

Abstract
Quantitative proteomic measurements are of significant interest in studies aimed at discovering disease biomarkers and providing new insights into biological pathways. A quantitative cysteinyl-peptide enrichment technology (QCET) can be employed to achieve higher efficiency, greater dynamic range, and higher throughput in quantitative proteomic studies based upon the use of stable-isotope labeling techniques combined with high-resolution capillary or nano-scale liquid chromatography (LC)-mass spectrometry (MS) measurements. The QCET approach involves specific 16O/18O labeling of tryptic peptides, high-efficiency enrichment of cysteinyl-peptides, and confident protein identification and quantification using high mass accuracy LC-Fourier transform ion cyclotron resonance mass spectrometry (FTICR) measurements and a previously established database of accurate mass and LC elution time information for the labeled peptides. This methodology has been initially demonstrated by using proteome profiling of naïve and in vitro-differentiated human mammary epithelial cells (HMEC) as an example, which initially resulted in the identification and quantification of 603 proteins in a single LC-FTICR analysis. QCET provides not only highly efficient enrichment of cysteinyl-peptides for more extensive proteome coverage and improved labeling efficiency for better quantitative measurements, but more importantly, a high-throughput strategy suitable for quantitative proteome analysis where extensive or parallel proteomic measurements are required, such as in time course studies of specific pathways and clinical sample analyses for biomarker discovery.
Keywords: Quantitative proteomics, QCET, 18O labeling, cysteinyl-peptide enrichment, FTICR, AMT
1. Introduction
Quantitative proteomic measurements play a significant role in studies aimed at discovering disease biomarkers and providing new insights into biological pathways. A common strategy for obtaining these measurements is to combine stable-isotope labeling techniques with automated liquid chromatography (LC) - tandem mass spectrometric analyses (MS/MS) (1-4). Inherent with this strategy, however, are a number of analytical challenges that stem from sample complexity and the wide dynamic range of protein abundances, in addition to the low analysis throughput that results from extensive chromatographic fractionation often needed to minimize MS/MS undersampling issues (5) and improve overall proteome coverage.
In this chapter, we detail a quantitative proteomics approach—the quantitative cysteinyl-peptide enrichment technology (QCET)—that was developed in response to these challenges. The QCET approach involves specific 16O/18O labeling of tryptic peptides, high-efficiency enrichment of cysteinyl-peptides, and confident protein identification and quantification using high mass accuracy LC-Fourier transform ion cyclotron resonance mass spectrometry (FTICR) measurements and a previously established database of accurate mass and elution time information. This technology enables higher efficiency, greater dynamic range, and higher throughput quantitative proteomic analyses than previous quantitation technologies (6).
This chapter is organized as follows with the materials required for the QCET approach being presented in Section 2. Following an overview of the technological approach and some of its attractive features, Section 3 provides stepwise methods that exemplify the QCET approach for protein profiling, using naïve and in vitro-differentiated human mammary epithelial cells (HMEC) as our specific example. Section 4 concludes with notes that pertain to both materials and methods.
2. Materials
In our example of protein profiling using QCET, naïve and in vitro-differentiated HMEC nontumorigenic strain 184A1 were routinely cultured in DFCI-1 medium (Gibco BRL, Gaithersburg, MD) until 90% confluence was achieved (7). Cells in 8 dishes (1×107 cells/dish) were treated with 200 nM PMA for 24 h to provide in vitro-differentiated cells, and cells in another 8 dishes were cultured under normal conditions to provide naïve cells. The materials required for this example are listed below by activity.
2.1. Protein digestion and clean-up
Common phosphate-buffered saline.
Cell lysis buffer: 10 mM Tris, 150 mM NaCl, 1% NP-40 (v/v), 1mM NaVO3, 10 mM NaF, and protease inhibitor cocktail (Roche, Indianapolis, IN), pH 7.4 (see Note 1).
Bicinchoninic acid (BCA) protein assay (Pierce, Rockford, IL).
Reducing buffer: 50 mM Tris-HCl, pH 8.2.
Solid high purity urea (Sigma, St. Louis, MO) for denaturing the proteins.
200 mM tributyl phosphine (TBP) solution (Sigma) for reducing the proteins (see Note 2).
Digestion buffer: 20 mM Tris-HCl, pH 8.2.
Sequencing grade modified porcine trypsin (Promega, Madison, WI), freshly dissolved in digestion buffer to a final concentration of 1 μg/μL.
1-mL SPE C18 column per cell state (Supelco, Bellefonte, PA).
SPE conditioning solution: 0.1% trifluoroacetic acid (TFA).
SPE washing solution: 5% acetonitrile and 0.1% TFA.
SPE eluting solution: 80% acetonitrile and 0.1% TFA.
2.2. Post-digestion 16O- to-18O exchange
Immobilized trypsin (Applied Biosystems, Foster City, CA), used as supplied by the manufacturer (see Note 3).
1 M NH4HCO3 stock solution in 18O water: dissolve 79 mg (solid) NH4HCO3 in 1 mL 18O-enriched water (95%; Isotec, Miamisburg, OH).
1 M CaCl2 stock solution prepared with regular (16O) water.
Thermal mixer (Model Thermomixer R; Eppendorf, Westbury, NY).
60% methanol prepared with regular (16O) water.
Handee Mini-Spin column (Pierce).
2.3. Cysteinyl-peptide enrichment
Thiopropyl Sepharose 6B affinity resin (Amersham Biosciences, Uppsala, Sweden) (see Note 4).
Handee Mini-Spin column kit (Pierce) (see Note 5).
Coupling buffer: 50 mM Tris-HCl, pH 7.5, 21 mM EDTA.
Washing buffer: 50 mM Tris-HCl, pH 8.0, 1 mM EDTA.
100 mM dithiothreitol (DTT) in coupling buffer: made fresh from aliquots of 1M DTT stock stored at −80 °C (see Note 6).
2 M NaCl.
80% acetonitrile/0.1% TFA solution.
20 mM DTT in washing buffer: made fresh from aliquots of 1M DTT stock stored at −80 °C.
1 M iodoacetamide solution: made fresh from solid iodoacetamide.
2.4. Strong Cation Exchange (SCX) fractionation
Agilent 1100 series HPLC system (Agilent, Palo Alto, CA).
Polysulfoethyl A 200 mm×2.1 mm column (PolyLC, Columbia, MD, USA) preceded by a 10 mm×2.1 mm guard column (PolyLC) with a flow rate of 0.2 mL/min.
Solvent A: 10 mM ammonium formate, 25% acetonitrile, pH 3.0.
Solvent B: 500 mM ammonium formate, 25% acetonitrile, pH 6.8.
2.5. Capillary LC-MS/MS and LC-FTICR analyses
High pressure capillary LC system (8).
LCQ ion trap mass spectrometer (ThermoFinnigan, San Jose, CA).
Apex III 9.4-Tesla FTICR mass spectrometer (Bruker Daltonics, Billerica, MA).
5-μm Jupiter C18 bonded particles (Phenomenex, Torrence, CA).
65-cm-long, 150 μm-i.d.×360 μm-o.d. fused silica capillary (Polymicro Technologies, Phoenix, AZ).
2-μm retaining stainless steel screen (Valco Instruments Co., Houston, TX).
On-line vacuum degasser (Jones Chromatography Inc., Lakewood, CO).
Mobile phase A: 0.2% acetic acid and 0.05% TFA in water.
Mobile phase B: 0.1% TFA in 90% acetonitrile/10% water.
3. Methods
Quantitative proteomics analysis using the QCET approach involves two stages (see Fig. 1). In the first stage, proteins from different cell states are extracted, mixed, and digested (to improve the proteome coverage). The cysteinyl-peptides from the tryptic digest are covalently enriched using a thiol-affinity resin (see Fig. 2) and further fractionated using strong cation exchange (SCX) chromatography. LC-MS/MS analysis of each SCX fraction provides accurate calculated mass and normalized elution time (NET) information for each peptide that serve as an effective “look-up table” of markers or “accurate mass and time tags” (AMT tags) for future peptide identifications (9).
Fig. 1. Strategy for quantification of differential protein expression using QCET.
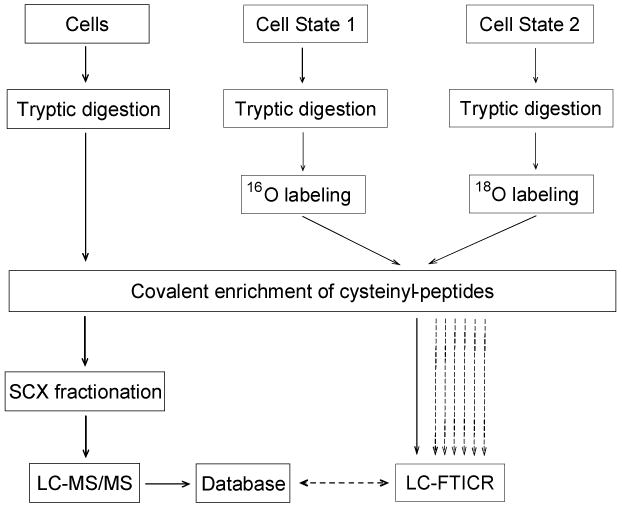
In the first stage, proteins from different cell states are mixed and digested by trypsin, followed by cysteinyl-peptide enrichment using thiol-affinity resin. The enriched cysteinyl-peptides are fractionated by SCX chromatography with each fraction analyzed by LC-MS/MS. An AMT tag database is generated based on the calculated masses and normalized elution times for all identified peptides. In the second stage, the two protein mixtures representing two different cell states are digested by trypsin separately. The resulting tryptic peptides are labeled by trypsin-catalyzed oxygen exchange using 16O- and 18O-enriched water, respectively. The two samples are combined and cysteinyl-peptides are selectively enriched and analyzed by LC-FTICR without pre-fractionation. Peptide features are identified and quantified by matching to the AMT tag database without the need for additional LC-MS/MS analyses. Once an AMT tag database is established for a biological system, the system can be extensively investigated in a high throughput manner by analyzing samples generated under different conditions using LC-FTICR. (Modified from ref. 6 with permission from the American Chemical Society.)
Fig. 2. Enrichment of cysteinyl-peptides using thiol-specific covalent resin.
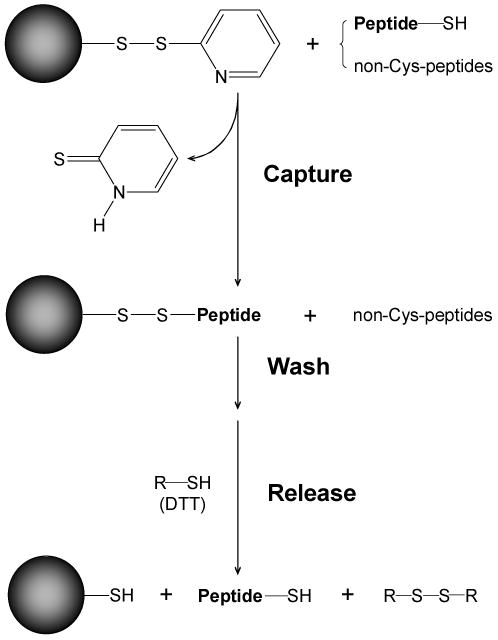
Cysteinyl-peptides were captured by the resin through the formation of a mixed disulfide bond. Stringent washes were applied to remove the non-specifically, noncovalently bound peptides, after which a low molecular weight reducing reagent (e.g., DTT) was added to release the bound cysteinyl-peptides. (Reproduced from ref. 10 with permission from John Wiley & Sons, Inc.)
In the next (typically second) stage, equal masses of protein from two different cell states are separately digested by trypsin under identical conditions, and the tryptic peptides from each sample are labeled with either 16O or 18O by trypsin-catalyzed oxygen exchange in either regular (16O) or 18O-enriched water. The highly efficient post-digestion 18O labeling strategy incorporates two 18O atoms in essentially all tryptic peptides. The 16O/18O labeled peptide pairs co-elute during LC separations. As a result, errors potentially introduced by differences in electrospray ionization suppression effects are minimized, and a framework for accurate quantitation is established.
The differentially labeled peptide samples are combined and subjected to cysteinyl-peptide enrichment. Enrichment of cysteinyl-peptides by the thiol-specific covalent resin is reproducible, highly efficient, and amenable to automation for high throughput studies. The reversible capture and release reaction of cysteinyl-peptides has no side reactions, and the enriched cysteinyl-peptide does not have a labile tag, which eliminates the problem of fragment ion production from the tags (e.g., ICAT and ICAT-like reagents) during collision induced dissociation (CID). Use of high efficiency cysteinyl-peptide enrichment along with global analysis has been demonstrated to significantly improve overall proteome coverage (10), and the significantly reduced (generally 5 fold) sample complexity (i.e., samples containing cysteinyl-enriched peptides only) makes the AMT tag approach even more effective for profiling complicated mammalian systems.
The enriched cysteinyl-peptides are then separated using LC conditions identical to those used in the first stage and analyzed by high mass accuracy Fourier transform ion cyclotron resonance mass spectrometry (FTICR), which provides increased sensitivity and dynamic range, in addition to higher throughput proteomic measurements (11). The greater sensitivity improves identification of lower abundance peptides, and thus leads to better overall proteome coverage without the need for SCX fractionation, and higher overall throughput. An LC-FTICR detected peptide “feature” is identified when both the measured mass and NET values match those of a particular AMT tag in the database within a defined error tolerance (see Fig 3). Once peptides are identified, relative abundance differences are quantified based on the MS peak intensities of the 16O/18O labeled peptide pairs.
Fig. 3. Experimental steps involved in establishing and using an AMT tag and the identification and quantification of LC-FTICR-detected features using the AMT tag strategy.
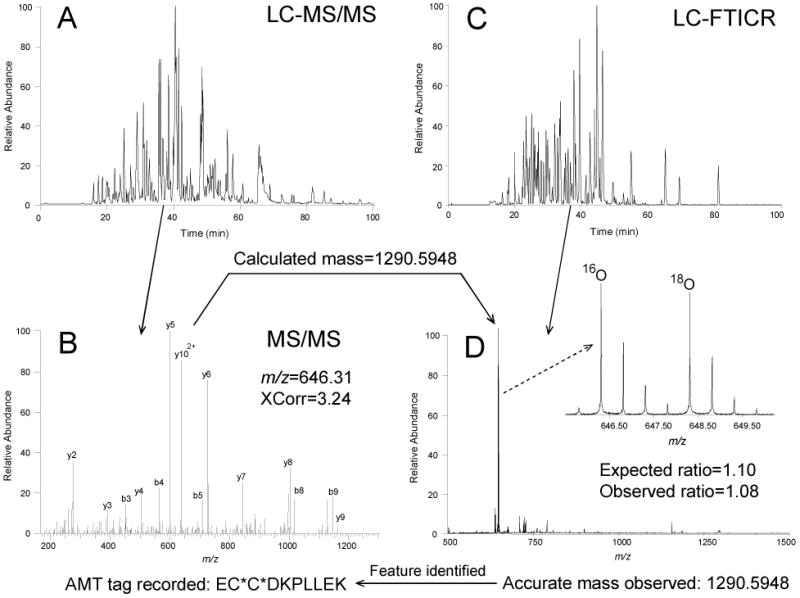
Protein mixtures A and B, each containing the following 5 proteins but at different concentrations are prepared using 16O/18O labeling and cysteinyl-peptide enrichment: bovine serum albumin (BSA; 1:1, A/B); bovine ribonuclease A (1:6); chicken lysozyme (6:1); chicken ovalbumin (3:1); and rabbit glyceraldehyde-3-phosphate dehydrogenase (1:3). (A) Enriched cysteinyl-peptides from the protein mixture are analyzed by LC-MS/MS. (B) A tryptic peptide from BSA (EC*C*DKPLLEK, where C* represents alkylated cysteine residues) is identified by LC-MS/MS. The calculated mass of this peptide based on its sequence (i.e. 1290.5948 Da) and its observed elution time are recorded in the AMT tag database. (C) In the second stage, the same sample is analyzed under the same LC conditions using a FTICR mass spectrometer. (D) A doubly charged feature was observed at the same elution time (36 min), having a mass within 1 ppm (i.e. 1290.5948 Da) of the calculated mass of this AMT tag. This feature is then identified as the corresponding peptide from BSA. The 16O/18O ratio for this peptide was estimated as 1.08 using the maximum intensities of paired monoisotopic peaks (inset). The expected ratio was calculated from the known amount of BSA present in each mixture. (Modified from ref. 6 with permission from the American Chemical Society.)
The combination of cysteinyl-peptide enrichment and AMT tag strategy can be readily combined with other labeling methods, such as 15N labeling (2) and labeling using amino acids in cell culture (3). Another advantage of this two-stage strategy is that once a database has been established for a particular biological system, time-consuming LC-MS/MS analyses are replaced in subsequent studies by high throughput LC-FTICR analyses that enable extensive investigations of that biological system. In practice, the QCET approach for high throughput quantitative proteome profiling can be effectively used with any high resolution MS (e.g., quadrupole time-of-flight).
In our initial demonstration of HMEC protein profiling using QCET (6), 603 proteins were identified and quantified in a single LC-FTICR analysis, with a number of proteins displaying either up- or down-regulation following PMA treatment (see Fig. 4). The histograms of mass and NET errors for peptides identified from AMT tag identifications (see Fig. 4B) display a typical Gaussian distribution. Note that the majority of these identifications are distributed within a mass error of 2 ppm and a NET error of 2%. Moreover, no peptide pairs are identified as C-terminal open tryptic peptides (peptides lacking a C-terminal lysine or arginine) when applies 5 ppm for mass and 5% for NET criteria. Trypsin-catalyzed 18O labeling has specificity for only peptides with K or R at the C terminus, thus any peptide pairs matching to C-terminal open partial tryptic peptides represent false positive hits. This result shows that the 5 ppm mass and 5% NET tolerances provide highly confident peptide identifications.
Fig. 4. Quantitative profiling of labeled and enriched cysteinyl-peptides from naïve and PMA-treated HMEC cells using a single LC-FTICR analysis.
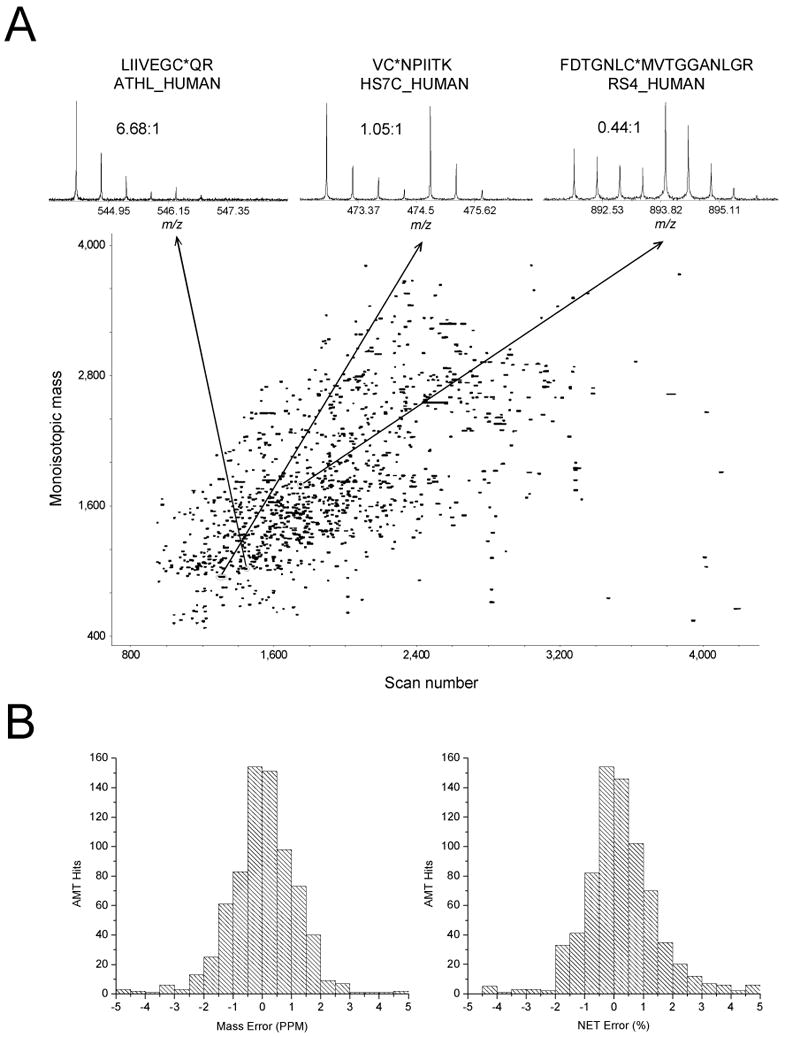
(A) A two-dimensional display of 1348 peptide pairs from which 935 pairs were identified as unique AMT tags corresponding to 603 proteins identified and quantified. Insets show three samples of peptide pairs with their sequences, corresponding proteins, and the 16O/18O ratios. (B) The mass error (left) and NET error (right) distributions of the 935 AMT tag hits. (Reproduced from ref. 6 with permission from the American Chemical Society.)
The stepwise methods employed for this demonstration of QCET protein profiling are detailed below.
3.1. Preparation of HMEC protein digests
Naïve and PMA-treated HMEC cells are placed in 50-mL Falcon tubes and spun at 1,000×g for 10 min. Each cell pellet is washed 3 times with ice-cold phosphate-buffered saline.
Add cell lysis buffer (0.4 mL/dish) to the cell pellets and lyse the cells using intermittent sonication on ice for 1 min.
Centrifuge the lysates for 20 min at 4 °C and 14,000×g to pellet any cellular debris. Collect the supernatants into separate containers and then split the supernatant from each cell state into two aliquots: the first aliquot contains 1.5 mg of protein for generating the AMT tag database and the second contains 100 μg of protein for quantitative analysis. The protein concentrations are ∼10 mg/mL using the BCA protein assay. All aliquots are stored at −80 °C until further use.
Mix 1.5 mg of protein from naïve cells with 1.5 mg of protein from PMA-treated cells and digest as described in steps 6 and 7.
Digest 100 μg of protein from each of the two lysates separately as described in steps 6 and 7.
First, dilute each sample by a factor of 2 by adding the reducing buffer. Then, add 200 mM TBP stock solution and solid urea to final concentrations of 10 mM and 8 M, respectively. Incubate samples at 37 °C for 1 h with gentle mixing.
Dilute the reduced protein mixture by a factor of 10 using the digestion buffer. Add sequencing grade modified porcine trypsin to obtain a trypsin: protein ratio of 1:50 (w/w) and incubate at 37 °C for 3 h.
Pre-condition 1-mL SPE C18 columns by slowly passing 3 mL methanol and then 2 mL SPE conditioning buffer through the column.
Load each of the tryptic digests onto separate pre-conditioned SPE C18 columns; pass each sample through, and wash each column with 4 mL of SPE washing buffer.
Elute the peptides from each SPE C18 column with 1 mL of SPE eluting buffer and dry each of the samples under reduced vacuum using a Speed-Vac.
Store samples at −80 °C until further use.
3.2. Post-digestion 16O-to-18O exchange
Treat each 16O- and 18O-labeled sample (100 μg each) separately until Step 8. Dissolve the dried peptide samples in 100 μL of 50 mM NH4HCO3, and boil for 10 min using water bath, immediately cool the samples on ice for 5 min to eliminate the residual trypsin activity (see Note 7).
Dry samples completely using a Speed-Vac.
Dissolve the dried peptide samples in 20 μL of acetonitrile plus 100 μL of 50 mM NH4HCO3 in either 18O-enriched water (made freshly from the 1 M stock in 18O-enriched water) or regular 16O water, depending on cell type. Peptides from naïve HMEC are to be labeled with 16O and those from the PMA-treated HMEC with 18O.
Add 1 μL of 1 M CaCl2 and 5 μL of immobilized trypsin resin to each of the peptide samples, then mix with constant shaking for 24 h at 30 °C using a thermal mixer (see Note 8).
Centrifuge the samples for 5 min at 15,000×g, and collect each supernatant in a separate, new microcentrifuge tube (see Note 9).
Add 100 μL of 60% methanol to the remaining pellet of immobilized trypsin resin; thoroughly mix the pellet into solution; using a pipette, transfer the suspension into an empty Handee Mini-Spin column (with frit) in 1.7-mL receiving tube; and collect the flow-through by centrifuging at 1000×g.
Repeat step 6 to wash the immobilized trypsin one more time.
Now, combine the supernatants that correspond to the 16O- and 18O-labeled samples from step 6 and the flow-through from step 7 (i.e., mix the naïve HMEC-16O peptide sample with the corresponding PMA-treated HMEC-18O peptide sample).
Dry the combined sample using a Speed-Vac and store at −80 °C until further use (see Note 10).
3.3. Cysteinyl-peptide enrichment by Thiopropyl Sepharose 6B
Degas the coupling and washing buffers and 100 mL of water for 20 min to prevent oxidation of the thiol content.
With the exception of the sample to be used to generate the AMT tag database, dissolve the 16O/18O-labeled sample in 20 μL of coupling buffer, and add 1 μL of 100 mM DTT (5 mM) to the sample; incubate at 37 °C for 1 h to reduce any possible mixed disulfide formation in the sample. Because of the larger size of the peptide sample that is being used to generate the AMT tag database, dissolve this sample in 80 μL of coupling buffer, and add 4 μL of 100 mM DTT.
Place 5 × 35 mg (5 × 2 μmol disulfide exchange capacity) of dried Thiopropyl Sepharose 6B resin into individual 1.7-mL tubes. Add 1 mL of water to each tube, and rehydrate the resin for 15 min at room temperature. Suspend the resin well using the pipette and modified 1-mL tips (see Note 11), and then place in a rack for ∼10 min. The final volume of rehydrated resin should be around 100 μL.
Remove and discard 0.5 mL of supernatant (water) off the top of each tube and resuspend the resin with the solution remaining in the tube. Use a pipette and the modified 1-mL tip to carefully transfer each suspension to a Handee Mini-Spin column (without the bottom cap on). Place each spin column in a 2-mL receiving tube and centrifuge at 1000×g for 30 sec to remove the water.
Add 0.5 mL of water to the spin column, which will readily resuspend the resin when water is added. Spin at 1000×g for 30 sec to remove the water. Repeat this washing step 5 more times.
Wash the resin 10 times each with 0.5 mL of coupling buffer in the spin column. After the last wash, put the bottom caps on, tightly.
Dilute the reduced samples to 100 μL by adding coupling buffer to a final DTT concentration of 1 mM. Dilute the peptide sample for generating the AMT tag database to 400 μL and then split the sample into 4×100 μL aliquots.
Add each 100 μL sample to a spin column that contains ∼100 μL of thiol-affinity resin, and put the top caps on the spin columns. Place each spin column with sample into a 1.7-mL tube (with caps cut off) and shake at medium speed for 1 h at room temperature to allow for cysteinyl-peptide capture by the resin (see Note 12).
Place each spin column into a new 1.7-mL tube with both top and bottom caps removed and spin at 1500×g for 1 min to collect the unbound portion (non-cysteinyl-peptides, which can be retained for global quantitative analysis based on non-cysteinyl-peptides).
Extensively wash the resin using 0.5 mL×6 of: 1) washing buffer, 2) 2 M NaCl, 3) 80% acetonitrile/0.1% TFA, and 4) washing buffer as in steps 5 and 6 above to remove the peptides bound non-specifically to the resin via ionic interactions or hydrophobic interactions.
Put the bottom caps back on the spin columns. Add 100 μL of 20 mM DTT to the resin; close the top caps; and incubate the spin columns at room temperature for 30 min with shaking. Collect the released cysteinyl-peptides as flow-through by centrifuging the spin columns at 1500×g for 1 min with both top and bottom caps removed. Use 2-mL receiving tubes for easy pooling of the flow-through with subsequent eluates.
Add another 100 μL of 20 mM DTT to the resin and mix using pipette tips (see Note 13). After incubating at room temperature for 10 min with shaking, place the spin columns into the same collection tubes and centrifuge at 1500×g for 1 min to collect the released cysteinyl-peptides. Repeat this step once.
Repeat step 12, but this time use 80% acetonitrile instead of 20 mM DTT.
Immediately alkylate the released cysteinyl-peptides by adding 32 μL (80 mM) of 1 M iodoacetamide solution to the pooled eluates (a total of 400 μL) and incubate for 1 h at room temperature in the dark.
Dilute the alkylated samples to 2 mL by adding washing buffer, followed by SPE C18 clean up as described in 3.1 above.
Dry the SPE C18 eluates using a Speed-Vac and store at −80 °C until further use.
3.4. SCX fractionation of enriched Cysteinyl-peptides
Reconstitute the enriched cysteinyl-peptides from the large peptide sample (for generating the AMT tag database) with 900 μL of solvent A and inject onto the SCX column with a flow rate of 0.2 mL/min.
Once loaded, maintain an isocratic gradient with 100% solvent A for 10 min. Separate peptides by applying a gradient from 0% to 50% B over 40 min, followed by a gradient of 50% to 100% B over 10 min. Hold the gradient isocratically at 100% solvent B for an additional 10 min.
Collect a total of 70 fractions using the automatic fraction collector on the HPLC, and then combine into a final total of 35 fractions. Dry each fraction and store at −80 °C until time for analysis.
3.5. Capillary LC-MS/MS and LC-FTICR analyses
In our laboratory, peptide samples are analyzed using a custom-built capillary LC system (8) coupled online to either a LCQ Deca XP ion trap mass spectrometer (for generating the AMT tag database) or to an Apex III 9.4-Tesla FTICR mass spectrometer equipped with an ESI interface for subsequent measurements.
The reversed-phase capillary column is prepared by slurry packing 5-μm Jupiter C18 bonded particles into a 65-cm-long, 150 μm-i.d. fused silica capillary.
Mobile phases are degassed on-line using a vacuum degasser. After injecting 10 μL of peptide sample onto the reversed-phase capillary column, hold the mobile phase at 100% A for 20 min. Attain exponential gradient elution by increasing the mobile-phase composition to ∼70% B over 150 min, using a stainless steel mixing chamber. Apply this same gradient for both LC-MS/MS and LC-FTICR analyses.
The ion trap and the FTICR mass spectrometers are operated under normal conditions. Details of the LC-MS/MS and LC-FTICR operating parameters employed for this demonstration are available in references (10, 12).
3.6. Data analysis
In our laboratory, data analysis and processing steps are automated and proceed as follows:
For LC-MS/MS analyses, peptides are identified using SEQUEST and static modification of cysteine residues (+57 Da) to search the MS/MS spectra against a normal non-redundant human International Protein Index (IPI) database (consisting of 47,306 protein entries at the time of our analysis; available online at http://www.ebi.ac.uk/IPI) and its sequence-reversed version.
The false positive rate of the SEQUEST search results is evaluated using a reversed protein sequence database (13). From this evaluation, the following set of criteria are determined for filtering the raw SEQUEST data with an overall confidence of >95% (ΔCn ≥ 0.1): for the 1+ charge state, Xcorr ≥ 1.5 for fully tryptic peptides and Xcorr ≥ 3.1 for partially tryptic peptides; for the 2+ charge state, Xcorr ≥ 1.9 for fully tryptic peptides and Xcorr ≥ 3.8 for partially tryptic peptides; and for the 3+ charge state, Xcorr ≥ 2.9 for fully tryptic peptides and Xcorr ≥ 4.5 for partially tryptic peptides. Nontryptic peptides are not included.
Peptides that meet the criteria in step 2 are included in the AMT tag database. The peptide retention times from each LC-MS/MS analysis are normalized to a range of 0 to 1 by using a predictive peptide LC-NET model and linear regression (14). An average NET value and NET standard deviation are assigned to each identified peptide, provided the same peptide was observed in multiple runs. Both the calculated accurate monoisotopic mass and the NET of the identified peptides are included in the AMT tag database.
The initial analysis of raw LC-FTICR data involves a mass transformation or de-isotoping step using ICR2LS, an analysis tool based on the THRASH algorithm (15). ICR2LS data analysis generates a text file report for each LC-FTICR dataset that includes both the monoisotopic masses and the corresponding intensities for all detected species for each spectrum.
Following ICR2LS analysis, data are processed to yield a two-dimensional mass and LC elution time data set. Automated data processing steps include filtering data, finding features (i.e., a peak with a unique mass and elution time signature) and pairs of features, computing abundance ratios for pairs of features, normalizing LC elution times, and matching the accurately measured masses and NET values of each feature to the corresponding AMT tag in the database to identify peptide sequences. The peptide sequences of a given feature or pair of features are assigned when the measured mass and NET match the calculated mass and NET in the AMT tag database within 5 ppm mass error and 5% NET error.
-
The abundance ratios (18O/16O) for labeled peptide pairs are computed by using an equation similar to that previously reported (4)
(1) where I0, I2, and I4 are the measured intensities for the monoisotopic peak for the peptide without 18O label, the peak with mass 2 Da higher, and the peak with 4 Da higher mass, respectively. M0, M2, and M4 are the predicted relative abundances for the monoisotopic peak for the peptide, the peak with mass 2 Da higher, and the peak with mass 4 Da higher, respectively. The and ratios are estimated based on the following two equations (Eq. 2 and 3) (16) where Mr represents peptide molecular weight.(2) (3) Ratios from multiple observations of the same peptide across different analyses are averaged to give one ratio per peptide. All quantified peptides are rolled up to non-redundant protein groups using ProteinProphet (17) (see Note 14), and the abundance ratio for each protein group is calculated by averaging the ratio of multiple unique peptides stemming from the same protein group.
Acknowledgments
The authors would like to thank Drs. Wan-Nan U. Chen and Brian Thrall for providing naïve and PMA-treated HMEC cells. The research discussed in this chapter is supported by the NIH National Center for Research Resources (RR18522) located in the Environmental Molecular Science Laboratory (a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory). Pacific Northwest National Laboratory is a multiprogram national laboratory operated by Battelle Memorial Institute for the U.S. Department of Energy under contract DE-AC05-76RLO-1830.
Footnotes
Unless otherwise stated, all solutions should be prepared in deionized water that has a resistivity of 18.2 MΩ-cm and total organic content of <5 ppb (parts per billion). This standard is referred as “water” or regular (16O) water” in the text.
TBP is received in sealed Ampere vials. Once opened, TBP may decline in quality. Thus, it is best used if stored at 4 °C under nitrogen gas for no longer than one week.
Immobilized trypsin is in the form of a fine resin and is received as a white suspension. Mix well or shake briefly before use.
Thiopropyl Sepharose 6B resin is received as dried powder. Once opened, it is best stored at 4 °C to 8 °C in a desiccator.
The Handee Mini-Spin column kit contains an empty spin column (0.5 mL capacity, with frit), top cap, and bottom cap. Make sure the bottom cap is fitting tightly with the bottom of the spin column to prevent leakage during incubation with shaking.
Because of the frequent use of DTT in the cysteinyl-peptide enrichment experiment, it is convenient to make a 1 M DTT stock solution in water and then split it into 10 μL aliquots. This stock solution can be stored at −80 °C for more than 3 months without decline in quality.
Failure to eliminate residual trypsin activity will result in 16O and 18O back-exchange. Alternatively, immobilized trypsin can be used for protein digestion, but additional sample handling may required (e.g., washing the immobilized trypsin resin following the digestion).
White precipitates are observed once the CaCl2 and immobilized trypsin are added. It is necessary to shake the suspension at high speed (e.g., 1,300 rpm) to keep the immobilized trypsin homogeneously distributed to improve the reaction efficiency.
Keep corresponding 16O- and 18O-labeled samples separate in different tubes until immobilized trypsin resin is filtered using the empty spin column. We recommend mixing differentially labeled samples from the same biological system as the last step to prevent trypsin-catalyzed 16O- and 18O back-exchange. If a precipitate is still observed in any of the final combined samples, we recommended centrifuging again and transferring the supernatant to a new tube.
There will be a light yellowish or whitish layer on the bottom of tubes after completely dried, but it can be readily dissolved in the coupling buffer in the cysteinyl-peptide enrichment step, or in other common buffers, such as 25 mM NH4HCO3.
The resin tends to get sticky at the beginning of the rehydration and thus, forms chunks that make pipetting difficult using regular tips. Cut 5 mm off the end of the tip to make the hole larger for easier mixing and transfer.
We recommend shaking the spin column at no less than 800 rpm to keep the resin homogeneously distributed. However, the use of a speed of higher than 1,000 rpm may cause spillage of the solution through the top cap, resulting in sample loss. Also, we recommend checking the bottom caps after shaking for 5 min. Finding a potential leakage problem earlier minimizes the chance of failure in the cysteinyl-peptide enrichment experiment.
Once incubated with DTT, the resin tends to get packed and sticky after the low-speed centrifugation step since its structure is changed with the elution of cysteinyl-peptides. Use pipette tips to completely mix the freshly added DTT with the resin.
All peptides that pass the filtering criteria are given the identical probability score of 1, and entered into the Protein Prophet program only for clustering analysis to generate a final non-redundant list of proteins or protein groups.
References
- 1.Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 2.Veenstra TD, Martinovic S, Anderson GA, Pasa-Tolic L, Smith RD. Proteome analysis using selective incorporation of isotopically labeled amino acids. J Am Soc Mass Spectrom. 2000;11:78–82. doi: 10.1016/S1044-0305(99)00120-8. [DOI] [PubMed] [Google Scholar]
- 3.Ong SE, Kratchmarova I, Mann M. Properties of 13C-substituted arginine in stable isotope labeling by amino acids in cell culture (SILAC) J Proteome Res. 2003;2:173–181. doi: 10.1021/pr0255708. [DOI] [PubMed] [Google Scholar]
- 4.Yao XD, Freas A, Ramirez J, Demirev PA, Fenselau C. Proteolytic 18O labeling for comparative proteomics: Model studies with two serotypes of adenovirus. Anal Chem. 2001;73:2836–2842. doi: 10.1021/ac001404c. [DOI] [PubMed] [Google Scholar]
- 5.Tabb DL, MacCoss MJ, Wu CC, Anderson SD, Yates JR., 3rd Similarity among tandem mass spectra from proteomic experiments: Detection, significance, and utility. Anal Chem. 2003;75:2470–2477. doi: 10.1021/ac026424o. [DOI] [PubMed] [Google Scholar]
- 6.Liu T, Qian WJ, Strittmatter EF, Camp DG, 2nd, Anderson GA, Thrall BD, Smith RD. High-throughput comparative proteome analysis using a quantitative cysteinyl-peptide enrichment technology. Anal Chem. 2004;76:5345–5353. doi: 10.1021/ac049485q. [DOI] [PubMed] [Google Scholar]
- 7.Chen WN, Woodbury RL, Kathmann LE, Opresko LK, Zangar RC, Wiley HS, Thrall BD. Induced autocrine signaling through the epidermal growth factor receptor contributes to the response of mammary epithelial cells to tumor necrosis factor alpha. J Biol Chem. 2004;279:18488–18496. doi: 10.1074/jbc.M310874200. [DOI] [PubMed] [Google Scholar]
- 8.Shen Y, Zhao R, Belov ME, Conrads TP, Anderson GA, Tang K, Pasa-Tolic L, Veenstra TD, Lipton MS, Smith RD. Packed capillary reversed-phase liquid chromatography with high-performance electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry for proteomics. Anal Chem. 2001;73:1766–1775. doi: 10.1021/ac0011336. [DOI] [PubMed] [Google Scholar]
- 9.Smith RD, Anderson GA, Lipton MS, Pasa-Tolic L, Shen Y, Conrads TP, Veenstra TD, Udseth HR. An accurate mass tag strategy for quantitative and high throughput proteome measurements. Proteomics. 2002;2:513–523. doi: 10.1002/1615-9861(200205)2:5<513::AID-PROT513>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 10.Liu T, Qian WJ, Chen WN, Jacobs JM, Moore RJ, Anderson DJ, Gritsenko MA, Monroe ME, Thrall BD, Camp DG, 2nd, Smith RD. Improved proteome coverage by using high efficiency cysteinyl peptide enrichment: The human mammary epithelial cell proteome. Proteomics. 2005 doi: 10.1002/pmic.200401055. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian WJ, Camp DG, 2nd, Smith RD. High throughput proteomics using Fourier transform ion cyclotron resonance (FTICR) mass spectrometry. Expert Review of Proteomics. 2004;1:89–97. doi: 10.1586/14789450.1.1.87. [DOI] [PubMed] [Google Scholar]
- 12.Belov ME, Anderson GA, Wingerd MA, Udseth HR, Tang K, Prior DC, Swanson KR, Buschbach MA, Strittmatter EF, Moore RJ, Smith RD. An automated high performance capillary liquid chromatography-Fourier transform ion cyclotron resonance mass spectrometer for high-throughput proteomics. J Am Soc Mass Spectrom. 2004;15:212–232. doi: 10.1016/j.jasms.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Qian WJ, Liu T, Monroe ME, Strittmatter EF, Jacobs JM, Kangas LJ, Petritis K, Camp DG, 2nd, Smith RD. Probability-based evaluation of peptide and protein identifications from tandem mass spectrometry and SEQUEST analysis: The human proteome. J Proteome Res. 2005;4:53–62. doi: 10.1021/pr0498638. [DOI] [PubMed] [Google Scholar]
- 14.Petritis K, Kangas LJ, Ferguson PL, Anderson GA, Pasa-Tolic' L, Lipton MS, Auberry KJ, Strittmatter E, Shen Y, Zhao R, Smith RD. Use of artificial neural networks for the prediction of peptide liquid chromatography elution times in proteome analyses. Anal Chem. 2003;75:1039–1048. doi: 10.1021/ac0205154. [DOI] [PubMed] [Google Scholar]
- 15.Horn DM, Zubarev RA, McLafferty FW. Automated reduction and interpretation of high resolution electrospray mass spectra of large molecules. J Amer Soc Mass Spectrom. 2000;11:320–332. doi: 10.1016/s1044-0305(99)00157-9. [DOI] [PubMed] [Google Scholar]
- 16.Johnson KL, Muddiman DC. A method for calculating 16O/18O peptide ion ratios for the relative quantification of proteomes. J Am Soc Mass Spectrom. 2004;15:437–445. doi: 10.1016/j.jasms.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 17.Nesvizhskii AI, Keller A, Kolker E, Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]