

Abstract
Tau hyperphosphorylation is thought to play an important role in the etiology of Alzheimer’s disease by facilitating the formation of neurofibrillary tangles. Reducing phosphorylation through kinase inhibition has therefore emerged as a target for drug development, but despite considerable efforts to develop therapeutic kinase inhibitors, success has been limited. An alternative approach is to develop pharmaceuticals to enhance the activity of the principal phospho-tau phosphatase, phosphoprotein phosphatase 2A (PP2A). In this article we review evidence that this mechanism is pharmacologically achievable and has promise for delivering the next generation of Alzheimer’s disease therapeutics. A number of different chemotypes have been reported to lead to enhanced PP2A activity through a range of proposed mechanisms. Some of these compounds appear to act directly as allosteric activators of PP2A, while others act indirectly by inhibiting the binding of PP2A inhibitors or by altering post-translational modifications that act in turn to regulate PP2A activity towards phospho-tau. These results indicate that PP2A may provide a useful target that can be safely, selectively and effectively modulated through pharmaceutical intervention to treat Alzheimer’s disease.
The identification of disease-modifying agents for neurodegenerative disorders, particularly Alzheimer’s disease (AD), is an area of active pursuit that remains an elusive goal for the scientific and drug-discovery communities. Recent clinical disappointments have led to a re-evaluation of the amyloid cascade hypothesis and engendered considerable interest in novel targets for AD therapeutics [1-3], with a clear shift towards addressing tau rather than β-amyloid pathology [4-8]. It has been shown that hyperphosphorylation of tau leads to aggregation, formation of neurofibrillary tangles (NFTs), microtubule disruption and neurodegeneration [9,10]. NFTs are a cardinal pathological hallmark of AD that have been observed to be composed of hyperphosphorylated tau [11]. NFT formation is promoted by tau hyperphosphorylation [12,13]. This strong disease-relevant tau biology has provided a new therapeutic paradigm that focuses on developing small molecules targeted at this aspect of AD pathology, either by inhibiting tau phosphorylation [14], blocking tau aggregation [15-18] or otherwise rescuing tau-associated microtubule dysfunction [19,20]. An alternative approach involves activation of phosphoprotein phosphatase 2A (PP2A), the key tau phosphatase. This could be a viable alternative to kinase inhibition for developing novel therapeutics for Alzheimer disease [21,22]. Recent results suggest that this is a pharmacologically tractable approach. In contrast with inhibiting tau kinases, upregulating PP2A may prove to be a more practical approach for developing a single therapeutic agent: multiple distinct protein kinases (e.g., GSK-3, MAPK, cdk5, CK-1, PKA, CaMKII and MARK) have been implicated in tau hyperphosphorylation [23-25], while PP2A alone accounts for over 70% of tau dephosphorylation [26]. Furthermore, PP2A activation would be expected to have beneficial effects that extend beyond simply reducing the level of tau phosphorylation [27], impacting a number of faulty signaling pathways that underlie neurodegeneration, thus providing superior therapeutic efficacy. The design of phosphatase inhibitors is well established in medicinal chemistry [28-30], but mechanisms of activation are only beginning to be explored.
PP2A & neurodegeneration in Alzheimer’s disease
Reduced PP2A mRNA [31], protein [32] and phosphatase activity [32-34] have been observed in postmortem brains from AD patients. There have also been reports of increased levels of endogenous inhibitors of PP2A, such as inhibitor 2 (I2), along with their cleavage and redistribution [35], which when overexpressed in an in vivo model result in cardinal features of AD, including amyloid-β deposition, tau hyperphosphorylation, neurodegeneration and cognitive deficits [36]. Decreases in PP2A carboxyl methylation have also been observed [37,38]. Together, the end result is decreased PP2A activity in AD brains, resulting in reduced dephosphosylation of tau and other phosphoprotein substrates. Inhibition of PP2A in rodent brains by okadaic acid [39] or calyculin A [40] recapitulates many of the hallmarks of neurodegeneration in AD, including amyloid deposition, development of NFTs, neurodegeneration and cognitive deficits. Decreased PP2A activity results in increased levels of protein phosphorylation. In AD, multiple phosphorylation-dependent signal transduction pathways go awry. The most prominent evidence of this is the hyperphosphorylated tau in NFTs. In addition to its tau phosphatase activity [26], PP2A acts to dephosphorylate and inactivate kinases that phosphorylate tau [41]. The principal hallmark of AD, deposition of amyloid plaques, is also subject to PP2A regulation. Generation of amyloid β (Aβ) from amyloid precursor protein (APP) via secretases is modulated by phosphorylation at APP-Thr-668 [42], at least in part via JNK [43]. Increased APP phosphorylation, either directly through decreased activity of PP2A towards phospho-APP, or indirectly through reduced phosphatase activity towards phospho-JNK, can result in increased Aβ production and ultimately amyloid plaque formation. Terminal stages of AD are characterized by neuronal cell death. Decreased PP2A methylation has been shown to trigger apoptosis in neuroblastoma cell lines, and PP2A deficiency leads to activation of kinases, including JNKs that can lead to AD-relevant neuronal cell death [44]. PP2A can also influence other key apoptotic proteins including Bcl2 [45]. Cell-cycle progression is highly dependent on PP2A, particularly in the G2/M phase transition, and in postmitotic neurons, cell cycle re-entry can be induced by PP2A activation [46]. Triggering cell cycle entry in neurons using SV40 in a conditional murine transgenic model resulted in the three major pathological hallmarks of AD: amyloid plaques, NFTs and neurodegeneration [47]. SV40 small t-antigen bound to PP2A and inhibited its activity [48,49]. These data suggest that PP2A dysfunction could trigger abnormal cell cycle re-entry that contributes to AD pathogenesis. PP2A’s additional roles in cellular signaling pathways can all be connected to possible mechanisms underlying AD [50]. Clearly, the reduced PP2A activity observed in AD can be a root cause of multiple abnormalities observed in patients and a means to maintain and enhance PP2A function may provide a useful therapeutic strategy.
PP2A structure & endogenous regulation of its activity
The PP2A holoenzyme is composed of a 36-kDa catalytic C subunit, a 65-kDa scaffold A subunit and one of a series of regulatory B subunits that serve to regulate PP2A specificity, selectivity and localization (Figure 1) [50,51]. PP2A also interacts with inhibitory subunits that block phosphatase activity, most notably I2 (also known as SET) [52]. PP2A also appears to be regulated by kinase-dependent phosphorylation of the C subunit at Thr and Tyr side chains [53]. Tyr-307 near the C-terminus has been the most thoroughly characterized, its phosphorylation inhibits PP2A [53]. The C-terminal residue, Leu-309, is the site of carboxyl methylation [54]. This unusual protein-modification chemistry is regulated by two converter enzymes that appear to be completely specific for PP2A. The PP2A methyltransferase (PPMT, also termed leucine carboxyl methyl transferase [LCMT]) transfers a methyl group from S-adenosylmethionine (SAM) to the C-terminal α-carboxyl of the catalytic C subunit of PP2A. The demethylating enzyme, PP2A methylesterase (PME), is a PP2A-specific methylesterase that catalyzes the hydrolysis of the Leu-309 methylester with the formation of unmethylated PP2A and methanol [55]. The PME active site, which only forms when PME binds to PP2A, contains a classic Ser-His-Asp catalytic triad [56]. Methylation has been shown to regulate PP2A phosphatase-specific activities and to facilitate the association of different B subunits with the conserved PP2A AC catalytic core [57,58].
Figure 1. PP2A is a heterotrimeric holoenzyme that is regulated by multiple endogenous mechanisms.

Catalytic C subunit (green), scaffolding A unit (red) and regulatory B subunit (blue) form the trimeric enzyme.
Three major classes of endogenous small molecules have been shown to regulate PP2A activity: metal cations, ceramides and polyamines. PP2A has a bimetallic phosphatase active site that accommodates Mg(II) or Mn(II). The activity and specificity of PP2A depends on the nature of the bound metal. PP2A is one of the prime targets for ceramide regulation, and ceramide levels have been shown to increase three- to four-fold in AD [59]. In addition, polyamines such as protamine and polylysine appear to stabilize PP2A and stimulate its activity [60]. These multiple regulatory interactions tightly control PP2A function. These various loci for PP2A control may provide entry points for pharmacological modulation of PP2A activity.
PP2A pharmacology
Increasing the activity of an enzyme is typically a challenging endeavor; however, the regulatory interactions of PP2A do provide opportunities. Broadly, there are three potential strategies:
-
▪
Inhibition of an inhibitory interaction;
-
▪
Modulation of post-translation modification enzymes;
-
▪
Allosteric activation.
In considering activators, it is important to keep in mind that because of the comprehensive role PP2A plays in cellular phosphoregulatory networks, agents that are highly potent are likely to be toxic. In this review we have subdivided known PP2A activators by structural features into the chemical classes sphingoids, phenolics, cations, anions and others (Figure 2).
Figure 2. Major classes of PP2A activating compounds.
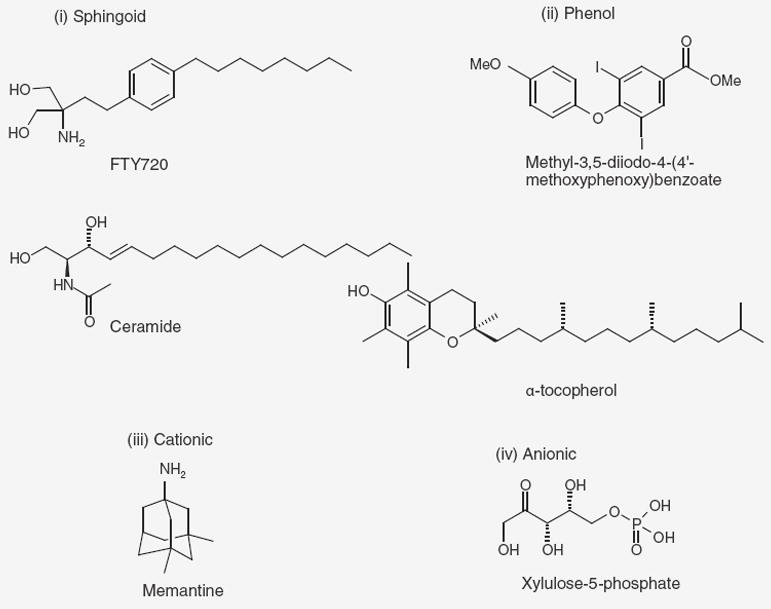
Exemplifying structures of compounds in each class.
Sphingoid activators
C2, C6 and C10 ceramides, pyridinium analogs of C6 and C18 ceramides, FTY720, N,N-dimethyl sphingosine, sphingosine and phytosphingosine comprise the sphingoid class (Table 1). PP2A has been shown to be a principal target of ceramide signaling. Since the initial report of ceramide’s direct allosteric activation of PP2A [61], it has subsequently been demonstrated that ceramides promote PP2A activity towards some phosphoprotein substrates [61,62], inhibit PP2A demethylation [63] and block I2 inhibitor binding [64]. Although d-erythro-C2-ceramide activates heterotrimeric (ABC) forms of PP2A, without affecting the activities of either PP2A AC dimers or isolated PP2A catalytic C subunits. In contrast with d-erythro-C2-ceramide, d-erythro-C6-ceramide activates monomeric PP2A composed of only the C subunit [65]. The specific chemical variations within this class have differing effects depending on the multimeric form of PP2A. While B56 (B′α)-containing PP2A was erythro-selective, B55 (Bα)-containing isoforms could be activated by both erythro- and threo-isomers. From a screen of various lipids and sphingosine derivatives, the key features required for PP2A activation appear to be a trans double bond, an aliphatic amide (C10 > C6 > C2 > C18) and an unsubstituted terminal hydroxy group. d-erythro-C18-ceramide appears to be highly stereospecific and increased PP2A activity two- to six-fold towards phosphorylated myelin basic protein (pMBP) at high micromolar concentrations. In the presence of Mn2+, activation by ceramide was increased by 175%, and could be increased further by manipulating solvent conditions. Conversely, at low micromolar concentrations, d-erythro-C18-ceramide had an inhibitory effect on heterotrimeric PP2A complexes and isolated catalytic C subunits [66].
Table 1.
Sphingoid class activators.
| Name (proposed mode of action) | Chemical structure | Biochemical assay
|
Cell-based assay
|
||||
|---|---|---|---|---|---|---|---|
| Activity | Substrate | PP2A | Selectivities | Activity | Cell type | ||
| FTY720 (direct) |
|
175% ABC and 120% AC (10 μM) | Phosphorylase a | ABC and AC | PP2A > PP1 | 150–300% (10 μM) | pERK1/2 in CLL (no Akt, STAT3,Bcl-2 or Bad); Akt and p70S6K (Jurkat) |
|
| |||||||
| Ceramide (direct, inhibitor 2, PP2A methylesterase) |
|
350% (20 μM) | Phosphorylated myelin basic protein | ABC AB′C but not AC or C | Some B subunit specificity C10 > C6 > C2 | 1000% (40 μM) | pBcl-2 in 10I |
|
| |||||||
| C6-pyr-ceramide† direct, inhibitor 2) |
|
115–130% (2–5 μM; without inhibitor 2) | Phosphorylated myelin basic protein | Heterotrimer | Not reported | c-myc dephosphorylation/degradation | A546 cells |
|
| |||||||
| N,N-dimethylsphingosine (ANP32A) |
|
Rescue (1.25 μM) | p-histone | AC | Not reported | 226% (10 μM) | HUVEC |
Also a member of cationic class.
PP: Phosphoprotein phosphatase.
The mechanism of activation of heterotrimeric PP2A by ceramides is still unclear. In vitro studies indicate that ceramide binding may promote the dissociation of B subunits, causing a two- to seven-fold increase in Vmax and a 1.6- to five-fold increase in Km of the resulting AC dimers [67]. In cells, however, d-erythro-C2-ceramide inhibits PP2A demethylation, thereby increasing the affinity of AC dimers for Bα or B′α subunits [63]. d-erythro-C6-ceramide has been shown to specifically bind to I2, but not to inhibitor 1 (I1) [63]. The ceramide binding site was mapped by site-directed mutagenesis and the VIK/SSS mutant of I2, which does not bind d-erythro-C6-ceramide, also does not affect PP2A activity. Interestingly, I2 has shown a preference for binding C6 and C18, but not C16 ceramides [64]. There has been no biochemical evidence of rescue or activation of PP2A by C6-ceramide in the presence of I2 [64]. On the other hand, the positively charged C6-ceramide pyridinium analog, d-erythro-C6-pyr-ceramide, is an effective activator of PP2A [64] that interacts with PP2A as both a direct allosteric activator and as an inhibitor of I2 inhibition. The cationic nature of these analogs may contribute to PP2A activation [68]. The data indicate that the d-erythro geometry as well as the double bond, amide and unprotected terminal hydroxyl moieties are critical.
Sphingosine, sphingosine phosphate, ceramide phosphate, 1-O-methyl-C6-ceramide, or dihydro analogs of C6-pyr-ceramide did not prevent I2 inhibition of PP2A [64]. The compounds share structural features with ceramides and similar to ceramides, they serve as allosteric activators of PP2A [61,65]. The observed range of PP2A activation was from 1.2- to 3.5-fold in biochemical assays [64], and cellular assays indicate as much as tenfold activation towards some phosphoprotein targets [63]. FTY720, a sphingosine analog that was developed as an immunosuppressive agent, was found to activate PP2A phosphatase activity towards phosphorylase a, with selectivity over PP1 [69]. The substrate specificity of FTY720-activated PP2A in cellular assays indicates reduced phosphorylation of ERK, but no effect on the phosphorylation status of Akt and STAT3 [70]. This selectivity depends on cell type, since in Jurkat cells Akt and p70(S6k) were dephosphorylated in response to treatment with FTY720 [69]. Treatment with 10 μM FTY720 failed to dephosphorylate pBcl-2 in cells [70], contrasting with effects of d-erythro-C2-ceramide [63]. Another mechanism of relevance to this class of compounds is through binding to ANP32A (acidic nuclear phosphoprotein 32 family member A), a protein inhibitor of PP2A. N,N-dimethylsphingosine (DSM) binds with high affinity to ANP32A [71]. This mechanism is similar to ceramide binding to I2; however, there is selectivity since DSM, sphingosine and phytosphingosine bind to ANP32A, while ceramide does not. DSM effectively rescues PP2A activity when incubated with PP2A AC dimers in the presence of ANP32A using phosphorylated histone as a substrate. Limited SAR data suggests that a terminal unprotected hydroxyl group is important as sphingosine-1-phosphate does not bind to ANP32A. The importance of a double bond is somewhat unclear since dihydrosphingosine does not bind to ANP32A but phytosphingosine, which also lacks this moiety, does. This may provide an explanation for the activation of PP2A by sphingosine and phytosphingosine previously observed in cells [72].
While data suggest that inhibiting ANP32A is the mechanism of PP2A activation for DMS, sphingosine and phytosphingosine, so far there is no solid evidence that these compounds could activate PP2A directly or share a binding site with other molecules from the sphingoid class. Nevertheless, it appears that sphingoid PP2A activators share the same mode of action by targeting protein–protein interactions between the phosphatase and its inhibitory interacting partners - I2, ANP32A or PME. Furthermore, given obvious structural similarities, and reported SAR, it would not be surprising if the sphingoids have a distinct PP2A binding site that has yet to be fully characterized. Finally, these small molecules have been shown to activate PP2A towards various pathways involved in cancer and inflammation; however some of them also activate the B55-containing isoform of PP2A – the major phosphatase for tau, warranting future evaluation of sphingoids in the context of AD.
Phenolic activators
The class of phenolic activators loosely encompasses certain quinolones and tetralones [73], methyl-3,5-diiodo-4-(4′-methoxyphenoxy) benzoate (DIME) [74], dihydroxy phenylethanol [75], α-tocopherol [76] and eicosanoyl-5-hydroxytryptamide (Table 2) [77].
Table 2.
Phenolic class activators.
| Name (proposed mode of action) | Chemical structure | Biochemical assay
|
Cell-based assay
|
||||
|---|---|---|---|---|---|---|---|
| Activity | Substrate | PP2A | Selectivities | Activity | Cell type | ||
| Tetralone (direct in the presence of ceramide) |
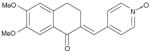
|
520% (20 μM; in the presence of C2-cer) | Phosphorylated myelin basic protein | Purified heterotrimer from Jurkat cells | Yes vs PP1 | Not reported | Not reported |
|
| |||||||
| Quinolone (direct in the presence of ceramide) |
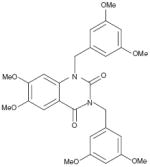
|
400% (20 μM; in the presence of C2-cer) | Phosphorylated myelin basic protein | Purified heterotrimer from Jurkat cells | Yes vs PP1 | Not reported | Not reported |
|
| |||||||
| (-)-epigallocatechin gallate (PP2A methylesterase) |
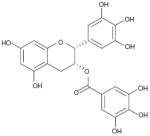
|
129% (100 μM) Antogonistic to OA IC50~80 μM ABC | No effect (p-histoneH1) | AC | Not reported | Not reported | Not reported |
|
| |||||||
| Dihydroxyphenylethano (direct) |

|
500% (400 μM) | Phosphopeptide | Recombinant C | Yes vs PP1 | 780% (400 μM) | HT-29 (synthetic phosphopeptide) |
|
| |||||||
| Methyl-3,5-diiodo-4-(4’-methoxyphenoxy) benzoate (direct) |

|
250% (10 μM) | [32P]-histone H1 | Purified red cell PP2A | Substrate specific, but activates ALP | 300% (10 μM) | Eras 20 ([32P]-histone H1) |
|
| |||||||
| β-tocopherol (direct, synergistic with ceramide, PME) |
|
190% (50 μM) | Phosphorylase a | Purified red cell PP2A | Yes vs PP1 | 500% (50 μM) | A7r5 ([32P]-PKC) |
PP: Phosphoprotein phosphatase.
Exploration of quinolone and tetralone analogs indicate that, depending on substitution pattern, these compounds exhibit three distinct PP2A activities [73]:
-
▪
Inhibition of basal PP2A activity;
-
▪
Inhibition of C2-ceramide activation;
-
▪
Amplification of C2-ceramide activation.
Inhibition of basal PP2A activity by these compounds has been suggested to be driven by a simple switch from methylated to free phenolic moieties. It is, however, likely that other structural features might also be important because similar polyphenols, such as quercetin and genistein, have no inhibitory activity [73]. Remarkably, with the exception of a few PP2A polyhydroxy inhibitors, no other reported quinolone and tetralone derivatives had any effect on PP2A activity in the absence of C2 ceramide, with the majority of methylated phenolic derivatives activating PP2A up to fivefold in the presence of ceramide. 2-(3,4-dihydroxyphenyl) ethanol (DPE) has the least number of structural features required for selective PP2A activation over PP1 [75]. Recombinant C subunit treated with DPE showed dose-dependent activation of PP2A, while there was no effect of DPE on the catalytic subunit of PP1. Methyl-3,5-diiodo-4-(4′-methoxyphenoxy) benzoate (DIME), but not its 4′-propoxy analog has been reported to activate cell-derived PP2A up to 250% with phosphorylated histone as a substrate [74].
Effects of tocopherols (vitamin E) on PP2A are intriguing and complex [76]. There appear to be at least three ways that tocopherols influence PP2A activity:
-
▪
Ceramide-independent activation [76];
-
▪
Ceramide-dependent phosphatase activation [78];
-
▪
Altered states of C-subunit carboxyl methylation through inhibition of methylating and demethylating enzymes [78].
While tocopherols may arguably be similar in molecular shape to sphingoids, and in other structural features to phenolic activators, we include them in the phenolic class because they exhibit cooperativity with ceramide to activate PP2A, similar to tetralones. All these effects of tocopherol on PP2A may have contributed to variable effects of vitamin E treatment in clinical trials for AD [79]. Eicosanoyl-5-hydroxytryptamide (EHT) shares lipid-like and phenol-like properties similar to tocopherols. It appears to activate phosphatase activity by inhibiting interactions with PME1 in vitro and in vivo [77].
Cationic activators
PP2A is activated by basic amines, including protamine (EC50 ~1 μM) and polylysine (EC50 0.2-0.3 μM) [60], which are positively charged at physiological pH (Table 3). While these interactions could, at least to some degree, reflect dependence of PP2A activity on ionic strength [68,66], this class can be further defined by the recent discoveries of positively charged modulators of PP2A such as C6-pyr-ceramide [64], certain ApoE peptide derivatives [201] and memantine [81]. These compounds activate PP2A by blocking its interaction with I2. Memantine reduces tau hyperphosphorylation in vitro by blocking I2 inhibition of PP2A. In the absence of I2, memantine does not activate PP2A, but in the presence of 5-nM I2, memantine restored phosphatase activity towards phosphorylated tau [80]. Memantine’s structure does not offer many clues as to how it interacts with PP2A other than a basic amino group that is charged at physiological pH and highly lypophilic adamantane moiety.
Table 3.
Cationic class PP2A activators.
| Name (proposed mode of action) | Chemical structure | Biochemical assay
|
Cell-based assay
|
||||
|---|---|---|---|---|---|---|---|
| Activity | Substrate | PP2A | Selectivities | Activity | Cell type | ||
| Memantine (I2) |
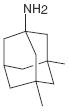
|
0.1 μM reversed I2 inhibition | p-Tau (Ser199) | Brain holoenzyme | Not commented | Rescue at 10 μM | tau441-PC12 transfected with I2 |
|
| |||||||
| C6-pyr-ceramide† (direct, I2) |
|
Rescue (0.25-1 μM; with I2) | Phosphorylated myelin basic protein | Heterotrimer | Inhibition of I2 is stereospecific for D-C6-erythro geometry | c-Myc dephosphorylation/degradation | A546 cells |
Also a member of sphingoids class.
Anionic activators
This class of PP2A activators encompasses xylulose-5-phosphate [81], heparine [60] and sodium selenate (Table 4) [82].
Table 4.
Anionic class activators.
| Name (proposed mode of action) | Chemical structure | Biochemical assay
|
Cell-based assay
|
||||
|---|---|---|---|---|---|---|---|
| Activity | Substrate | PP2A | Selectivities | Activity | Cell type | ||
| Sodium selenate (direct) | Na2SeO4 | 350% (50 μM) | Phosphothreonine peptide | Human AC | Yes vs phosphoprotein phosphatase 1 | Significant p-Tau reduction | BE2M17 and SH-SY5Y |
|
| |||||||
| Xylulose-5-phosphate (direct) |
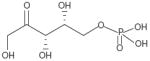
|
220% (10 μM) | RL2K:2P | Heterotrimer (with Bα-isoform) | Substrate-specific, no effect on phosphorylase a or pyruvate kinase | Not reported | Not reported |
Xylulose-5-phosphate, but not other sugar phosphates, was reported to activate PP2A isolated from rat liver [81]. The activation of liver PP2A by xylulose-5-phosphate appears to be part of a specific mechanism to regulate carbohydrate metabolism. Xylulose-5-phosphate is a highly polar anionic molecule whose structural features are very different from other PP2A activators and, therefore, it possibly has a distinct binding site. Heparine, a polymer consisting of repeated highly anionic amino sugar sulfate units, stimulates PP2A phosphatase activity towards phosphorylase A by PP2A [60]. Sodium selenate is another negatively charged anionic compound that activates PP2A in vitro and in vivo [82]. Furthermore, sodium selenate reverses memory deficits and reduces tau phosphorylation in models of AD [82,83]. Sodium selenate appears to reduce phosphorylated tau via PP2A activation in vivo, supporting the proposed role for selenium in AD and cognitive decline [84-86]. PP2A activation by sodium selenate appears to be selective towards phosphorylated tau in vivo and has no apparent adverse effects that might be associated with indiscriminate PP2A activation.
Other potential PP2A activators
More than a dozen small molecules known to activate PP2A in cellular assays have also been identified. While some have structural similarities with the PP2A activators discussed above, they have not been characterized in vitro, and it is unclear whether they act on PP2A directly or indirectly. Among cell-based PP2A activators, the following have been reported: palmitic acid [87], melatonin [88], troglitazone [89], progesterone [90], dithiolethione [91], taurolidine [92], forskolin and related compounds [93,94], 4-hydroxynonenal [95,96], ebelactone B [97], epigallocatechine gallate [98] and 1,8-naphthyridines [99]. The mechanisms underlying the effects of these compounds on PP2A could be variable, for example, thiaziolodinediones (e.g., troglitazone) may act by enhancing PP2A gene expression [100].
General properties & considerations of PP2A activators
Apart from tetralones and ceramides, very few SAR studies have been reported for PP2A activators. Generalizations of structural requirements for different activator chemotypes are based on meta-analyses of data from biochemical assays carried out by different research groups using a range of different conditions, phosphorylated substrates and PP2A isoforms. As PP2A is a ubiquitous regulator of many biological processes, selectivity of small-molecule activators is critical. Studies of PP2A activation have employed a wide range of phosphoprotein, phosphopeptide and small-molecule substrates. The most commonly used substrates, p-nitrophenyl phosphate (pNPP) and phosphorylase A, are not physiologically relevant to PP2A function in neurons. Despite these complications in interpretation, it is clear that there are a number of PP2A-activating compounds and mechanisms indicating the pharmaceutical potential of targeting PP2A for therapeutic intervention in AD.
Most activators share some structural features, such as distinctly lipophilic ridges or side chains with H-bond donors. While it is tempting to interpret some of these properties as defining a binding site, reports suggest that there is likely to be more than one activation site on PP2A [73]. Furthermore, molecules that activate PP2A by blocking the effects of inhibitors such as I2 or PME may not share the same binding sites as direct PP2A activators. With the exception of sodium selenate and xylulose-5-phosphate, physicochemical properties of molecules directly interacting with PP2A are generally defined by a rather narrow range of surface area (29.5 < TPA < 74.6) and high lipophilicity (0.81 < cLogP < 10.51) (Figure 3). These properties indicate that they have reasonable expectations to be brain–blood barrier permeable (LogBBB > -1) [101], and therefore able to reach PP2A in the target compartment in humans.
Figure 3. Physiochemical properties of molecules interacting directly with PP2A.
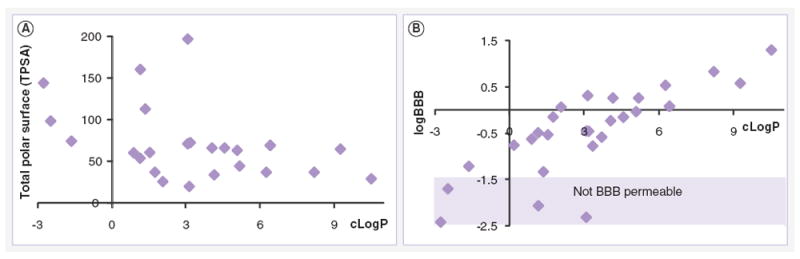
(A) Compound distribution according to total polar surface area, and cLogP. (B) Compound distribution predicts BBB permeability, by LogBBB against cLogP, total polar surface area and cLogP were calculated using JChem software. LogBBB was calculated as in [101].
BBB: Blood–brain barrier.
Future perspective
Activation of PP2A by small molecules offers a new therapeutic strategy to treat AD and other neurodegenerative disorders, including tauopathies such as frontotemporal dementias that may provide more straightforward development paths. While the field of PP2A activators is only beginning to be explored, and well-defined comprehensive and systematic analyses are lacking, the currently available literature offers substantial evidence of the drugability of this target. There are clearly defined mechanisms through which this can be achieved, through direct interaction with PP2A, inhibition of interactions with proteins such as I2, and modulation of post-translational modifications such as carboxyl methylation. Therefore, traditional medicinal chemistry can be deployed to identify PP2A-enhancing molecules. The ubiquitous distribution of PP2A and its broad range of substrates raise safety concerns for the use of pharmaceuticals that target PP2A. Such considerations suggest that, rather than seeking higher specificity and potency, a novel ‘systems pharmacology’ approach may be desirable, employing agents that act in more subtle ways to modulate PP2A function. Moreover, the findings outlined in this review indicate that specificity can be achieved by drugs that perturb the protein–protein interactions that control the balance between different PP2A holoenzyme subtypes. This raises the possibility that selective pharmaceutical activation of PP2A towards specific classes of phosphoprotein substrates may provide safe and efficacious therapeutic opportunities for the prevention and treatment of AD and related neurodegenerative diseases.
Executive summary.
-
▪
Phosphoprotein phosphatase 2A (PP2A) is a target strongly linked through pathological findings and mechanistic relevance to Alzheimer’s disease. Deficiencies in PP2A’s phosphatase activity are associated with multiple pathological hallmarks of the disorder, therefore enhancing its activity is an attractive target for drug development.
-
▪
Tight regulation of PP2A occurs due to its critical roles in cellular function. Mechanisms to modulate its function provide unique opportunities for pharmacological intervention that can upregulate PP2A’s activity. Direct interaction at allosteric sites, disruption of inhibitory protein–protein interactions and modulation of post-translational modifications provide entry points for pharmacological enhancers of PP2A function.
-
▪
Multiple chemical classes of PP2A enhancers have been identified, including sphingoid, phenolic, anionic and cationic classes. Pharmacological tractability of PP2A enhancement is demonstrated by multiple chemotypes being efficacious and selective towards specific PP2A isoforms.
-
▪
Comparison of data across studies is complicated by differences in assay systems, PP2A composition and substrates. A systematic evaluation of chemistries across defined conditions will further illuminate the relative value of identified PP2A modulators.
-
▪
Properties of identified PP2A enhancers indicate that they are generally likely to be able to cross the blood–brain barrier and, thus, able to reach the target compartment for CNS conditions such as Alzheimer’s disease.
-
▪
Demonstrating that PP2A enhancers can be safely, selectively and effectively deployed in the clinic is the next hurdle to prove the value of this mechanism for neurodegenerative disorders.
-
▪
Enhancement of PP2A function has the potential to provide the next generation of Alzheimer’s disease therapeutics acting through a novel mechanism to truly modify the course of disease progression.
Acknowledgments
Michael Voronkov and Steven P Braithwaite are employees of Signum Biosciences Inc. Jeffry B Stock is a director of Signum Biosciences Inc. Michael Voronkov and Steven P Braithwaite are funded in part by NIH SBIR grant 1R43AG035448-01 and a grant from the Alzheimer’s Drug Discovery Foundation. Jeffry B Stock is supported in part by a grant from Rohto Pharmaceuticals to Princeton University.
Glossary
- Alzheimer’s disease
The most common neurodegenerative disorder and cause of dementia with a prevalence of over 26 million cases worldwide
- Tau
Microtubule-associated protein that regulates protein trafficking and neuronal function. Tau dysfunction through hyperphosphorylation is a primary defect in Alzheimer’s disease
- Neurodegeneration
Progressive loss of neurons in the brain through cell death that leads to conditions such as Alzheimer’s disease
- Phosphoprotein phosphatase 2A
The major serine/threonine dephosphorylating enzyme present in the brain
- Ceramides
Sphingolipids present at high concentrations in cells and involved in cellular signaling events
Footnotes
Financial & competing interests disclosure The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
Bibliography
Papers of special note have been highlighted as:
-
▪
of interest
-
▪▪
of considerable interest
- 1.Kidd PM. Alzheimer’s disease, amnestic mild cognitive impairment, and age-associated memory impairment: current understanding and progress toward integrative prevention. Altern Med Rev. 2008;13(2):85–115. [PubMed] [Google Scholar]
- 2.Stewart AJ, Fox A, Morimoto BH, Gozes I. Looking for novel ways to treat the hallmarks of Alzheimer’s disease. Expert Opin Investig Drugs. 2007;16(8):1183–1196. doi: 10.1517/13543784.16.8.1183. [DOI] [PubMed] [Google Scholar]
- 3.Seabrook GR, Ray WJ, Shearman M, Hutton M. Beyond amyloid: the next generation of Alzheimer’s disease therapeutics. Mol Interv. 2007;7(5):261–270. doi: 10.1124/mi.7.5.8. [DOI] [PubMed] [Google Scholar]
- 4.Small SA, Duff K. Linking Aβ and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008;60(4):534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dickey CA, Petrucelli L. Current strategies for the treatment of Alzheimer’s disease and other tauopathies. Expert Opin Ther Targets. 2006;10(5):665–676. doi: 10.1517/14728222.10.5.665. [DOI] [PubMed] [Google Scholar]
- 6.Roder HM, Hutton ML. Microtubule-associated protein tau as a therapeutic target in neurodegenerative disease. Expert Opin Ther Targets. 2007;11(4):435–442. doi: 10.1517/14728222.11.4.435. [DOI] [PubMed] [Google Scholar]
- 7.Schneider A, Mandelkow E. Tau-based treatment strategies in neurodegenerative diseases. Neurotherapeutics. 2008;5(3):443–457. doi: 10.1016/j.nurt.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323(Pt 3):577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alonso AC, Zaidi T, Grundke-Iqbal I, Iqbal K. Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91(12):5562–5566. doi: 10.1073/pnas.91.12.5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li B, Chohan MO, Grundke-Iqbal I, Iqbal K. Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated tau. Acta Neuropathol. 2007;113(5):501–511. doi: 10.1007/s00401-007-0207-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA. 1986;83(13):4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alonso A, Zaidi T, Novak M, Grundke-Iqbal I, Iqbal K. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci USA. 2001;98(12):6923–6928. doi: 10.1073/pnas.121119298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alonso AD, Di Clerico J, Li B, et al. Phosphorylation of tau at Thr212, Thr231, and Ser262 combined causes neurodegeneration. J Biol Chem. 2010;285(40):30851–30860. doi: 10.1074/jbc.M110.110957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mazanetz MP, Fischer PM. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov. 2007;6(6):464–479. doi: 10.1038/nrd2111. [DOI] [PubMed] [Google Scholar]
- 15.Bulic B, Pickhardt M, Khlistunova I, et al. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew Chem Int Ed Engl. 2007;46(48):9215–9219. doi: 10.1002/anie.200704051. [DOI] [PubMed] [Google Scholar]
- 16.Chang E, Congdon EE, Honson NS, Duff KE, Kuret J. Structure–activity relationship of cyanine tau aggregation inhibitors. J Med Chem. 2009;52(11):3539–3547. doi: 10.1021/jm900116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bulic B, Pickhardt M, Schmidt B, Mandelkow EM, Waldmann H, Mandelkow E. Development of tau aggregation inhibitors for Alzheimer’s disease. Angew Chem Int Ed Engl. 2009;48(10):1740–1752. doi: 10.1002/anie.200802621. [DOI] [PubMed] [Google Scholar]
- 18.Ballatore C, Brunden KR, Piscitelli F, et al. Discovery of brain-penetrant, orally bioavailable aminothienopyridazine inhibitors of tau aggregation. J Med Chem. 2010;53(9):3739–3747. doi: 10.1021/jm100138f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trojanowski JQ, Lee VM. Pathological tau: a loss of normal function or a gain in toxicity? Nat Neurosci. 2005;8(9):1136–1137. doi: 10.1038/nn0905-1136. [DOI] [PubMed] [Google Scholar]
- 20.Trojanowski JQ, Smith AB, Huryn D, Lee VM. Microtubule-stabilising drugs for therapy of Alzheimer’s disease and other neurodegenerative disorders with axonal transport impairments. Expert Opin Pharmacother. 2005;6(5):683–686. doi: 10.1517/14656566.6.5.683. [DOI] [PubMed] [Google Scholar]
- 21.Wang JZ, Grundke-Iqbal I, Iqbal K. Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci. 2007;25(1):59–68. doi: 10.1111/j.1460-9568.2006.05226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22▪▪.Iqbal K, Grundke-Iqbal I. Pharmacological approaches of neurofibrillary degeneration. Curr Alzheimer Res. 2005;2(3):335–341. doi: 10.2174/1567205054367810. Decrease in the activity of protein phosphatase PP2A proposed as a cause for the abnormal hyperphosphorylation in Alzheimer’s disease brains. [DOI] [PubMed] [Google Scholar]
- 23.Drewes G. MARKing tau for tangles and toxicity. Trends Biochem Sci. 2004;29(10):548–555. doi: 10.1016/j.tibs.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 24.Haddad JJ. Mitogen-activated protein kinases and the evolution of Alzheimer’s: a revolutionary neurogenetic axis for therapeutic intervention? Prog Neurobiol. 2004;73(5):359–377. doi: 10.1016/j.pneurobio.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Ferrer I, Gomez-Isla T, Puig B, et al. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr Alzheimer Res. 2005;2(1):3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- 26.Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22(8):1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 27.Tian Q, Wang J. Role of serine/threonine protein phosphatase in Alzheimer’s disease. Neurosignals. 2002;11(5):262–269. doi: 10.1159/000067425. [DOI] [PubMed] [Google Scholar]
- 28.Mccluskey A, Sim AT, Sakoff JA. Serine-threonine protein phosphatase inhibitors: development of potential therapeutic strategies. J Med Chem. 2002;45(6):1151–1175. doi: 10.1021/jm010066k. [DOI] [PubMed] [Google Scholar]
- 29.Combs AP. Structure-based drug design of new leads for phosphatase research. IDrugs. 2007;10(2):112–115. [PubMed] [Google Scholar]
- 30.Lavecchia A, Coluccia A, Di Giovanni C, Novellino E. Cdc25B phosphatase inhibitors in cancer therapy: latest developments, trends and medicinal chemistry perspective. Anticancer Agents Med Chem. 2008;8(8):843–856. doi: 10.2174/187152008786847783. [DOI] [PubMed] [Google Scholar]
- 31.Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol. 2001;168(2):402–412. doi: 10.1006/exnr.2001.7630. [DOI] [PubMed] [Google Scholar]
- 32.Sontag E, Luangpirom A, Hladik C, et al. Altered expression levels of the protein phosphatase 2A ABaC enzyme are associated with Alzheimer disease pathology. J Neuropathol Exp Neurol. 2004;63(4):287–301. doi: 10.1093/jnen/63.4.287. [DOI] [PubMed] [Google Scholar]
- 33.Gong CX, Shaikh S, Wang JZ, Zaidi T, Grundke-Iqbal I, Iqbal K. Phosphatase activity toward abnormally phosphorylated tau: decrease in Alzheimer disease brain. J Neurochem. 1995;65(2):732–738. doi: 10.1046/j.1471-4159.1995.65020732.x. [DOI] [PubMed] [Google Scholar]
- 34.Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem. 1993;61(3):921–927. doi: 10.1111/j.1471-4159.1993.tb03603.x. [DOI] [PubMed] [Google Scholar]
- 35.Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am J Pathol. 2005;166(6):1761–1771. doi: 10.1016/S0002-9440(10)62486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang X, Blanchard J, Kohlbrenner E, et al. The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J. 2010;24(11):4420–4432. doi: 10.1096/fj.10-158477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37▪.Sontag E, Hladik C, Montgomery L, et al. Downregulation of protein phosphatase 2A carboxyl methylation and methyltransferase may contribute to Alzheimer disease pathogenesis. J Neuropathol Exp Neurol. 2004;63(10):1080–1091. doi: 10.1093/jnen/63.10.1080. Postmortem evidence linking PP2A demethylation and changes in its heteromultimeric composition and substrate specificity with Alzheimer’s disease pathogenesis. [DOI] [PubMed] [Google Scholar]
- 38.Zhou XW, Gustafsson JA, Tanila H, et al. Tau hyperphosphorylation correlates with reduced methylation of protein phosphatase 2A. Neurobiol Dis. 2008;31(3):386–394. doi: 10.1016/j.nbd.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 39.Arendt T, Holzer M, Fruth R, Bruckner MK, Gartner U. Paired helical filament-like phosphorylation of tau, deposition of β/A4-amyloid and memory impairment in rat induced by chronic inhibition of phosphatase 1 and 2A. Neuroscience. 1995;69(3):691–698. doi: 10.1016/0306-4522(95)00347-l. [DOI] [PubMed] [Google Scholar]
- 40.Sun L, Liu SY, Zhou XW, et al. Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience. 2003;118(4):1175–1182. doi: 10.1016/s0306-4522(02)00697-8. [DOI] [PubMed] [Google Scholar]
- 41.Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24(5):186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- 42.Lee MS, Kao SC, Lemere CA, et al. APP processing is regulated by cytoplasmic phosphorylation. J Cell Biol. 2003;163(1):83–95. doi: 10.1083/jcb.200301115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Colombo A, Bastone A, Ploia C, et al. JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol Dis. 2009;33(3):518–525. doi: 10.1016/j.nbd.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 44.Braithwaite SP, Schmid RS, He DN, et al. Inhibition of c-Jun kinase provides neuroprotection in a model of Alzheimer’s disease. Neurobiol Dis. 2010;39(3):311–317. doi: 10.1016/j.nbd.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ruvolo PP, Deng X, Ito T, Carr BK, May WS. Ceramide induces Bcl2 dephosphorylation via a mechanism involving mitochondrial PP2A. J Biol Chem. 1999;274(29):20296–20300. doi: 10.1074/jbc.274.29.20296. [DOI] [PubMed] [Google Scholar]
- 46.Trinkle-Mulcahy L, Lamond AI. Mitotic phosphatases: no longer silent partners. Curr Opin Cell Biol. 2006;18(6):623–631. doi: 10.1016/j.ceb.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 47.Park KH, Hallows JL, Chakrabarty P, Davies P, Vincent I. Conditional neuronal simian virus 40 T antigen expression induces Alzheimer-like tau and amyloid pathology in mice. J Neurosci. 2007;27(11):2969–2978. doi: 10.1523/JNEUROSCI.0186-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y, Xu Y, Bao Q, et al. Structural and biochemical insights into the regulation of protein phosphatase 2A by small t antigen of SV40. Nat Struct Mol Biol. 2007;14(6):527–534. doi: 10.1038/nsmb1254. [DOI] [PubMed] [Google Scholar]
- 49.Pallas DC, Shahrik LK, Martin BL, et al. Polyoma small and middle T antigens and SV40 small t antigen form stable complexes with protein phosphatase 2A. Cell. 1990;60(1):167–176. doi: 10.1016/0092-8674(90)90726-u. [DOI] [PubMed] [Google Scholar]
- 50.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353(Pt 3):417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lechward K, Awotunde OS, Swiatek W, Muszynska G. Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochim Pol. 2001;48(4):921–933. [PubMed] [Google Scholar]
- 52.Tsujio I, Zaidi T, Xu J, Kotula L, Grundke-Iqbal I, Iqbal K. Inhibitors of protein phosphatase-2A from human brain structures, immunocytological localization and activities towards dephosphorylation of the Alzheimer type hyperphosphorylated tau. FEBS Lett. 2005;579(2):363–372. doi: 10.1016/j.febslet.2004.11.097. [DOI] [PubMed] [Google Scholar]
- 53.Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257(5074):1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- 54.Lee J, Stock J. Protein phosphatase 2A catalytic subunit is methyl-esterified at its carboxyl terminus by a novel methyltransferase. J Biol Chem. 1993;268(26):19192–19195. [PubMed] [Google Scholar]
- 55.Lee J, Chen Y, Tolstykh T, Stock J. A specific protein carboxyl methylesterase that demethylates phosphoprotein phosphatase 2A in bovine brain. Proc Natl Acad Sci USA. 1996;93(12):6043–6047. doi: 10.1073/pnas.93.12.6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xing Y, Li Z, Chen Y, Stock JB, Jeffrey PD, Shi Y. Structural mechanism of demethylation and inactivation of protein phosphatase 2A. Cell. 2008;133(1):154–163. doi: 10.1016/j.cell.2008.02.041. [DOI] [PubMed] [Google Scholar]
- 57▪▪.Tolstykh T, Lee J, Vafai S, Stock JB. Carboxyl methylation regulates phosphoprotein phosphatase 2A by controlling the association of regulatory B subunits. EMBO J. 2000;19(21):5682–5691. doi: 10.1093/emboj/19.21.5682. Changes in methylation indirectly regulate PP2A phosphatase activity by controlling the binding of regulatory B subunits to AC dimers. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu J, Tolstykh T, Lee J, Boyd K, Stock JB, Broach JR. Carboxyl methylation of the phosphoprotein phosphatase 2A catalytic subunit promotes its functional association with regulatory subunits in vivo. Embo J. 2000;19(21):5672–5681. doi: 10.1093/emboj/19.21.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cutler RG, Kelly J, Storie K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101(7):2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pelech S, Cohen P. The protein phosphatases involved in cellular regulation 1 Modulation of protein phosphatases-1 and 2A by histone H1, protamine, polylysine and heparin. Eur J Biochem. 1985;148(2):245–251. doi: 10.1111/j.1432-1033.1985.tb08832.x. [DOI] [PubMed] [Google Scholar]
- 61▪.Dobrowsky RT, Kamibayashi C, Mumby MC, Hannun YA. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem. 1993;268(21):15523–15530. First example of PP2A activation by a small molecule. [PubMed] [Google Scholar]
- 62.Ruvolo PP, Clark W, Mumby M, Gao F, May WS. A functional role for the B56 α-subunit of protein phosphatase 2A in ceramide-mediated regulation of Bcl2 phosphorylation status and function. J Biol Chem. 2002;277(25):22847–22852. doi: 10.1074/jbc.M201830200. [DOI] [PubMed] [Google Scholar]
- 63.Chen CL, Lin CF, Chiang CW, Jan MS, Lin YS. Lithium inhibits ceramide- and etoposide-induced protein phosphatase 2A methylation, Bcl-2 dephosphorylation, caspase-2 activation, and apoptosis. Mol Pharmacol. 2006;70(2):510–517. doi: 10.1124/mol.106.024059. [DOI] [PubMed] [Google Scholar]
- 64.Mukhopadhyay A, Saddoughi SA, Song P, et al. Direct interaction between the inhibitor 2 and ceramide via sphingolipid-protein binding is involved in the regulation of protein phosphatase 2A activity and signaling. FASEB J. 2009;23(3):751–763. doi: 10.1096/fj.08-120550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine-threonine protein phosphatases. J Lipid Res. 2004;45(3):496–506. doi: 10.1194/jlr.M300347-JLR200. [DOI] [PubMed] [Google Scholar]
- 66.Chalfant CE, Kishikawa K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem. 1999;274(29):20313–20317. doi: 10.1074/jbc.274.29.20313. [DOI] [PubMed] [Google Scholar]
- 67.Kamibayashi C, Estes R, Slaughter C, Mumby MC. Subunit interactions control protein phosphatase 2A. Effects of limited proteolysis, N-ethylmaleimide, and heparin on the interaction of the B subunit. J Biol Chem. 1991;266(20):13251–13260. [PubMed] [Google Scholar]
- 68.Cheng Q, Erickson AK, Wang ZX, Killilea SD. Stimulation of phosphorylase phosphatase activity of protein phosphatase 2A1 by protamine is ionic strength dependent and involves interaction of protamine with both substrate and enzyme. Biochemistry. 1996;35(48):15593–15600. doi: 10.1021/bi960709b. [DOI] [PubMed] [Google Scholar]
- 69.Matsuoka Y, Nagahara Y, Ikekita M, Shinomiya T. A novel immunosuppressive agent FTY720 induced Akt dephosphorylation in leukemia cells. Br J Pharmacol. 2003;138(7):1303–1312. doi: 10.1038/sj.bjp.0705182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Q, Zhao X, Frissora F, et al. FTY720 demonstrates promising preclinical activity for chronic lymphocytic leukemia and lymphoblastic leukemia/lymphoma. Blood. 2008;111(1):275–284. doi: 10.1182/blood-2006-10-053884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Habrukowich C, Han DK, Le A, et al. Sphingosine interaction with acidic leucine-rich nuclear phosphoprotein-32A (ANP32A) regulates PP2A activity and cyclooxygenase (COX)-2 expression in human endothelial cells. J Biol Chem. 2010;285(35):26825–26831. doi: 10.1074/jbc.M110.147058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoon CH, Kim MJ, Park MT, et al. Activation of p38 mitogen-activated protein kinase is required for death receptor-independent caspase-8 activation and cell death in response to sphingosine. Mol Cancer Res. 2009;7(3):361–370. doi: 10.1158/1541-7786.MCR-08-0069. [DOI] [PubMed] [Google Scholar]
- 73.Leoni LM, Shih HC, Deng L, et al. Modulation of ceramide-activated protein phosphatase 2A activity by low molecular weight aromatic compounds. Biochem Pharmacol. 1998;55(7):1105–1111. doi: 10.1016/s0006-2952(97)00685-0. [DOI] [PubMed] [Google Scholar]
- 74.Bauer PI, Kirsten E, Kun E. Mechanisms of antitumor action of methyl-3,5-diiodo-4-(4′-methoxyphenoxy)benzoate: drug-induced protein dephosphorylations and inhibition of the permissive action of ceramide on growth factor induced cell proliferation. Oncol Rep. 2005;13(3):465–468. [PubMed] [Google Scholar]
- 75.Guichard C, Pedruzzi E, Fay M, et al. Dihydroxyphenylethanol induces apoptosis by activating serine/threonine protein phosphatase PP2A and promotes the endoplasmic reticulum stress response in human colon carcinoma cells. Carcinogenesis. 2006;27(9):1812–1827. doi: 10.1093/carcin/bgl009. [DOI] [PubMed] [Google Scholar]
- 76.Ricciarelli R, Tasinato A, Clement S, Ozer NK, Boscoboinik D, Azzi A. α-tocopherol specifically inactivates cellular protein kinase C α by changing its phosphorylation state. Biochem J. 1998;334(Pt 1):243–249. doi: 10.1042/bj3340243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kang-Woo Lee WC, Junn E, Grosso H, et al. Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J Neurosci. 2011;31(19):6963–6971. doi: 10.1523/JNEUROSCI.6513-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Li Z, Semmelhack V, Braithwaite SP, Stock JB. Competition between α-tocopherol and ceramide determines protein phosphatase 2A catalytic activity. Presented at Society for Neuroscience Annual Meeting; San Diego, CA, USA. 13–17 November 2010. [Google Scholar]
- 79.Usoro OB, Mousa SA. Vitamin E forms in Alzheimer’s disease: a review of controversial and clinical experiences. Crit Rev Food Sci Nutr. 2010;50(5):414–419. doi: 10.1080/10408390802304222. [DOI] [PubMed] [Google Scholar]
- 80▪.Chohan MO, Khatoon S, Iqbal IG, Iqbal K. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett. 2006;580(16):3973–3979. doi: 10.1016/j.febslet.2006.06.021. Memantine, a clinically prescribed uncompetitive inhibitor of NMDA receptors, inhibits abnormal phosphorylation of tau and cell death and prevents the I2PP2A-induced inhibition of PP2A activity in vitro. [DOI] [PubMed] [Google Scholar]
- 81.Nishimura M, Uyeda K. Purification and characterization of a novel xylulose 5-phosphate-activated protein phosphatase catalyzing dephosphorylation of fructose-6-phosphate,2-kinase:fructose-2,6-bisphosphatase. J Biol Chem. 1995;270(44):26341–26346. doi: 10.1074/jbc.270.44.26341. [DOI] [PubMed] [Google Scholar]
- 82▪▪.Van Eersel J, Ke YD, Liu X, et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc Natl Acad Sci USA. 2010;107(31):13888–13893. doi: 10.1073/pnas.1009038107. Treatment with sodium selenate, an orally bioavailable PP2A activator, reduces tau hyperphosphorylation, completely abrogates neurofibrillary tangle formation, improves contextual memory and motor performance, and prevents neurodegeneration in tau transgenic mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Corcoran NM, Hovens CM, Michael M, Rosenthal MA, Costello AJ. Open-label, Phase I dose-escalation study of sodium selenate, a novel activator of PP2A, in patients with castration-resistant prostate cancer. Br J Cancer. 2010;103(4):462–468. doi: 10.1038/sj.bjc.6605798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Benton D. Selenium intake, mood and other aspects of psychological functioning. Nutr Neurosci. 2002;5(6):363–374. doi: 10.1080/1028415021000055925. [DOI] [PubMed] [Google Scholar]
- 85.Akbaraly TN, Hininger-Favier I, Carriere I, et al. Plasma selenium over time and cognitive decline in the elderly. Epidemiology. 2007;18(1):52–58. doi: 10.1097/01.ede.0000248202.83695.4e. [DOI] [PubMed] [Google Scholar]
- 86.Cardoso BR, Ong TP, Jacob-Filho W, Jaluul O, Freitas MI, Cozzolino SM. Nutritional status of selenium in Alzheimer’s disease patients. Br J Nutr. 2009;103(6):803–806. doi: 10.1017/S0007114509992832. [DOI] [PubMed] [Google Scholar]
- 87.Wu Y, Song P, Xu J, Zhang M, Zou MH. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282(13):9777–9788. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 88.Yang X, Yang Y, Fu Z, et al. Melatonin ameliorates alzheimer-like pathological changes and spatial memory retention impairment induced by calyculin A. J Psychopharmacol. 2010 doi: 10.1177/0269881110367723. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 89.Cho DH, Choi YJ, Jo SA, et al. Troglitazone acutely inhibits protein synthesis in endothelial cells via a novel mechanism involving protein phosphatase 2A-dependent p70 S6 kinase inhibition. Am J Physiol Cell Physiol. 2006;291(2):C317–C326. doi: 10.1152/ajpcell.00491.2005. [DOI] [PubMed] [Google Scholar]
- 90.Liu B, Arbogast LA. Progesterone decreases tyrosine hydroxylase phosphorylation state and increases protein phosphatase 2A activity in the stalk-median eminence on proestrous afternoon. J Endocrinol. 2010;204(2):209–219. doi: 10.1677/JOE-09-0335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Switzer CH, Ridnour LA, Cheng RY, et al. Dithiolethione compounds inhibit Akt signaling in human breast and lung cancer cells by increasing PP2A activity. Oncogene. 2009;28(43):3837–3846. doi: 10.1038/onc.2009.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Aceto N, Bertino P, Barbone D, et al. Taurolidine and oxidative stress: a rationale for local treatment of mesothelioma. Eur Respir J. 2009;34(6):1399–1407. doi: 10.1183/09031936.00102308. [DOI] [PubMed] [Google Scholar]
- 93.Neviani P, Santhanam R, Trotta R, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8(5):355–368. doi: 10.1016/j.ccr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 94.Perrotti D, Neviani P. Protein phosphatase 2A (PP2A), a drugable tumor suppressor in Ph1(+) leukemias. Cancer Metastasis Rev. 2008;27(2):159–168. doi: 10.1007/s10555-008-9119-x. [DOI] [PubMed] [Google Scholar]
- 95.Nakashima I, Liu W, Akhand AA, et al. 4-hydroxynonenal triggers multistep signal transduction cascades for suppression of cellular functions. Mol Aspects Med. 2003;24(4-5):231–238. doi: 10.1016/s0098-2997(03)00018-9. [DOI] [PubMed] [Google Scholar]
- 96.Liu W, Akhand AA, Takeda K, et al. Protein phosphatase 2A-linked and -unlinked caspase-dependent pathways for downregulation of Akt kinase triggered by 4-hydroxynonenal. Cell Death Differ. 2003;10(7):772–781. doi: 10.1038/sj.cdd.4401238. [DOI] [PubMed] [Google Scholar]
- 97.Kowluru A, Seavey SE, Rabaglia ME, Nesher R, Metz SA. Carboxylmethylation of the catalytic subunit of protein phosphatase 2A in insulin-secreting cells: evidence for functional consequences on enzyme activity and insulin secretion. Endocrinology. 1996;137(6):2315–2323. doi: 10.1210/endo.137.6.8641181. [DOI] [PubMed] [Google Scholar]
- 98.Kitano K, Nam KY, Kimura S, Fujiki H, Imanishi Y. Sealing effects of (-)-epigallocatechin gallate on protein kinase C and protein phosphatase 2A. Biophys Chem. 1997;65(2-3):157–164. doi: 10.1016/s0301-4622(96)02254-5. [DOI] [PubMed] [Google Scholar]
- 99.De Los Rios C, Egea J, Marco-Contelles J, et al. Synthesis, inhibitory activity of cholinesterases, and neuroprotective profile of novel 1,8-naphthyridine derivatives. J Med Chem. 2010;53(14):5129–5143. doi: 10.1021/jm901902w. [DOI] [PubMed] [Google Scholar]
- 100.Altiok S, Xu M, Spiegelman BM. PPARγ induces cell cycle withdrawal: inhibition of E2F/DP DNA-binding activity via down-regulation of PP2A. Genes Dev. 1997;11(15):1987–1998. doi: 10.1101/gad.11.15.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Norinder U, Haeberlein M. Computational approaches to the prediction of the blood-brain distribution. Adv Drug Deliv Rev. 2002;54(3):291–313. doi: 10.1016/s0169-409x(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 201.Vitek MPC, Dale J, Oddo J. US0144627. 2010