and X.C. were responsible for providing the bacterially purified fragments of Tet1 and Mecp2. K.E.S., X.L. and P.J. wrote the manuscript. All of the authors discussed the results and commented on the manuscript.
Accession codes. NCBI GEO: GSE32188.
Note: Supplementary information is available on the Nature Neuroscience website.
COMPETING FINANCIAL INTERESTS The authors declare no competing financial interests.
References
- 1.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 2.Ito S, et al. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature. 2010;466:1129–1133. doi: 10.1038/nature09303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tahiliani M, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valinluck V, et al. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2) Nucleic Acids Res. 2004;32:4100–4108. doi: 10.1093/nar/gkh739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ko M, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468:839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koh KP, et al. Tet1 and tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell. 2011;8:200–213. doi: 10.1016/j.stem.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu W, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate–dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ficz G, et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398–402. doi: 10.1038/nature10008. [DOI] [PubMed] [Google Scholar]
- 10.Song CX, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams K, et al. TET1 and hydroxymethylcytosine in transcription and DNA methylation fidelity. Nature. 2011;473:343–348. doi: 10.1038/nature10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu H, et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679–684. doi: 10.1101/gad.2036011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen WG, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 14.Ma DK, et al. Neuronal activity–induced Gadd45b promotes epigenetic DNA demethylation and adult neurogenesis. Science. 2009;323:1074–1077. doi: 10.1126/science.1166859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinowich K, et al. DNA methylation–related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 16.Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci. 2010;13:1319–1323. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 18.Muotri AR, et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010;468:443–446. doi: 10.1038/nature09544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skene PJ, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riccio A. Dynamic epigenetic regulation in neurons: enzymes, stimuli and signaling pathways. Nat Neurosci. 2010;13:1330–1337. doi: 10.1038/nn.2671. [DOI] [PubMed] [Google Scholar]
- 21.Globisch D, et al. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS ONE. 2010;5:e15367. doi: 10.1371/journal.pone.0015367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 24.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG–binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 25.Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 26.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 27.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 28.Hutnick LK, et al. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum Mol Genet. 2009;18:2875–2888. doi: 10.1093/hmg/ddp222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem. 1987;262:9948–9951. [PubMed] [Google Scholar]
- 30.Pastor WA, et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394–397. doi: 10.1038/nature10102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szulwach KE, et al. Integrating 5-hydroxymethylcytosine into the epigenomic landscape of human embryonic stem cells. PLoS Genet. 2011;7:e1002154. doi: 10.1371/journal.pgen.1002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lister R, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lengner CJ, et al. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell. 2010;141:872–883. doi: 10.1016/j.cell.2010.04.010. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuemerle B, Zanjani H, Joyner A, Herrup K. Pattern deformities and cell loss in Engrailed-2 mutant mice suggest two separate patterning events during cerebellar development. J Neurosci. 1997;17:7881–7889. doi: 10.1523/JNEUROSCI.17-20-07881.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song DL, Chalepakis G, Gruss P, Joyner AL. Two Pax-binding sites are required for early embryonic brain expression of an Engrailed-2 transgene. Development. 1996;122:627–635. doi: 10.1242/dev.122.2.627. [DOI] [PubMed] [Google Scholar]
- 37.Lister R, et al. Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature. 2011;471:68–73. doi: 10.1038/nature09798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muotri AR, et al. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 39.Coufal NG, et al. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu H, et al. Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature. 2011;473:389–393. doi: 10.1038/nature09934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hellman A, Chess A. Gene body–specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 42.Deininger PL, Moran JV, Batzer MA, Kazazian HH., Jr Mobile elements and mammalian genome evolution. Curr Opin Genet Dev. 2003;13:651–658. doi: 10.1016/j.gde.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 43.Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Z, et al. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth and spine maturation. Neuron. 2006;52:255–269. doi: 10.1016/j.neuron.2006.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson ED, Kavalali ET, Monteggia LM. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J Neurosci. 2008;28:395–406. doi: 10.1523/JNEUROSCI.3796-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGraw CM, Samaco RC, Zoghbi HY. Adult neural function requires MeCP2. Science. 2011;333:186. doi: 10.1126/science.1206593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collins AL, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13:2679–2689. doi: 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 48.Szulwach KE, et al. Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J Cell Biol. 2010;189:127–141. doi: 10.1083/jcb.200908151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Emig D, et al. AltAnalyze and DomainGraph: analyzing and visualizing exon expression data. Nucleic Acids Res. 2010;38:W755–762. doi: 10.1093/nar/gkq405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.