

Decreased diffusion anisotropy correlated with decreased compound action potential. Decreased axial diffusivity correlated with histology determined axonal injury. Increased radial diffusivity correlated with histology determined myelin damage.
Abstract
Purpose.
This study evaluated the function of mouse optic nerves after transient retinal ischemia using in vitro electrophysiologic recordings of compound action potentials (CAPs) correlated with diffusion tensor imaging (DTI) injury markers with confirmation by immunohistochemistry-determined pathology.
Methods.
Retinal ischemia was induced in 7- to 8-week-old female C57BL/6 mice by elevating intraocular pressure to 110 mm Hg for 60 minutes. At 3 and 7 days after retinal ischemia, optic nerves were removed for CAP measurements. The CAP amplitude was recorded using suction electrodes in isolated control and injured optic nerves followed by ex vivo DTI evaluation. After DTI, optic nerves were embedded in paraffin and cut for immunohistochemical analyses.
Results.
Consistent with previous in vivo DTI measurements, a 25% decrease in axial diffusivity with normal radial diffusivity was seen at 3 days after retinal ischemia, suggesting axonal injury without myelin damage. At 7 days, there was no additional change in axial diffusivity compared with that at 3 days, but radial diffusivity significantly increased by 50%, suggestive of significant myelin damage due to sustained axonal injury. The relative anisotropy (RA) progressively decreased after retinal ischemia when compared with that of the controls. The CAP amplitude in injured nerves also progressively decreased after retinal ischemia, which correlated with the reduced RA (r = 0.80).
Conclusions.
This study suggests that CAP amplitude reflects both axonal and myelin integrity and RA is an optimal parameter for functional assessment compared with axial or radial diffusivity alone in murine optic nerves after retinal ischemia.
Optic nerve dysfunction is a common complication in both neuroinflammatory and neurodegenerative diseases such as multiple sclerosis1 and glaucoma.2 In multiple sclerosis, optic neuritis is caused by an autoimmune response with symptoms including sudden blurred or vision loss and eye movements accompanied by pain. In addition to inflammatory changes, axonal or myelin damage of the optic nerve is also often observed.3 Glaucoma is a leading cause of blindness as a result of progressive degeneration of the retinal ganglion cell (RGC) and its axons.4 The mechanism underlying RGC loss has not been clearly elucidated.4 Recent histologic studies have provided evidence of early axonal damage in an animal model of glaucoma, highlighting the neuropathy and axonopathy paradox.5–7 Although current clinical management includes imaging of the optic disc/retinal nerve fiber layer to provide an early diagnosis of glaucoma before visual field defects,8–11 these managements may not be sufficient for early assessment of RGC axon damage.
Few in vivo techniques are available to characterize the early changes in optic nerve axons that correlate with long-term vision recovery in optic neuritis or glaucomatous optic neuropathy. The versatility of magnetic resonance imaging (MRI) provides an opportunity to quantify the optic nerve pathology noninvasively. Among MRI methods, diffusion tensor imaging (DTI)12 has been widely used to study the underlying pathology of CNS white matter injury since its inception.13–16 As a pure white matter tract, the optic nerve represents an ideal model for pathophysiologic studies in response to CNS injuries for evaluating DTI-derived parameters.14,17,18 Retinal ischemic injury to the optic nerve has previously been shown to elicit a progressive pattern of neuropathology with axonal injury followed by myelin degeneration.14,19,20 DTI-derived directional diffusivities have been proposed to serve as biomarkers of both axon and myelin injury.15 Specifically, decreased axial diffusivity (λ‖, describing diffusion parallel to the axonal tracts) has been demonstrated to reflect axonal injury,21 whereas the increased radial diffusivity (λ⊥, describing diffusion perpendicular to the axonal tracts) correlates with the myelin damage.22 In addition to directional diffusivities, DTI-derived relative anisotropy (RA) or fractional anisotropy and mean diffusivity have also been shown as sensitive markers of white matter injury.23,24 In spite of the advances in the use of DTI to assess white matter injury, it is still unknown how DTI parameters correlate with functional changes within white matter tracts.
As a robust functional measurement, electrophysiologically recorded compound action potentials (CAPs) have shown great promise in investigating cellular mechanisms underlying white matter pathology in isolated rodent optic nerves.17,25,26 CAPs recorded in vitro in isolated optic nerve represent a summated response of all unitary action potentials evoked by electrical stimulation of individual axons within the myelinated nerve bundle.17 Thus, the maximal CAP amplitude represents the total excitatory response of all axons capable of generating an action potential. A decrease in CAP amplitude is ascribed primarily to a reduction in the number of normally conducting axons. Decreased peak CAP velocity may result from decrements in conductivity of unitary action potentials in individual axons and/or from reduced action potential synchronicity within the nerve bundle. Both decrements may be due to selective axonal and/or myelin damage.27–29 Thus, CAP measurement is an effective functional end point of neuronal fiber tract integrity.
We hypothesized that the loss of structural integrity in the optic nerve would result in diminishing functional responses reflected as a reduced CAP response. To prove this hypothesis, we measured in vitro CAP changes in optic nerves from mice with transient (1 hour) retinal ischemia, followed by DTI and postimaging immunohistochemical staining. We demonstrate that decrements in CAP amplitudes correlated with optic nerve integrity as measured by DTI indices and immunohistochemical staining at 3 days (axonal injury without myelin degeneration14,19,20) and 7 days (the coexistence of axon and myelin damage14,20) after retinal ischemia.
Methods
Animal Model
Seven- to 8-week-old female C57BL/6 mice (n = 24; Jackson Laboratory, Bar Harbor, ME) were used in the study. All procedures were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the ARVO statement on the Use of Animals in Ophthalmic Research and approved by the Loma Linda University Institutional Animal Care and Use Committee. Retinal ischemia was induced in mice anesthetized with intraperitoneal injection of a cocktail containing ketamine (87 mg/kg) and xylazine (13 mg/kg). The anterior chamber of the right eye was cannulated with a 32-gauge needle attached to a saline-filled reservoir raised above the animal to increase intraocular pressure to 110 mm Hg for 60 minutes. Animal body temperature was maintained at 37°C and monitored using a small animal heating and monitoring system (SA Instruments, Stony Brook, NY). At the end of the ischemic period, the needle was removed from the anterior chamber, allowing reperfusion to the retina.
Based on our previous findings, two postischemic time points were selected representing two distinct time points after injury: (1) 3 days after retinal ischemia, where only axonal damage is observed without myelin damage; and (2) 7 days after retinal ischemia, where both axonal injury and myelin damage have been reported.19,20 Mice were divided into three groups (n = 8 each), where two groups underwent retinal ischemia and optic nerves were removed either at 3 or at 7 days later. The third group of mice did not undergo ischemia and served as controls (at 3 and 7 days).
Electrophysiology
Mice were deeply anesthetized with 3.5% isoflurane and decapitated. Mouse heads were chilled with ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl, 130; KCl, 3.5; KH2PO4, 1.25; MgSO4, 1.5; CaCl2, 2.0; NaHCO3, 24; and glucose, 10. The ACSF was saturated with carbogen (95% O2 + 5% CO2) with pH of 7.4. An incision was made posterior to eyeballs (outside the skull). Skulls were then cut along the midline and opened to remove the brain. Both nerves from the ischemic and nonischemic eyes were pulled back from their optic canals while they remained attached at the level of optic chiasm. Optic chiasm was then cut free from the brain and optic nerves (still attached) were then placed into an incubation chamber for approximately 20 minutes at 25°C before being transferred into a recording chamber superfused with ACSF at a flow rate of 2 to 3 mL/min. Nerves were allowed to equilibrate for at least 20 minutes before electrophysiologic assessment, allowing the cut ends of the axons to heal, which was later confirmed by the presence of biphasic CAP.30 The nerve from the nonischemic eye was used to handle and to position the ischemic nerve into the electrode system, thereby minimizing mechanical damage to the ischemic nerve due to manipulation. After the ischemic nerve was placed into the stimulation suction electrode for recording (see the following text), the nonischemic nerve was cut free from the optic chiasm and discarded. We opted not to remove the dural sheets from the nerves31 because this procedure would likely further injure the axons, thus preventing comparison of subtle axonal or myelin changes. Optic nerves from control, nonischemic animals underwent the identical procedures.
The rostral end (approximately 1 mm long) of the nerve was inserted loosely into a glass custom-made stimulating suction electrode filled with ACSF. The electrode was connected through Ag/AgCl electrode system to a stimulus isolation unit (AMPI, Jerusalem, Israel) and pulses were generated using a commercial timer (Master 8; AMPI). The caudal end (∼1 mm long) of the optic nerve was tightly sucked into the recording electrode. The resistance of the resultant connection between the recording electrode and bath solution was approximately 1 MΩ. All recordings were orthodromic and the interelectrode distance was set to approximately 1 mm. The recording suction electrode was attached to a headstage connected to a commercial amplifier (Axoscope 2A; Axon Instruments, Foster City, CA). The voltage traces were digitized and stored on a PC for off-line analyses using commercial software (Clampfit pClamp 9.20; Axon Instruments). The evoked CAPs were monitored for at least 20 minutes to assess stability of the preparation. In our preparation, the excised nerve length varied slightly. Thus, the exact origin of CAP initiation within the stimulation electrode was unknown, and despite the carefully controlled interelectrode distance we observed variability in the CAP latency. In contrast, CAP amplitude is less susceptible to this variability and, as such, we chose to assess CAP amplitude as the functional outcome for correlating with DTI structural measures.
Input–output (I/O) curves were constructed to determine the optic nerve excitability profile. Incremental stimulation intensity using constant-current pulses (0.05 ms), ranging from 0.5 to 10 mA, were applied to determine the threshold, half-maximal, and the maximal amplitude of the evoked CAP. On completion of the electrophysiologic recordings the nerves were fixed in 4% PBS-buffered paraformaldehyde for subsequent DTI assessment.
MRI
MRI of control and ischemic optic nerves was performed using a 4.7-T small animal scanner (Varian; Agilent Technologies, Santa Clara, CA) using a custom-built solenoid coil to transmit and receive. Optic nerves were inserted into small tubing filled with PBS sealed with wax. A Stejskal–Tanner spin-echo diffusion-weighted sequence32 was used to acquire diffusion-weighted images with the following parameters: repetition time, 1 second; spin-echo time 55 ms; duration of the time between application of gradient pulses (Δ), 45 ms; duration of the diffusion gradients (δ), 4 ms; slice thickness, 0.5 mm; field of view, 0.5 × 0.5 cm2; and data matrix, 96 × 96 (zero filling 192 × 192). Diffusion sensitizing gradients were applied in six directions: (Gx, Gy, Gz) = (1, 1, 0), (1, 0, 1), (0, 1, 1) (−1, 1, 0), (0, −1, 1), and (1, 0, −1), and diffusion factors, b-values, were 0 and 1805 s/mm2. The total acquisition time of DTI data collection was 20 minutes.
Data Analysis: Electrophysiology and DTI
The CAP amplitude was evaluated by determining the voltage difference between the baseline (0 mV), measured at the beginning of the stimulation artifact, and the most positive peak of the CAP. I/O curves were generated and fitted to a nonlinear Boltzmann sigmoid model. At maximal SI, the CAP peak often overlapped with the stimulation artifact, which obstructed the precise quantification of maximal CAP amplitude. However, the half-maximal CAP amplitudes derived from I/O curves (Prism 4.02; GraphPad Software, San Diego, CA) proved to be the most consistent measure of nerve excitability and, thus, this end point was used to compare differences between groups as well as for correlations with the DTI data.
The six independent elements of the diffusion tensor were calculated from diffusion-weighted images. The eigenvalues (λ1, λ2, and λ3) of the diffusion tensor were derived by matrix diagonalization.33 On a pixel-by-pixel basis, DTI indices including axial diffusivity (λ‖), radial diffusivity (λ⊥), and RA were derived using software written in a programming software (Matlab; The MathWorks, Natick, MA) as we have described previously.20 Regions of interest (ROIs; Fig. 1, black circle) were chosen near the center of optic nerves based on the image contrast of DTI indices maps to avoid partial volume effects from the surrounding PBS (Fig. 1).
Figure 1.
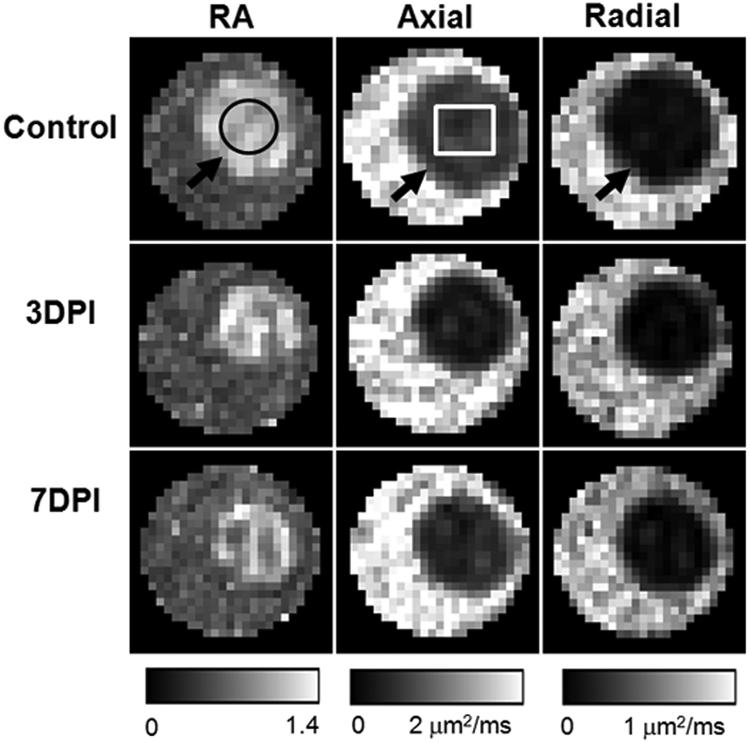
DTI-derived RA, axial diffusivity, and radial diffusivity of control (top row), 3 days postischemia (DPI) (middle row), and 7DPI (bottom row) experiment groups. The optic nerve (black arrows) was embedded in a tube filled with PBS. The optic nerve was readily differentiated from the PBS in all DTI parameter maps, which simplified region of interest (ROI) placement. The black circle is the ROI for data analysis. The white rectangle signifies the region for quantitative immunohistochemistry staining.
Immunohistochemistry
After DTI acquisition, the optic nerves were fixed and embedded in paraffin. Transverse 3-μm-thick sections matching the part of the optic nerve used for DTI maps were obtained for immunohistochemical examination. Sections were deparaffinized and rehydrated. Antigen retrieval was performed by incubating sections in 1 mM EDTA in a water bath at 95–100°C. To prevent nonspecific binding of antibodies, sections were blocked in 2% blocking buffer (Invitrogen, Carlsbad, CA) for 1 hour at room temperature. Sections were then incubated at 4°C overnight with primary antibodies, including mouse anti–myelin basic protein (anti-MBP; 1:1000; Abcam, Cambridge, UK) to assess myelin integrity, mouse anti-phosphorylated neurofilament H (SMI-31; 1:2000; Sternberger Monoclonals, Lutherville, MD) to assess the distribution of intact axons, mouse anti-nonphosphorylated epitope in neurofilament H (SMI-32; 1:2000; Sternberger Monoclonals) to assess the distribution of injured axons, or mouse anti-panaxonal neurofilament (SMI-312; 1:400; Sternberger Monoclonals) to assess the distribution of the total axons including both normal and injured axons. After rinsing in PBS, goat anti-mouse IgG conjugated with cyanine 3 (1:1000; Jackson ImmunoResearch, West Grove, PA) was used to visualize the immunoreaction. After washing, sections were coverslipped (Vectashield Mounting Medium, with 4′, 6′-diamidino-2-phenylindole; Vector Laboratories, Inc., Burlingame, CA).
Immunostained sections were examined with a fluorescence microscope equipped with a ×60 oil objective (Eclipse 80i, Nikon Instruments Inc., Melville, NY), and images were captured with a black–white charge-coupled device camera using image-capture and analysis software (MetaMorph; Universal Imaging Corp., Downingtown, PA). The absolute number of axons stained with SMI-31, SMI-32, SMI-312, and integral myelin sheath labeled with MBP were counted by image-analysis software (CellC; http://www.cs.tut.fi/sgn/csb/cellc/).34
Statistics
All statistical analyses were performed using statistical analysis software (SAS Software; SAS Institute, Cary, NC).
Quantitative data are expressed as mean ± SD. For comparisons between two experimental groups, the significance of the difference between the means was calculated. One-way ANOVA was used to compare data of electrophysiology, DTI, and immunohistochemistry of optic nerves from mice at control and 3 and 7 days after retina ischemia. Statistical significance was accepted at P < 0.05. Spearman's rank correlation was used to test for the presence of monotone increasing or decreasing association between electrophysiologic, DTI, and immunohistochemical measures.
Results
The optic nerve was readily differentiated from the surrounding PBS on all DTI parameter maps (Fig. 1). A 25% decrease in axial diffusivity at 3 days after retinal ischemia (days postischemia [DPI]): 3DPI; Table 1) was observed when compared with controls. No further changes in axial diffusivity were detected at 7DPI, compared with those measured at 3DPI. There was no difference in radial diffusivity between controls and ischemic nerves at 3DPI. However, the radial diffusivity was significantly increased by 50% at 7DPI compared with that at 3DPI, suggestive of myelin damage at 7DPI. A progressive trend in decreased RA (Table 2) was also observed.
Table 1.
Measures of Axon and Myelin Integrity Measured by DTI and Histology
| Measure | Axon |
Myelin |
||||
|---|---|---|---|---|---|---|
| Axial Diffusivity (μm2/ms) | SMI-31 | SMI-32 | SMI-312 | Radial Diffusivity (μm2/ms) | MBP | |
| Control | 0.59 ± 0.06 | 5900 ± 800 | 0 ± 0 | 5900 ± 400 | 0.08 ± 0.1 | 5300 ± 900 |
| 3DPI | 0.45 ± 0.07* | 3000 ± 500† | 500 ± 200† | 3400 ± 400† | 0.09 ± 0.1 | 5400 ± 400 |
| 7DPI | 0.45 ± 0.06* | 1600 ± 600† | 1100 ± 300† | 2900 ± 800† | 0.12 ± 0.1† | 3200 ± 600† |
Data are presented as mean ± SD (n = 8 for DTI measurement, n = 5 for immunohistochemistry analysis).
P < 0.05;
P < 0.005, compared with controls.
Table 2.
DTI and Electrophysiology Measures of White Matter Integrity
| Measure | RA | 50% Amplitude (mV) |
|---|---|---|
| Control | 0.98 ± 0.03 | 12.80 ± 2.48 |
| 3DPI | 0.85 ± 0.03† | 8.40 ± 1.80* |
| 7DPI | 0.72 ± 0.07† | 5.61 ± 1.92† |
Data are presented as mean ± SD (n = 8 for DTI measurement, n = 5 for immunohistochemistry analysis).
P < 0.05;
P < 0.005, compared with controls.
Centers of the optic nerves corresponding to DTI maps (Fig. 1, white rectangle) were selected for immunohistochemical analyses of axonal and myelin changes. Staining of axons with SMI-31 (Fig. 2a), SMI-32 (Fig. 2b), SMI-312 (Fig. 2c), and myelin with MBP antibodies (Fig. 2d) clearly demonstrated axonal injury and myelin damage. Consistent with previous observations,14,19,20 we observed axonal injury, with a progressive decrease in SMI-31 staining of intact axons at 3DPI (3000 ± 500) and 7DPI (1600 ± 600), compared with those in controls (5900 ± 800) (Table 1). In accordance with the decreased numbers of axons, we observed a significantly (P < 0.005) increased number of injured axons using SMI-32 staining at 3 and 7DPI (500 ± 200 and 1100 ± 300, respectively). Control optic nerves did not exhibit any SMI-32 staining (0 ± 0) (Table 1). In control nerves, these immunohistochemical data confirmed that the isolation procedures and subsequent incubation of the specimens for electrophysiology did not compromise axonal integrity. However, in ischemic nerves at 3DPI the total number of neurofilaments labeled by the SMI-312 antibody was significantly reduced (3400 ± 400), compared with that of controls (5900 ± 400; Table 1, P < 0.005), suggesting axonal loss already being present at 3DPI. At 7DPI, the total neurofilament number (2900 ± 800) was not decreased and was comparable with those at 3DPI. MBP staining at 3DPI was comparable to that in controls. At 7DPI, MBP staining significantly decreased, reflecting myelin degeneration after axonal injury and loss. These results support our previously published in vivo DTI measurements that showed a significant axonal injury at 3 days but without myelin damage.20
Figure 2.

Immunohistochemistry revealed changes of intact axons staining with SMI-31 (a), injured axons with SMI-32 (b), total axon content with SMI-312 (c), and myelinated axons using MBP (d). With the progress of the injury, there were decreased numbers of intact axons labeled by SMI-31 in 3 and 7DPI compared with the control group (a, P < 0.005). Injured axons in the retina ischemia optic nerves were labeled with SMI-32. In the control optic nerve, no positive SMI-32 staining was seen, suggesting no injured axons in the control optic nerve. However, SMI-32 staining increased in 7DPI compared with that at 3DPI (b, P < 0.005). The total neurofilaments labeled by SMI-312 in 3DPI optic nerves were significantly decreased compared with control optic nerves (c, P < 0.005). The total neurofilaments at 7DPI are comparable to those at 3DPI. MBP staining in 3DPI was comparable with that in controls and were significantly higher than those at 7DPI (d, P < 0.05). Scale bar: 10 μm.
A typical evoked CAP response in the mouse optic nerve is shown in Figure 3b. The half-maximal amplitude (CAP 50%) of the CAP was used to compare optic nerve excitability between groups and to correlate with DTI measurements (Fig. 3c, Table 2). Incrementally increasing stimulation resulted in increased CAP amplitudes until they reached maximal CAP of a plateau (Fig. 3c). A significant reduction of the average half-maximal CAP (as well as the maximal CAP) amplitude was observed at 3DPI (8.40 ± 1.80 mV), compared with that of controls (12.80 ± 2.48 mV, P < 0.05). The half-maximal CAP amplitudes from injured nerves were further significantly decreased at 7DPI (5.61 ± 1.92 mV), consistent with corresponding DTI measurements (Table 2). The trend of decreased CAP amplitude over our range of stimulation intensities in injured nerves demonstrates progressive functional impairment, possibly due to reduced numbers of functional axons at 3DPI and/or reduced conductivity of the remaining partially myelinated axons at 7DPI. This progressive, time-dependent trend was similar to the trend of reduced RA determined by DTI (Table 2). Indeed, a significant correlation between half-maximal CAP amplitude and RA was seen (r = 0.80, P < 0.0001; Fig. 4). A similar correlation between RA and maximal amplitude of CAP (r = 0.81, P < 0.0001; data not shown) was also found.
Figure 3.

Electrophysiologic measurements of CAP were performed using suction electrodes. (a) The optic nerve was loosely inserted into a glass stimulating electrode (left) and tightly “sucked” into the recording electrode. (b) A typical CAP trace where the amplitude was determined as the voltage difference between the baseline (dotted line) at the beginning of the stimulation artifact (*) and the most positive peak of the trace. (c) Input/output curves of CAP amplitude from increasing stimulation intensities, where the CAP amplitude gradually increased until maximal amplitudes in each group reached a plateau. The 50% amplitude as well as the maximal CAP amplitudes was significantly decreased at 3DPI. Further significant decrements were observed at 7DPI.
Figure 4.

The correlation between RA and 50% amplitude of CAP (r = 0.80, P < 0.0001). A significant correlation between RA and maximal amplitude of CAP (r = 0.81, P < 0.0001) was also observed.
The relationship between half-maximal CAP amplitudes and SMI-31– (r = 0.58, P < 0.05) and SMI-312–positive (r = 0.78, P < 0.001) axon counts revealed a significant positive correlation (Table 3). In contrast, the increased number of SMI-32–stained axons negatively correlated with the half-maximal CAP amplitude (r = −0.52, P < 0.05). The MBP-positive axon counts also negatively correlated with the half-maximal CAP amplitude (r = 0.27, P < 0.5).
Table 3.
Correlations between Immunohistochemical, half-CAP Amplitudes, and Axial and Radial Diffusivities
| Factor | SMI-31 | SMI-32 | SMI-312 | MBP |
|---|---|---|---|---|
| 50% amplitude | r = 0.56, P < 0.005 | r = −0.66, P < 0.005 | r = 0.8, P < 0.005 | r = 0.27, P < 0.5 |
| Axial diffusivity | r = 0.57, P < 0.05 | r = −0.52, P < 0.05 | r = 0.78, P < 0.001 | N/A |
| Radial diffusivity | N/A | N/A | N/A | r = −0.71, P < 0.005 |
n = 8 for DTI measurement; n = 5 for immunohistochemistry analysis. N/A, not applicable.
Discussion
In the present study, the evolving optic nerve injury induced by retinal ischemia was examined by DTI in correlation with electrophysiologic and immunohistochemical measures. Consistent with our previous DTI findings, axial diffusivity at 3DPI was significantly decreased compared with that of controls, reflecting axonal injury followed by increased radial diffusivity at 7DPI consistent with myelin damage.20 The CAP electrophysiologic assessments confirmed the functionally progressive impairment at 3 and 7 days that correlated well with decreased RA, a structural integrity measure, from our DTI measurements. Our results suggest that optic nerve function is likely impaired by cumulative mechanisms, initially by axonal damage then followed by deterioration of their myelin sheaths.35 Importantly, decreased diffusion anisotropy (RA) may optimally reflect optic nerve function after retinal ischemia.
Our measured axial and radial diffusivity substantiated previous in vivo DTI reports in detecting and differentiating axonal injury and myelin damage in the retinal ischemia mouse model.14,19,20 However, axial diffusivity (0.59 ± 0.06 μm2/ms) and radial diffusivity (0.08 ± 0.01 μm2/ms) of controls in our study were smaller than those in a previously reported ex vivo study (axial diffusivity: 0.74 ± 0.12 μm2/ms; radial diffusivity: 0.1 ± 0.04 μm2/ms).20 In the present study, optic nerve DTI was acquired with 52 × 52 μm2 in-plane resolution compared with 117 × 117 μm2 by Sun et al.20 The ROI in our study was placed in the middle of the optic nerves (Fig. 1, black circle) to avoid the PBS contamination, whereas the rectangular ROI extended to the circumference of the optic nerve in the study of Sun et al., resulting in partial volume effects that may account for the observed differences between the studies. In addition, the previous study by Sun et al. did not take into account the diffusion effect of the imaging gradients, where diffusion effects of all gradients were considered in the present study. Although these effects are expected to be minimal, the higher diffusivity observed by Sun and colleagues resulting from the imaging gradient effects cannot be ruled out.
Visual evoked potentials (VEPs) have been widely used clinically to evaluate visual function by measuring action potential conduction along the visual pathway. Decreased VEP amplitude has been reported to reflect axonal dysfunction, whereas a decrease in conduction velocity or an increase in peak latency suggested myelin damage.36,37 Studies correlating DTI parameters with VEP amplitude have provided a new approach to evaluate the capabilities of DTIs to reflect the axonal conductivity38 and to predict clinical outcome.39,40 However, VEP does not specifically localize or distinguish the underlying optic nerve pathology.
Unlike VEPs recorded in vivo, which reflect the integrity of the whole visual pathway from the retina to the primary visual cortex,36 the CAP measured in vitro from isolated optic nerves evaluates axonal conductance of only the optic nerve.17
The CAP reflects the synchronous transduction of neuronal impulses (action potentials) along the nerve fibers. Reduced CAP amplitude may be caused by several processes, including: (1) reduced amplitude of unitary action potentials in all affected fibers (e.g., by axonal membrane damage and/or ionic imbalance), (2) a complete absence of action potential conductivity in a portion of axons, (3) a loss of axons,37 and/or (4) myelin damage.38 An initial approximately 30% decrease in CAP was observed at 3DPI, where immunohistochemical data revealed both axonal loss (43%) and axonal injury. These combined results (electrophysiology, immunohistochemistry) suggested that the impairment of optic nerve function at 3DPI resulted from both axonal loss (e.g., loss of most sensitive axonal population) and/or partial axonal injury of more resistant axonal populations. A further approximately 30% decrease in the CAP amplitude was observed at 7DPI. Although no further axonal loss was observed (SMI-312 staining), a significant increase in the number of injured axons was observed at 7DPI (compared with that at 3DPI). Thus, increased axonal injury likely contributed to the further reduction of the CAP amplitude at 7DPI. Alternatively, the decrease in MBP staining observed at 7DPI, which reflects myelin damage, could also cause an asynchronous conduction of action potentials, resulting in an overall decrease of CAP amplitude.38 Thus, the progressive reduction of the CAP amplitude (5.61 ± 1.92 mV) at 7DPI was likely a combined and synergistic effect of axonal injury and myelin damage.
The electrophysiologic recordings using suction electrodes in this study were performed similar to procedures reported by Stys et al.,39 with minor modifications. In contrast to previously published data by Evans and colleagues,31 we did not observe multiple CAP peaks that reflect axonal action potentials with different conduction velocities. These differences may be due to the short interelectrode distance (∼1 mm) in our experiments as well as the fact that we did not remove the epineurium, an approach we opted for to minimize the mechanical disturbance. We argue that such manipulation may trigger additional axonal damage and prevent quantitative comparisons between experimental groups.
The cumulative damage triggered by two pathologic mechanisms (axonal injury followed by myelin loss) is supported by the progressive decrease in RA. In white matter, water diffuses preferentially along the fiber tracts, whereas the diffusion perpendicular to the fiber tracts is hindered by the axonal membrane and myelin sheath. White matter integrity is often assessed by diffusion anisotropy. As a scalar measure, RA is the quantitative index calculated from the three diffusion tensor eigenvalues (λ1, λ2, and λ3).40 With the decrease in axial diffusivity (axonal injury) and the increase in radial diffusivity (myelin degeneration), RA would decrease more significantly and reflect the combined effects of axon injury and myelin degeneration. In the present study, at 3DPI, the significantly decreased RA is a result of the decreased axial diffusivity, reflecting axonal injury but without myelin damage. At 7DPI, the further significant decrease in RA results from increased radial diffusivity, reflecting the combined effects of axonal and myelin damage. The significant correlation between decreased RA and decreased CAP (Fig. 4) strongly suggests that RA could be a suitable index to assess the optic nerve function.
As expected, axial and radial diffusivities correlated with CAP, respectively (r = 0.62, P < 0.005; r = −0.62, P < 0.005), suggesting that both axonal injury and myelin damage affect the CAP amplitude. As a combined index, RA correlated with CAP amplitude more significantly, favorably suggesting that it is a better marker than axial and radial diffusivity alone to predict the functional outcome. Immunohistochemical findings largely confirmed our DTI results, where axonal integrity correlated with half-maximal CAP amplitude (Table 3); on the other hand, myelin integrity exhibited a statistically significant but weaker correlation.
Previous studies have demonstrated that DTI-derived axial diffusivity reflects disease severity and predicts long-term outcomes in experimental autoimmune encephalomyelitis and spinal cord–injured mice.21,41 Similarly, studies also demonstrated the predictive value of DTI for motor outcome in patients who have had a stroke.42,43 In the present study, we hypothesized that the loss of white matter structural integrity in the optic nerve would result in diminishing functional responses reflected as reduced CAP amplitude. Our current results demonstrate that DTI-derived RA strongly correlated with CAP amplitude, a synchronized and summated response of individual nerve fibers to the stimulation. The strong correlation we observed between RA and CAP amplitude suggests that DTI could be used to evaluate optic nerve structural integrity and predict functional alterations.
In conclusion, the significant correlation between noninvasive DTI parameters and functional CAP amplitude measurements clearly demonstrates the utility of DTI to reflect the function of white matter tracts in progressive injury. Overall, functional assessment of the injured optic nerve is best evaluated by diffusion anisotropy, rather than axial or radial diffusivity alone.
Footnotes
Supported in part by National Aeronautics and Space Administration Specialized Centers of Research Grant NNJ04HC9OG and National Institutes of Health/National Institute of Neurological Disorders and Stroke Grants R01 NS-054194, R01 NS-047592, and P01 NS-059560.
Disclosure: Q. Wang, None; R. Vlkolinsky, None; M. Xie, None; A. Obenaus, None; S.-K. Song, None
References
- 1. Frohman EM, Frohman TC, Zee DS, McColl R, Galetta S. The neuro-ophthalmology of multiple sclerosis. Lancet Neurol. 2005;4:111–121 [DOI] [PubMed] [Google Scholar]
- 2. Morrison JC, Johnson EC, Cepurna W, Jia L. Understanding mechanisms of pressure-induced optic nerve damage. Prog Retinal Eye Res. 2005;24:217–240 [DOI] [PubMed] [Google Scholar]
- 3. Compston A. Mechanisms of axon-glial injury of the optic nerve. Eye. 2004;18:1182–1187 [DOI] [PubMed] [Google Scholar]
- 4. Kuehn MH, Fingert JH, Kwon YH. Retinal ganglion cell death in glaucoma: mechanisms and neuroprotective strategies. Ophthalmol Clin North Am. 2005;18:383–395, vi [DOI] [PubMed] [Google Scholar]
- 5. Buckingham BP, Inman DM, Lambert W, et al. Progressive ganglion cell degeneration precedes neuronal loss in a mouse model of glaucoma. J Neurosci. 2008;28:2735–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Crish SD, Sappington RM, Inman DM, Horner PJ, Calkins DJ. Distal axonopathy with structural persistence in glaucomatous neurodegeneration. Proc Natl Acad Sci USA. 2010;107:5196–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang X, Sun P, Wang J, Wang Q, Song S-K. Diffusion tensor imaging detects retinal ganglion cell axon damage in mouse model of optic nerve crush. Invest Ophthalmol Vis Sci. 2011;52:7001–7006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zangwill LM, Bowd C. Retinal nerve fiber layer analysis in the diagnosis of glaucoma. Curr Opin Ophthalmol. 2006;17:120–131 [DOI] [PubMed] [Google Scholar]
- 9. Bowd C, Zangwill LM, Berry CC, et al. Detecting early glaucoma by assessment of retinal nerve fiber layer thickness and visual function. Invest Ophthalmol Vis Sci. 2001;42:1993–2003 [PubMed] [Google Scholar]
- 10. Sommer A, Katz J, Quigley HA, et al. Clinically detectable nerve fiber atrophy precedes the onset of glaucomatous field loss. Arch Ophthalmol. 1991;109:77–83 [DOI] [PubMed] [Google Scholar]
- 11. Johnson CA, Sample PA, Zangwill LM, et al. Structure and function evaluation (SAFE): II. Comparison of optic disk and visual field characteristics. Am J Ophthalmol. 2003;135:148–154 [DOI] [PubMed] [Google Scholar]
- 12. Basser PJ, Mattiello J, LeBihan D. MR diffusion tensor spectroscopy and imaging. Biophys J. 1994;66:259–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Werring DJ, Clark CA, Barker GJ, Thompson AJ, Miller DH. Diffusion tensor imaging of lesions and normal-appearing white matter in multiple sclerosis. Neurology. 1999;52:1626–1632 [DOI] [PubMed] [Google Scholar]
- 14. Song SK, Sun SW, Ju WK, Lin SJ, Cross AH, Neufeld AH. Diffusion tensor imaging detects and differentiates axon and myelin degeneration in mouse optic nerve after retinal ischemia. Neuroimage. 2003;20:1714–1722 [DOI] [PubMed] [Google Scholar]
- 15. Song SK, Sun SW, Ramsbottom MJ, Chang C, Russell J, Cross AH. Dysmyelination revealed through MRI as increased radial (but unchanged axial) diffusion of water. Neuroimage. 2002;17:1429–1436 [DOI] [PubMed] [Google Scholar]
- 16. Sidaros A, Engberg AW, Sidaros K, et al. Diffusion tensor imaging during recovery from severe traumatic brain injury and relation to clinical outcome: a longitudinal study. Brain. 2008;131:559–572 [DOI] [PubMed] [Google Scholar]
- 17. Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci. 1992;12:430–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baltan S, Inman DM, Danilov CA, Morrison RS, Calkins DJ, Horner PJ. Metabolic vulnerability disposes retinal ganglion cell axons to dysfunction in a model of glaucomatous degeneration. J Neurosci. 2010;30:5644–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun SW, Liang HF, Cross AH, Song SK. Evolving Wallerian degeneration after transient retinal ischemia in mice characterized by diffusion tensor imaging. Neuroimage. 2008;40:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sun SW, Liang HF, Le TQ, Armstrong RC, Cross AH, Song SK. Differential sensitivity of in vivo and ex vivo diffusion tensor imaging to evolving optic nerve injury in mice with retinal ischemia. Neuroimage. 2006;32:1195–1204 [DOI] [PubMed] [Google Scholar]
- 21. Budde MD, Xie M, Cross AH, Song SK. Axial diffusivity is the primary correlate of axonal injury in the experimental autoimmune encephalomyelitis spinal cord: a quantitative pixelwise analysis. J Neurosci. 2009;29:2805–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Song SK, Yoshino J, Le TQ, et al. Demyelination increases radial diffusivity in corpus callosum of mouse brain. Neuroimage. 2005;26:132–140 [DOI] [PubMed] [Google Scholar]
- 23. Huisman TA, Schwamm LH, Schaefer PW, et al. Diffusion tensor imaging as potential biomarker of white matter injury in diffuse axonal injury. AJNR Am J Neuroradiol. 2004;25:370–376 [PMC free article] [PubMed] [Google Scholar]
- 24. Dineen RA, Vilisaar J, Hlinka J, et al. Disconnection as a mechanism for cognitive dysfunction in multiple sclerosis. Brain. 2009;132:239–249 [DOI] [PubMed] [Google Scholar]
- 25. Alix JJ, Fern R. Glutamate receptor-mediated ischemic injury of premyelinated central axons. Ann Neurol. 2009;66:682–693 [DOI] [PubMed] [Google Scholar]
- 26. Nikolaeva MA, Richard S, Mouihate A, Stys PK. Effects of the noradrenergic system in rat white matter exposed to oxygen-glucose deprivation in vitro. J Neurosci. 2009;29:1796–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bowe CM, Kocsis JD, Targ EF, Waxman SG. Physiological effects of 4-aminopyridine on demyelinated mammalian motor and sensory fibers. Ann Neurol. 1987;22:264–268 [DOI] [PubMed] [Google Scholar]
- 28. Waxman SG. Conduction in myelinated, unmyelinated, and demyelinated fibers. Arch Neurol. 1977;34:585–589 [DOI] [PubMed] [Google Scholar]
- 29. Nashmi R, Fehlings MG. Changes in axonal physiology and morphology after chronic compressive injury of the rat thoracic spinal cord. Neuroscience. 2001;104:235–251 [DOI] [PubMed] [Google Scholar]
- 30. Easton DM. Simple, inexpensive suction electrode system for the student physiology laboratory. Am J Physiol. 1993;265:S35–S46 [DOI] [PubMed] [Google Scholar]
- 31. Evans RD, Weston DA, McLaughlin M, Brown AM. A non-linear regression analysis method for quantitative resolution of the stimulus-evoked compound action potential from rodent optic nerve. J Neurosci Methods. 2010;188:174–178 [DOI] [PubMed] [Google Scholar]
- 32. Stejskal EO, Tanner J. E. Spin diffusion measurements: spin echoes in the presence of time-dependent field gradient. J Chem Phys. 1965;42:288–292 [Google Scholar]
- 33. Basser PJ, Pierpaoli C. Microstructural and physiological features of tissues elucidated by quantitative-diffusion-tensor MRI. J Magn Reson B. 1996;111:209–219 [DOI] [PubMed] [Google Scholar]
- 34. Selinummi J, Seppala J, Yli-Harja O, Puhakka JA. Software for quantification of labeled bacteria from digital microscope images by automated image analysis. BioTechniques. 2005;39:859–863 [DOI] [PubMed] [Google Scholar]
- 35. Adachi M, Takahashi K, Nishikawa M, Miki H, Uyama M. High intraocular pressure-induced ischemia and reperfusion injury in the optic nerve and retina in rats. Graefes Arch Clin Exp Ophthalmol. 1996;234:445–451 [DOI] [PubMed] [Google Scholar]
- 36. Ridder WH, 3rd, Nusinowitz S. The visual evoked potential in the mouse–origins and response characteristics. Vision Res. 2006;46:902–913 [DOI] [PubMed] [Google Scholar]
- 37. Yiu EM, Geevasinga N, Nicholson GA, Fagan ER, Ryan MM, Ouvrier RA. A retrospective review of X-linked Charcot-Marie-Tooth disease in childhood. Neurology. 2011;76:461–466 [DOI] [PubMed] [Google Scholar]
- 38. Roncagliolo M, Schlageter C, Leon C, Couve E, Bonansco C, Eguibar JR. Developmental impairment of compound action potential in the optic nerve of myelin mutant taiep rats. Brain Res. 2006;1067:78–84 [DOI] [PubMed] [Google Scholar]
- 39. Stys PK, Ransom BR, Waxman SG. Compound action potential of nerve recorded by suction electrode: a theoretical and experimental analysis. Brain Res. 1991;546:18–32 [DOI] [PubMed] [Google Scholar]
- 40. Le Bihan D, Mangin JF, Poupon C, et al. Diffusion tensor imaging: concepts and applications. J Magn Reson Imaging. 2001;13:534–546 [DOI] [PubMed] [Google Scholar]
- 41. Kim JH, Loy DN, Wang Q, et al. Diffusion tensor imaging at 3 hours after traumatic spinal cord injury predicts long-term locomotor recovery. J Neurotrauma. 2010;27:587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jang SH, Ahn SH, Sakong J, et al. Comparison of TMS and DTT for predicting motor outcome in intracerebral hemorrhage. J Neurol Sci. 2010;290:107–111 [DOI] [PubMed] [Google Scholar]
- 43. Konishi J, Yamada K, Kizu O, et al. MR tractography for the evaluation of functional recovery from lenticulostriate infarcts. Neurology. 2005;64:108–113 [DOI] [PubMed] [Google Scholar]