

This study examined the neuroprotective effects of T-cell receptor ligand (RTL) on autoimmune optic neuritis in humanized HLA-DR3 mice. Such immunotherapy significantly suppressed inflammation, inhibited demyelination with signs of myelin recovery, and prevented axonal loss in the optic nerves.
Abstract
Purpose.
Optic neuritis (ON) is a condition involving primary inflammation, demyelination, and axonal injury in the optic nerve and leads to apoptotic retinal ganglion cell (RGC) death, which contributes to the persistence of visual loss. Currently, ON has no effective treatment. The goal was to determine the effectiveness of immunotherapy with recombinant T-cell receptor ligand (RTL) in preventing ON in humanized HLA-DR2 transgenic mice.
Methods.
Experimental autoimmune encephalomyelitis (EAE) was induced with myelin oligodendrocyte glycoprotein in humanized HLA-DR2 (DRβ1*1501) transgenic mice. Five consecutive doses of RTL342M were administrated at the onset of ON. The development of autoimmune ON was assessed by histopathology at different time points. The levels of myelin loss, axonal loss, and RGC damage were examined by immunofluorescence.
Results.
HLA-DR2 mice developed chronic ON 2 days before EAE characterized by progressive neurodegeneration in both organs. RTL342M significantly suppressed inflammation in the optic nerve and spinal cord and provided protection for at least 30 days. Examination of myelin loss showed a marked suppression of demyelination and an increase in myelin recovery in the optic nerve. Moreover, RTL342M treatment revealed a neuroprotective effect on optic nerve axons and RGCs in retinas at postimmunization (PI) day 62.
Conclusions.
RTL342M suppressed clinical and histologic signs of EAE/ON by preventing the recruitment of inflammatory cells into the optic nerve and showed neuroprotective effects against ON. However, to achieve full therapeutic benefit, more doses may be needed. These findings suggest a possible clinical application of this novel class of T-cell-tolerizing drugs for patients with optic neuritis.
Optic neuritis (ON) is a condition involving primary inflammation of the optic nerve presenting with sudden onset of painful unilateral visual loss.1 The optic nerve is a well-defined white matter tract originating from retinal ganglion cells (RGCs). ON may occur in isolation or may co-exist with multiple sclerosis (MS) or a variety of systemic autoimmune disorders.2,3 The disease typically affects young adults ranging from 18 to 45 years of age, with a mean age of 30 to 35 years and also children as young as 4 years.4 The annual incidence of ON is approximately 5 in 100,000, with a prevalence estimated to be 115 in 100,000. Acute demyelinating ON is the presenting feature in 15% to 20% of patients with MS, and it occurs at some time during the course of the disease in 50% of patients. Since there is evidence of early axonal damage in acute demyelinating ON, disease-modifying drugs are considered in patients at high risk of developing MS as a prophylaxis against permanent neurologic impairment and vision loss.5,6 Permanent vision loss from RGC death occurs in 40% to 60% of patients. Recurrences of ON after a single isolated episode are quite common. The risk of later development of clinically definite MS correlates with white matter demyelinating lesions on magnetic resonance imaging (MRI). During a 10-year follow-up, MS was diagnosed in 38% of patients with a first episode of ON who were enrolled in the Optic Neuritis Treatment Trial (ONTT)7,8 An ON episode is also a strong predictor of the 15-year risk of MS in patients with the presence of brain MRI abnormalities. The risk of developing MS increases to 72% compared with 25% in patients with no lesions on baseline brain MRI.9 Multiple factors can influence the outcome of the disease.
One important factor in susceptibility to ON is the expression of human leukocyte antigen (HLA) class II alleles that also have a strong association with MS. The subjects expressing HLA-DR2 are four times more likely to develop MS after ON. Moreover, a significant increase in frequency of HLA-DR2 was found in subjects with isolated ON (P < 0.01; RR = 2.9) compared with the normal population.10 Subjects with ON who were HLA-DR3 positive had an increased risk for the development of MS (RR = 2.8) and this risk was further enhanced when DR3 occurred in combination with DR2 (RR = 6.7). The overall increased risk that MS would develop in patients with this combination was 26 times that in the normal population.
Experimental autoimmune encephalomyelitis (EAE), which serves as an animal model for MS, often co-presents with ON in some animal strains. EAE can be induced by immunization with a number of myelin antigens, including myelin basic protein (MBP), proteolipid protein (PLP), and myelin oligodendrocyte glycoprotein (MOG).11 Inflammatory demyelination is the pathologic hallmark of MS.12 In this study, we used a humanized HLA-DR2 (DRβ1*1501) transgenic mouse model, in which the animals develop MOG-induced EAE.13 As we determined in these studies, the mice also develop chronic ON. Remarkably, MOG is expressed in the outermost surface of the oligodendrocyte myelin in the optic nerve,14 which could become clinically significant in the context of suboptimal T-cell responses and T-cell-targeted treatments such as RTLs.
The goal of these studies was to determine whether recombinant T-cell receptor ligand (RTL) immunotherapy would protect HLA-DR2 mice from developing ON induced with the MOG35-55 peptide. RTLs are new biologics that target T-cell-mediated inflammatory responses in an antigen-specific manner.15,16 We selected RTL342M, which is a mouse version of the human DR2-restricted RTL1000, also containing the MOG35-55 peptide, because RTL1000 has been shown to be safe in MS patients in a phase 1 clinical trial.17 In these studies humanized HLA-DR2 transgenic mice that developed chronic ON associated with EAE and RTL342M suppressed inflammation, demyelination, and axonal loss in the optic nerve.
Methods
Animals
Inbred HLA-DR2 (DRA:DRβ1*1501) transgenic mice 8 to 10 weeks old were housed at animal facilities of the VA Medical Center (Portland, OR). All experimental animals were handled according to the regulations formulated by the Institutional Animal Care and Use Committee and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Recombinant T-Cell Receptor Ligand
HLA-DR2 (DRA:DRβ1*1501)-derived RTL covalently linked to the encephalitogenic mouse MOG35-55 peptide (MEVGWYRSPFSRVVHLYRNGK) was produced as previously described.18–20 RTL342M comprises the β1 and α1 domains of DR2 (DRβ1*1501;DRA*0101) covalently linked to the MOG35-55 peptide.
Induction and Assessment of ON
To induce EAE and ON, groups of seven 8- to 12-week-old male and female transgenic mice expressing the DR2 allele were immunized with 200 μg MOG35-55 peptide in CFA containing 400 μg Mycobacterium tuberculosis H37RA subcutaneously (SC) at four sites on the flanks.13,20 In addition, the mice received 70 ng pertussis toxin on days 0 and 2 PI. At the onset of ON, the mice were given five daily 20-μg SC doses of RTL342M. Control mice received five doses of buffer vehicle (Tris buffer) at the same times. The mice were assessed daily for clinical signs of EAE as previously published.21 At the end of the experiment, each mouse was perfused with 4% PFA, and the eyes with optic nerves and full vertebral column were dissected and fixed for 2 hours. The tissues were transferred to 30% sucrose for overnight incubation at 4°C. Next, the spinal cords were dissected from bone column, and all tissues were frozen in OCT and stored at −80°C until sectioning. Optic nerve cryosections were cut in 10-μm longitudinal (left eyes) and cross sections (right eyes). Spinal cords were cut in 10-μm cross sections only. The optic nerve sections were stained with hematoxylin/eosin (H&E) and evaluated for the presence of inflammatory cell infiltration in longitudinal sections of the optic nerve, according to a scale from 0 to 4 by a masked observer: 0, no infiltration; 1, mild cellular infiltration of the optic nerve or optic nerve sheath; 2, moderate infiltration; 3, severe infiltration; and 4, massive infiltration of the optic nerve parenchyma and nodule infiltration. We collected the eyes to determine a possible inflammation after immunization with MOG, but intraocular cellular infiltration was not observed in any of the mouse eyes.
Myelin Quantification
Frozen longitudinal sections of optic nerves from each treatment group were stained with Luxol fast blue without cresyl violet, to stain myelin only. Micrographs were then taken at 20× of each section with a light microscope (model BH-2; Olympus, Tokyo, Japan) with an attached camera (DP21; Olympus) and were cropped to 0.119 × 0.506 mm. The cropped images were then pixilated by the ImageJ program with the Hessian plug-in feature, as described by Grider et al.22 (ImageJ, developed by Wayne Rasband, National Institutes of Health, Bethesda, MD; available at http://rsb.info.nih.gov/ij/index.html). Pixilation intensity was counted from the whole assigned area in a masked fashion using Image ProPlus and graphed (Prism; GraphPad, San Diego, CA). The area was normalized to 0.1 mm2.
Immunofluorescent Staining
Frozen long or cross sections were postfixed with 4% paraformaldehyde in PBS for 10 minutes, washed, and blocked with 10% normal goat serum in PBS containing 1% BSA and 0.2% Tween for 1 hour. After the primary antibodies were blocked in PBS with 1% BSA and 0.2% Tween, the sections were incubated for 1 hour at room temperature. The following antibodies were used: anti-MOG (1:200; Abnova, Walnut, CA), anti-CD11b (1:100; Abcam, Cambridge, MA), anti-RT97 (1:500; Millipore, Billerica, MA), anti-MBP (1:200; Abnova,), anti-NeuN (1:100; Chemicon, Temecula, CA), and anti-GFAP (1:100; BD Pharmingen, San Diego, CA) for overnight incubation at 4°C. Next, tissue was washed and incubated in appropriate secondary antibodies in PBS with 1% BSA, 0.2% Tween for 1 hour, including anti-mouse AlexaFluor 488 (1:400), anti-rabbit AlexaFluor 594 (1:400), or anti-goat AlexaFluor 594 (1:400; all from Invitrogen, Carlsbad, CA). After the tissues was washed, DAPI (Roche, Indianapolis, IN) counterstain was applied for 1 minute, and the slides were then mounted (Fluoromount-G; Southern Biotech, Birmingham, AL) to be photographed with a confocal fluorescence microscope (Fluoview1000; Olympus, or DM5000 Leica, Deerfield, IL). Pictures were then taken at 10× and 20× magnifications and analyzed (ImagePro; Media Cybernetics, Silver Spring, MD). A negative control contained secondary antibodies only.
Axon Quantification
Ten-micrometer cross sections of experimental and age-matched normal optic nerves were prepared every 30 μm for immunostaining with anti-RT97 antibodies. At least four sections were used from each animal. Anti-RT97 (Millipore) diluted at 1:500 were used for 1 hour followed by incubation with anti-mouse Alexa Fluor 488 diluted at 1:400 for another hour and then counterstain with DAPI (Roche) for mounting. Pictures were taken of each stained section at 40× using a confocal microscope (Fluoview1000; Olympus), and pseudocolor images were acquired for analysis. The images were then cropped to the same 220 × 130-μm area and counted in a masked fashion based on the intensity of the fluorescent green stain (ImagePro Plus; Media Cybernetics). The negative control was also stained and counted to subtract the background from all the counts.
RGC Evaluation
Retinas were removed from fixed left eyes, and whole mounts were made by four radial cuts representing the dorsal, ventral, temporal, and nasal sides. Cross sections (10 μm) were prepared from the right eye. The retinas were placed in a solution containing 100× diluted anti-NeuN antibodies that labeled RGCs specifically, followed by fluorescent anti-mouse Alexa488 diluted 400×. After a brief wash, the retinas were flat mounted on the slide, and retinal images were captured at 10× magnification with a fluorescence microscope (DM5000B; Leica) and photographed. RGCs were counted in a masked fashion with the imaging software.
Statistical Analysis
The data are expressed as the mean ± SEM. A two-tailed Student's t-test was used to determine the statistical significance. The significance between the controls and treatment groups was determined by one-way analysis of variance (ANOVA; Prism 3.0; GraphPad). Differences with a P < 0.05 were significant.
Results
Therapeutic Effect on Inflammation in Chronic ON in HLA-DR2 Mice
Our previous studies have shown that immunization of HLA-DR2 mice with MOG35-55 peptide induced chronic EAE,20 but, until now, it was unknown whether these mice would develop ON. In our studies, MOG immunization induced ON of a chronic nature, and the inflammatory lesions in the optic nerve correlated with the lesions in the spinal cord (not shown). Figure 1A shows that HLA-DR2 mice given MOG peptide developed chronic inflammation of the optic nerve that occurred on PI days 8 to 9 before the onset of EAE (Fig. 1B; 11–12 days), with inflammatory infiltrates in the optic nerve at onset on the long and cross sections stained with H&E. The incidence was 100%.
Figure 1.

Pathology of ON associated with EAE in HLA-DR2 mice induced with MOG35-55 peptide. (A) Time course of ON in HLA-DR2 mice based on three experiments determined using longitudinal sections of the optic nerve stained with H&E. (B) Histopathology of representative ON at onset. H&E staining of the cross- and longitudinal sections; arrows: inflammatory cells. (C) Clinical time course of EAE in vehicle- and RTL342M-treated HLA-DR2 mice. Top arrows: the time of tissue collection; bottom arrows: the time of treatment administration (five doses of 20 μg RTL342M/dose on consecutive days starting at onset of ON). (D) Severity of ON in vehicle- and RTL342M-treated mice.
In these studies, we tested whether RTL immunotherapy would suppress inflammation, demyelination, and loss of axons in the optic nerve of HLA-DR2 mice when RTL342M was administrated at the onset of ON. To evaluate the effectiveness of RTL342M therapy, the tissues were collected from treated and control mice on days 16 (peak), 30, and 62 PI. The treatment of the mice with five consecutive daily doses of RTL342M by subcutaneous administration at onset of ON (day 8) significantly suppressed development of histologic ON for the first 30 days and reduced inflammatory cell infiltrates in optic nerves (Fig. 2). The clinical scoring for EAE was performed daily and is shown in Figure 1C from a representative experiment. Figure 1D shows that the severity of ON was significantly reduced on day 16 PI with an average inflammatory score of 0.4 ± 0.24 for treated optic nerves compared with 2.8 ± 0.66 for the controls (P < 0.0094). After treatment stopped, this effect was later diminished, and the mice presented signs of ON progression. The results suggest that, although RTL342M was effective in stopping the influx of inflammatory cells, additional doses of RTL may be needed to inhibit further development of inflammation, to reach full therapeutic benefit in those mice.
Figure 2.
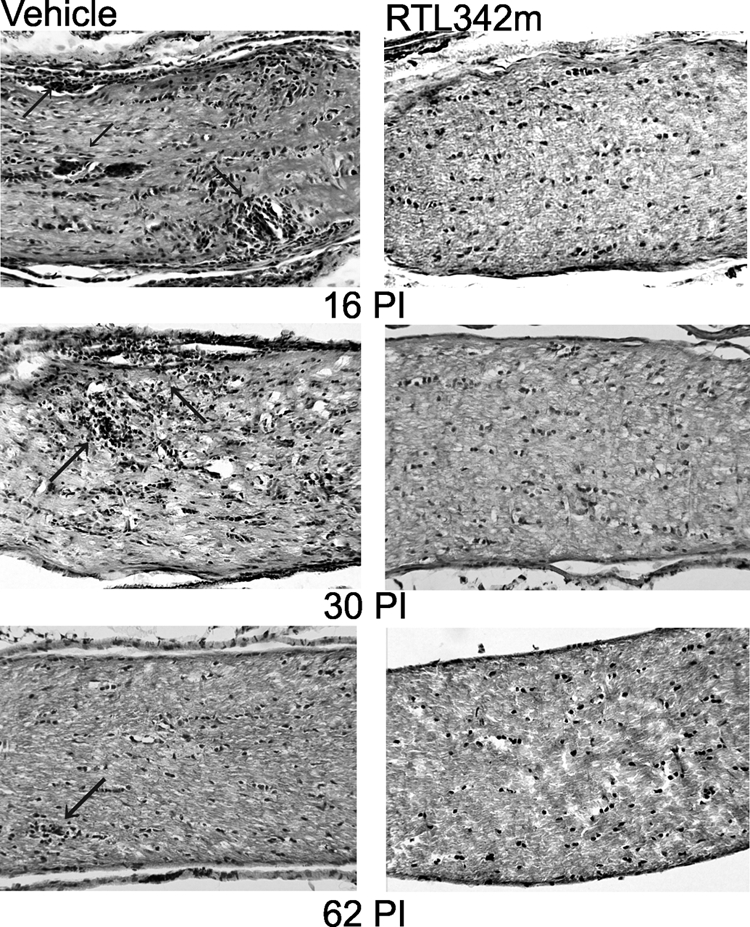
Histopathologic micrographs of HLA-DR2 mouse optic nerves collected on days 16, 30, and 62 PI. Long sections of optic nerves were stained with H&E. Arrows: inflammatory lesions.
Inhibition of Myelin Loss in Optic Nerves
The next experiment examined the effect of RTL342M on myelin loss in optic nerves. Figure 3 shows that demyelination of the optic nerve occurred around the time of inflammation, as determined by Luxol fast blue staining (Figs. 3A–C). The myelin loss in Figure 3 shows that myelin in normal untreated mice was nicely compact within the nerve, whereas mice with inflammation exhibited patchy loss of myelin. Moreover, the loss of myelin was profound around inflammatory lesions and was progressive in untreated mice over the 62-day observation period compared with that in normal optic nerves (P < 0.001). Figure 3D shows that although the mice lost myelin due to inflammation, RTL342M protected long-term demyelination of the optic nerve compared with the control, vehicle-treated mice (days 30 and 62 PI).
Figure 3.

Analysis of myelin loss in optic nerves of RTL342M- and vehicle-treated HLA-DR2 mice. (A–C) Representative images of long optic nerve sections at ON collected on days 16, 30, and 62 PI stained with Luxol fast blue to label myelin. (A) Normal optic nerve shows compact myelin staining; longitudinal sections of optic nerves from (B) vehicle- and (C) RTL342M-treated mice collected at different times after immunization with MOG. (D) Evaluation of demyelination of optic nerves by quantification of the intensity of the staining and adjusted to 0.1 mm2 showed significantly greater demyelination in vehicle-treated mice. Values are expressed as the mean ± SEM of staining intensity from four optic nerves per group.
Figure 4 shows representative early pathology in consecutive long sections of the optic nerve collected 16 days (peak) after immunization without treatment. The inflammation was associated with the influx of inflammatory cells and the accumulation of macrophages (mϕ)/microglia in inflammatory lesions (arrows), as determined by immunofluorescent staining with anti-CD11b antibodies of the optic nerve (Figs. 4B). The myelin loss was apparent around inflammatory lesions, as shown using cresyl violet to counterstain the inflammatory infiltrate (Fig. 4C). In addition, early axonal damage near inflammatory cells in optic nerves (Figs. 4D–F) was revealed by immunostaining with anti-axon RT97 antibodies (Fig. 4D) and by Bielschowsky silver impregnation of the long section of the optic nerve (Figs. 4E, 4F). The cross section of the affected nerve showed axonal injury compared with healthy looking axons in normal optic nerve (Fig. 4G, 4H). Together, these results from the peak of ON indicate that inflammation had an effect on myelin and axonal loss.
Figure 4.

Early demyelination of optic nerves and axonal degeneration in MOG-induced ON in HLA-DR2 transgenic mice at day 16 PI (peak). Representative sequential long sections of the same optic nerve with ON stained for inflammatory cells with H&E. (A) Myelin staining with H&E; (B) macrophages (mϕ)/microglia immunofluorescent staining with anti-CD11b antibodies (red); (C) myelin staining with Luxol fast blue with cresyl blue to label cells; and (D) immunofluorescent axonal labeling using RT97 antibodies (green). (A–D, arrows) An inflammatory lesion; (E–H) Bielschowsky silver impregnation for axonal staining; (E, G) normal, untreated optic nerves are populated by axons; (F, H, arrows) damaged axons.
Quantitative analysis of Luxol fast blue–stained myelin evaluated at 30 and 62 days PI revealed that RTL342M slowed, and perhaps reversed, myelin loss compared with vehicle treatment (Fig. 3D), and so we examined whether myelin recovery occurred after the initial loss due to inflammation and damage to the oligodendrocytes in optic nerves. The long sections were immunolabeled with anti-MOG antibodies, markers for oligodendrocytes, and RT97 antibodies to label axons. Figure 5 shows an increase in immunofluorescent labeling of MOG+ (white arrows) in the RTL-treated optic nerve on day 62 PI compared with vehicle-treated mice, which showed visibly fewer immunolabeled cells. Double immunofluorescent labeling with anti-CD11b and anti-MOG antibodies revealed a greater presence of mϕ/microglia (yellow arrows) in untreated optic nerve, whereas the RTL-treated optic nerve showed a reduction in microglia and an increase in MOG-stained cells. This lack of overlapping colors showing separated staining of microglia and MOG labeling may suggest oligodendrocyte recovery rather than an increase in microglia phagocytizing myelin from damaged cells. Also, there was a substantial increase in immunofluorescence of RT97-labeled axons at day 30 PI in contrast to those in vehicle controls. These findings strongly suggest oligodendrocyte regeneration and an increase in myelin in the optic nerve after initial degeneration (30 PI) due to inflammatory processes.
Figure 5.

Oligodendrocyte immunofluorescent labeling of the vehicle- and RTL342M-treated optic nerves. (A) Double-immunofluorescent labeling in the optic nerves collected at 30 and 62 PI with anti-MOG antibodies as a marker for oligodendrocytes and with anti-RT97 for labeling axons: anti-MOG (red), anti-RT97 (green) antibodies and colocalization (yellow). (A, B, white arrows) Red-labeled MOG+ myelin-producing cells. (B) Double immunofluorescent labeling of 62PI vehicle and RTL-treated optic nerves with anti-MOG (red) and anti-CD11b antibodies for labeling of mϕ/microglia (green); yellow arrows: microglia, nuclear labeling with DAPI (blue).
Protection from Axonal Degeneration
As shown in Figures 2 and 3, inflammation and demyelination of the optic nerve were associated with progressive axonal loss. We thus analyzed RGCs and axons that comprise the optic nerve. Figure 6A shows immunofluorescent axonal immunolabeling in the optic nerves at 62 PI by RT97 antibodies, which label neurofilaments. Normal optic nerves showed densely labeled axons (Fig. 6A), but the area of axonal immunofluorescent staining decreased in vehicle-treated optic nerves and showed a significant trend (Fig. 6B). In our experiments, some mice were fully protected by RTL342M, but other mice still showed signs of inflammation, and in those mice, there was more myelin loss and more neuronal damage. Figure 6B shows averaged results from both groups of mice and compared to the axonal immunolabeling for day 30 PI. In RTL342M-treated nerves at 62 PI, the number of labeled axons was significantly greater than in the vehicle-treated optic nerves (P = 0.047), suggesting neuroprotection. To further examine the axonal degeneration, we analyzed RGCs by preparing retinal whole mounts and retinal cross sections obtained from day 62 PI RTL342M- and vehicle-treated mice. The retinas were immunofluorescently labeled with anti-RGC antibodies against neuron-specific nuclear protein (NeuN). All retinal whole mounts from vehicle-treated mice showed a noticeable decline in the number of immunolabeled RGCs, compared with the count in RTL342M-treated mice (Fig. 6C), but analysis of the fluorescent cell count showed no statistical significance. An additional experiment of immunolabeling of RGCs with anti-NeuN antibodies on retinal cross sections also demonstrated a marked loss of RGCs in vehicle-treated retinas, which showed statistical significance (P = 0.0068; Fig. 6E). Immunolabeling with anti-GFAP antibodies specific for astrocytes revealed an increase in astrocyte labeling in vehicle-treated retinas in the ganglion cell layer (Fig. 6D) in contrast to RTL342M-treated retinas where more RGCs and fewer astrocytes were immunolabeled in the ganglion cell layer. These findings further suggest the protection of RGCs and their axons by the RTL treatment.
Figure 6.

Immunofluorescent labeling of surviving axons and RGCs in the optic nerve from vehicle- and RTL342M-treated mice collected on day 62 PI. (A) Immunostaining of cross sections with anti-RT97 antibodies for axons in normal and treatment groups collected on day 62 PI. Note a visible reduction in RT97-labeled axons in vehicle-treated optic nerves compared with normal and RTL342M-treated optic nerves; (B) quantitative analysis of fluorescence density of RT97 axonal labeling in vehicle- and RTL342M-treated optic nerves collected on days 30 and 62 PI. Results are expressed as the mean ± SEM of fluorescence density in four sections 30 μm apart in stained optic nerves (n = 4). (C) Representative images of retinal whole mounts demonstrate NeuN-labeled RGCs on day 62 PI taken from RTL342M- and vehicle-treated mice. (D) Double-immunofluorescence labeling in the retina with anti-NeuN as a marker for RGCs and anti-GFAP antibodies as marker for astrocytes: anti-GFAP (red), anti-NeuN (green), nuclear labeling with DAPI (blue); the pictures show the ganglion cell layer of representative cross sections of eyes collected from vehicle- and RTL342M-treated mice. (E) Quantitative analysis of immunofluorescence-labeled RGCs with NeuN antibodies in retinal cross-sections in vehicle- and RTL342M-treated mice collected on day 62 PI. Results are expressed as mean ± SEM of fluorescent cell counts from four areas of the retina (n = 4) and normalized to a 0.1-mm2 area.
Discussion
Our studies demonstrate that immunization of HLA-DR2 mice with MOG35-55 peptide-induced ON of a chronic nature that was associated with EAE; however, the onset of ON preceded the onset of EAE by at least 2 days. This observation is similar to human optic nerve disease in which ON is frequently the initial manifestation of MS.6,9 The RTL (RTL342M) used in these investigations demonstrated the potential to inhibit inflammation and promote neuroprotection and even the remyelination process. The RTL treatment at onset of ON (day 8) significantly reduced the development of both clinical EAE and histologic ON by preventing the recruitment of inflammatory cells into the optic nerve. RTL342M not only markedly suppressed inflammation, but also inhibited demyelination for up to 30 days. Moreover, the optic nerve from mice treated with RTL showed signs of myelin recovery. By 60 days, there was an increase in the number of MOG-labeled cells (oligodendrocytes) after an initial decline, which may be important in designing future treatment for human chronic or recurrent ON. Loss of myelin and axonal damage in optic nerves seemed to be the effect of inflammation that started in the proximity of inflammatory lesions. We do not have any direct evidence that axonal loss in optic nerves preceded inflammation, which is similar to previously reported studies using other animal models of ON.22–24 In our model, there was continuous demise of axons over the 62-day experimental period that could be a result of injured axons more vulnerable in the nerve regions lacking in myelin.25 It is likely that we have not reached the full therapeutic potential of RTL342M, because the treatment regimen included only five initial daily doses administered at the onset of ON. We believe that longer-lasting effects may be achieved with additional booster injections to enhance beneficial effects of RTL therapy.
There are several models of EAE with associated ON.11 We selected humanized HLA-DR2 mice, which developed EAE with MOG immunization13 and provided a clinically relevant model applicable for testing the efficacy of RTLs before clinical trials for subjects with ON. The humanized HLA-DR2 transgenic mice were susceptible to moderate to severe chronic ON, as determined for the first time in these studies. MOG is an important central nerve system (CNS) antigen, which is expressed in larger quantities in the optic nerve, prominently in the outermost surface of the oligodendrocyte myelin.14 Thus, inflammatory cells targeting MOG appeared first around the pia, at the onset of ON.
Some experimental therapies have been reported recently for treatment of autoimmune ON, including corticosteroid therapy, which suppresses ON and prevents RGC loss if treatment is initiated before onset of optic nerve inflammation, but is less effective after inflammation has begun.26 The Bowman-Birk inhibitor (BBI), a soybean-derived serine protease inhibitor, significantly reduces the incidence of ON and prevents loss of RGCs.27 Intraocular delivery of antioxidant genes such as superoxide dismutase or catalase cloned into recombinant AAV provides a long-lasting suppression and showed a decreased RGC loss by 29%, optic nerve demyelination by 36%, axonal loss by 44%, and cellular infiltration by 34%, compared with the control eye with ON at 6 months PI.28 Lipoic acid, also an antioxidant, reduces inflammation and protected axonal injury when delivered on day 8 or 13 PI in the C57BL/6 EAE model.29 RTL molecules are new biologics that consist of the membrane distal α1 and β1 domains of class II MHC molecules and contain covalently linked antigenic peptides,15,18,30 including the MOG35-55 peptide. By inhibiting autoreactive T-cell responses, RTLs have been shown to suppress clinical and histologic signs in various experimental autoimmune disease models, including uveitis,31,32 encephalomyelitis (EAE), arthritis,33 and stroke.16 The application of RTL342M is a new therapy for ON in humanized mice. Our studies demonstrated that RTL342M had a therapeutic effect on both ON and EAE, and its treatment led to the suppression of inflammatory responses, myelin recovery, and some axonal protection. A recent study of human ON suggested that all optic nerve inflammatory lesions tend to remyelinate regardless of the size of the initially demyelinated zone.34 This study showed that smaller lesions get more complete remyelination than larger lesions, and the extent of the initial inflammatory demyelinating assault was probably the single most important factor determining success of remyelination. Moreover, the first 5 to 6 months after the demyelinating event may be crucial for the remyelinating process to succeed.34 Therefore, by suppressing acute inflammation in the optic nerve, RTL therapy could be regarded as a remyelination-enhancing therapy that could accelerate myelin recovery in the CNS if applied almost immediately after onset. Recent animal studies using SJL/J mice with established EAE could support this notion. A successful treatment with a different myelin-specific RTL401 (I-As/PLP139-151 peptide) after the peak of EAE markedly reduces inflammation in the CNS associated with an increase in remyelinating axons and the presence of small, presumably regenerative axonal sprouts.35 These findings indicate that RTL therapy targeting pathogenic T cells may promote CNS neuroregenerative processes, and these findings may also have implications for clinical trials.
In summary, our study showed a successful therapy for ON with RTL342M administered at onset, resulting in markedly reduced inflammation, demyelination, and axonal loss. Such immunomodulatory therapy may prevent loss of myelin and thus RGC damage. Because the protection was not complete, we believe that additional doses over the course of the disease may be needed to increase the benefit of RTL treatment.
Footnotes
Supported by National Eye Institute Grant EY17781 (GA) and National Institutes of Health Grant NIH NS047661 (AAV) and an unrestricted grant from Research to Prevent Blindness (CEI). This material is based on work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Disclosure: G. Adamus, Artielle ImmunoTherapeutics, Inc. (I), P; L. Brown, None; S. Andrew, None; R. Meza-Romero, None; G.G. Burrows, Artielle ImmunoTherapeutics, Inc. (I), P; A.A. Vandenbark, Artielle ImmunoTherapeutics, Inc. (I), P
References
- 1. Margalit E, Sadda SR. Retinal and optic nerve diseases. Artif Organs. 2003;27:963–974 [DOI] [PubMed] [Google Scholar]
- 2. Balcer LJ. Clinical practice: optic neuritis. N Engl J Med. 2006;354:1273–1280 [DOI] [PubMed] [Google Scholar]
- 3. Pau D, Al Zubidi N, Yalamanchili S, Plant GT, Lee AG. Optic neuritis. Eye. 2011;25:833–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wilejto M, Shroff M, Buncic JR, Kennedy J, Goia C, Banwell B. The clinical features, MRI findings, and outcome of optic neuritis in children. Neurology. 2006;67:258–262 [DOI] [PubMed] [Google Scholar]
- 5. Shams PN, Plant GT. Optic neuritis: a review. Int MS J. 2009;16:82–89 [PubMed] [Google Scholar]
- 6. Arnold AC. Evolving management of optic neuritis and multiple sclerosis. Am J Ophthalmol. 2005;139:1101–1108 [DOI] [PubMed] [Google Scholar]
- 7. Beck RW, Trobe JD, Moke PS, et al. High- and low-risk profiles for the development of multiple sclerosis within 10 years after optic neuritis: experience of the optic neuritis treatment trial. Arch Ophthalmol. 2003;121:944–949 [DOI] [PubMed] [Google Scholar]
- 8. Volpe NJ. The optic neuritis treatment trial: a definitive answer and profound impact with unexpected results. Arch Ophthalmol. 2008;126:996–999 [DOI] [PubMed] [Google Scholar]
- 9. The Optic Neuritis Study Group: Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. Arch Neurol. 2008;65:727–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Francis DA, Compston DA, Batchelor JR, McDonald WI. A reassessment of the risk of multiple sclerosis developing in patients with optic neuritis after extended follow-up. J Neurol Neurosurg Psychiatry. 1987;50:758–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Furlan R, Cuomo C, Martino G. Animal models of multiple sclerosis. Methods Mol Biol. 2009:157–173 [DOI] [PubMed] [Google Scholar]
- 12. Trapp BD, Nave KA. Multiple sclerosis: an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269 [DOI] [PubMed] [Google Scholar]
- 13. Rich C, Link JM, Zamora A, et al. Myelin oligodendrocyte glycoprotein-35-55 peptide induces severe chronic experimental autoimmune encephalomyelitis in HLA-DR2-transgenic mice. Eur J Immunol. 2004;34:1251–1261 [DOI] [PubMed] [Google Scholar]
- 14. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med. 2003;197:1073–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang JW, Mechling DE, Bachinger HP, Burrows GG. Design, engineering, and production of human recombinant t cell receptor ligands derived from human leukocyte antigen DR2. J Biol Chem. 2001;276:24170–24176 [DOI] [PubMed] [Google Scholar]
- 16. Offner H, Sinha S, Wang C, Burrows GG, Vandenbark AA. Recombinant T cell receptor ligands: immunomodulatory, neuroprotective and neuroregenerative effects suggest application as therapy for multiple sclerosis. Rev Neurosci. 2008;19:327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Offner H, Sinha S, Burrows GG, Ferro AJ, Vandenbark AA. RTL therapy for multiple sclerosis: a phase I clinical study. J Neuroimmunol. 2011;231:7–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Burrows GG. Systemic immunomodulation of autoimmune disease using MHC-derived recombinant TCR ligands. Curr Drug Targets Inflamm Allergy. 2005;4:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Link JM, Rich CM, Korat M, Burrows GG, Offner H, Vandenbark AA. Monomeric DR2/MOG-35-55 recombinant TCR ligand treats relapses of experimental encephalomyelitis in DR2 transgenic mice. Clin Immunol. 2007;123:95–104 [DOI] [PubMed] [Google Scholar]
- 20. Vandenbark AA, Rich C, Mooney J, et al. Recombinant TCR ligand induces tolerance to myelin oligodendrocyte glycoprotein 35-55 peptide and reverses clinical and histological signs of chronic experimental autoimmune encephalomyelitis in HLA-DR2 transgenic mice. J Immunol. 2003;171:127–133 [DOI] [PubMed] [Google Scholar]
- 21. Sinha S, Subramanian S, Proctor TM, et al. A promising therapeutic approach for multiple sclerosis: recombinant T-cell receptor ligands modulate experimental autoimmune encephalomyelitis by reducing interleukin-17 production and inhibiting migration of encephalitogenic cells into the CNS. J Neurosci. 2007;27:12531–12539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grider MH, Chen Q, Shine HD. Semi-automated quantification of axonal densities in labeled CNS tissue. J Neurosci Meth. 2006;155:172–179 [DOI] [PubMed] [Google Scholar]
- 23. Hobom M, Storch MK, Weissert R, et al. Mechanisms and time course of neuronal degeneration in experimental autoimmune encephalomyelitis. Brain Pathol. 2004;14:148–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guan Y, Shindler KS, Tabuena P, Rostami AM. Retinal ganglion cell damage induced by spontaneous autoimmune optic neuritis in MOG-specific TCR transgenic mice. J Neuroimmunol. 2006;178:40–48 [DOI] [PubMed] [Google Scholar]
- 25. Shindler KS, Ventura E, Dutt M, Rostami A. Inflammatory demyelination induces axonal injury and retinal ganglion cell apoptosis in experimental optic neuritis. Exp Eye Res. 2008;87:208–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McConnell P, Berry M. Regeneration of axons in the mouse retina after injury. Bibl Anat 1982;(23):26–37 [PubMed] [Google Scholar]
- 27. Dutt M, Tabuena P, Ventura E, Rostami A, Shindler KS. Timing of corticosteroid therapy is critical to prevent retinal ganglion cell loss in experimental optic neuritis. Invest Ophthalmol Vis Sci. 2010;51:1439–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Touil T, Ciric B, Ventura E, Shindler KS, Gran B, Rostami A. Bowman-Birk inhibitor suppresses autoimmune inflammation and neuronal loss in a mouse model of multiple sclerosis. J Neurolog Sci. 2008;271:191–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Qi X, Sun L, Lewin AS, Hauswirth WW, Guy J. Long-term suppression of neurodegeneration in chronic experimental optic neuritis: antioxidant gene therapy. Invest Ophthalmol Vis Sci. 2007;48:5360–5370 [DOI] [PubMed] [Google Scholar]
- 30. Chaudhary P, Marracci G, Yu X, Galipeau D, Morris B, Bourdette D. Lipoic acid decreases inflammation and confers neuroprotection in experimental autoimmune optic neuritis. J Neuroimmunol. 2011;233:90–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Burrows GG, Chang JW, Bachinger HP, Bourdette DN, Offner H, Vandenbark AA. Design, engineering and production of functional single-chain T cell receptor ligands. Protein Eng. 1999;12:771–778 [DOI] [PubMed] [Google Scholar]
- 32. Adamus G, Burrows GG, Vandenbark AA, Offner H. Treatment of autoimmune anterior uveitis with recombinant TCR Ligands, Invest Ophthalmol Vis Sci. 2006;47:2555–2561 [DOI] [PubMed] [Google Scholar]
- 33. Adamus G, Karren L, Mooney J, Burrows G. A promising therapeutic approach for treatment of posterior uveitis: recombinant T cell receptor ligand protects Lewis rats from acute and recurrent experimental autoimmune uveitis, Ophthalmic Res. 2010;44:24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Huan J, Kaler LJ, Mooney JL, Subramanian S, et al. MHC class II derived recombinant T cell receptor ligands protect DBA/1LacJ mice from collagen-induced arthritis. J Immunol. 2008;180:1249–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Klistorner A, Arvind H, Garrick R, Yiannikas C, Paine M, Graham SL. Remyelination of optic nerve lesions: spatial and temporal factors. Multiple Sclerosis. 2010;16:786–795 [DOI] [PubMed] [Google Scholar]
- 36. Wang C, Gold BG, Kaler LJ, et al. Antigen-specific therapy promotes repair of myelin and axonal damage in established EAE. J Neurochem. 2006;98:1817–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]