

Abstract
Salivary gland acinar cells have two types of Ca2+-activated K channels required for fluid secretion: the intermediate conductance (IK1) channel and the large conductance (BK) channel. Activation of IK1 inhibits BK channels including in small, cell-free, excised membrane patches. As a first step toward understanding the mechanism underlying this interaction, we examined its voltage sensitivity. We found that the IK1-induced inhibition of BK channels was only weakly voltage dependent and not accompanied by alteration in BK gating kinetics. These actions of IK1 on BK channels are not consistent with a mechanism whereby activation of IK1 causes a shift of the BK channel's voltage dependence as occurs for many BK modulatory processes. In a search for other clues about the interaction mechanism, we noted that the N-terminus of the IK1 channel shares some chemical features with the N-terminal regions of two BK subunits known to inhibit BK activity by blocking the cytoplasmic end of the BK pore. Thus, we tested the idea that the N-terminus of IK1 channels may act similarly. We found that a peptide derived from the N-terminal region of the IK1 protein blocked BK channels. Significantly, we also found that the activation of IK1 channels competed with block by the N-terminus peptide. Thus, the activation of IK1 channels inhibits BK channels by a mechanism that involves block of the cytoplasmic pore, not an alteration in the voltage dependence of BK gating. The mediator of this cytoplasmic pore block may be the IK1 N-terminus.
Keywords: ion channels, Ca2+-activated K channels, maxi-K channels, IK1 channels, N-terminus, DCEBIO
Introduction
Ca2+-activated K channels shape the membrane potential of cells in response to intracellular Ca2+ signals. In neurons such channels control the duration of action potentials and control action potential firing rates.1 In non-excitable cells, these types of channels control a range of physiological functions including the regulation of T-cell activation,2,3 volume regulation in red blood cells4,5 and the control of fluid secretion.6–8 There are three classes of Ca2+-activated K channels and these differ in several characteristics including their single channel current levels. The founding member of the large conductance family, KCa1.1, is activated by voltage as well as by Ca2+ ions and this big conductance (BK) channel can be associated with various β subunits. The three small-conductance (SK) channels (KCa2.1, KCa2.2 and KCa2.3) share considerable amino acid identity and biophysical properties. The intermediate conductance channel (KCa3.1 or IK1) shares only about 50% amino acid homology with the members of the SK family9,10 and, like the SK channels and in contrast to BK channels, their activation has no voltage dependence.
BK and IK1 channels are both expressed in a variety of tissues including salivary glands,11–13 the colon,14–16 macrophages,17 endothelial cells,18 and vascular smooth muscle cells.19–21 We have recently shown that activation of IK1 channels inhibits BK channels in acinar cells from mouse and human parotid22,23 and from mouse submandibular glands.24 Muscarinic stimulation causes fairly rapid (near 0.2 or 0.3 Hz) global oscillations of Ca2+ in these cells,25,26 and the IK1-induced inhibition of BK channels causes the activity of these channels to be out of phase.22 The IK1-mediated inhibition of BK channels can be recapitulated in heterologous expression systems,22 which suggests it may occur in the other cells in which both channels coexist.
The IK1-induced inhibition of BK channels occurs in excised membrane patches (near 1–2 μm in size) suggesting a close interaction between the two channels that does not require water-soluble intermediaries. The IK1-induced inhibition of BK channels does not alter the BK single channel current level22 and the rapid reversibility of the inhibition during Ca2+ oscillations (see above) argues against a mechanism involving rapid insertion and removal of BK channels from the membrane. Thus, the IK1-induced inhibition of BK channels may involve a close interaction between the IK1 and BK channels.
As a first step toward determining the molecular mechanism underlying the IK1-mediated inhibition of BK channels, we tested the voltage dependence of the interaction. We found that IK1 activation inhibits BK channels with little or no voltage dependence. We also found little or no effect of IK1 activation on BK channel gating kinetics. These two results argue against an inhibition mechanism involving large voltage shifts of BK channel activation as happens with many BK channel modulators. The lack of a significant effect of voltage on the IK1-induced BK inhibition is suggestive of a mechanism whereby the activation of IK1 channels results in block of the cytoplasmic portion of the BK channel-outside of the transmembrane electric field. In pursuing this idea we noted that it has been shown that two BK channel β subunits (β2 and β3) produce BK inactivation by inserting their cytoplasmic N-terminal domains into the a subunit in such a way as to inhibit ion flow through the pore.27–30 The N-terminus of IK1 channels shares a general similarity with the N-termini of these two BK beta subunits so may act via a similar mechanism. We found that a peptide derived from the IK1 N-terminus was an effective open channel blocker of BK channels. Significantly, activation of IK1 channels competed with the peptide resulting in a significantly slowed peptide on-rate. This latter result demonstrates that the mechanism of IK1-induced inhibition of BK channels includes events involving the cytoplasmic end of the BK channel permeation pathway. The simplest model to account for these results is that IK1 channels are located sufficiently close to BK channels that activation of IK1 channels causes their N-termini to insert through cytoplasmic side portals in the BK channels and so inhibit ion permeation through the BK pore.
Results
BK channel inhibition is tightly linked to IK1 activation
As noted in Introduction, activation of the Ca2+-dependent K channel, IK1, in mouse parotid acinar (and other) cells inhibits another type of Ca2+-sensitive K channel, the large conductance, BK channel. An example of this behavior is illustrated in Figure 1. The leftmost panel of the upper inserts in part A of this figure illustrates the pulse protocol used—in this case the membrane voltage (Vm) was +50 mV. Since BK channels are activated by voltage as well as by Ca2+, this depolarization elicited the expected time-dependent BK current as illustrated (Control). The 80 nM of free Ca2+ in the pipette was too low to activate detectable IK1 current. Application of 10 μM of DCEBIO, a chemical activator for IK1,31 triggered the expected time- and voltage-independent current through these channels. The current trace labeled (DCEBIO) was obtained after the IK1 activation reached steady state. It is apparent that the time-dependent BK current was reduced when IK1 channels were activated. DCEBIO has no effects on BK channels in these same types of cells from mice with the IK1 gene ablated.22 The IK1-induced reduction of BK current was readily reversible (Recovery): 96% of control in this example; in a total of 9 such experiments the mean recovery was 94 ± 3% with only one recovery below 90%.
Figure 1.
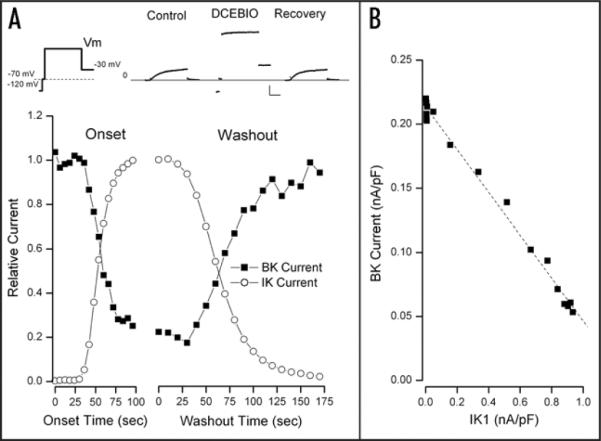
Time course of IK1-induced inhibition of BK current. This, and all cells were patched with a solution containing 80 nM free Ca2+. (A) Top: currents recorded with the pulse protocol illustrated on the left with the main test potential at +50 mV. Cells were held at −70 mV; a 5 msec step to −120 mV was followed by a 5 msec return to the −70 mV holding potential. This brief return to −70 mV serves to monitor for leak currents since −70 mV is slightly depolarized from the K+ equilibrium potential, K channel current will be positive at this voltage and leak current will be negative. The voltage protocol continues with a 70–100 msec pulse to the test potential followed by a step to −30 mV. The currents resulting from this protocol are shown before (Control) during (DCEBIO) and after (Recovery) application of 10 μM DCEBIO. Lines serve only to connect the data points and have no significance. Calib: 0.5 nA/pF, 20 msec. Main: IK1 (○) and BK (■) current components (see Methods) normalized to their maximum average values and shown during application (Onset) and washout of 10 μM DCEBIO. (B) The amount of BK current at the test voltage of +50 mV as a function of the IK1 current at this same potential. The dashed line is from the fit of a simple linear relationship between these two parameters (see text for details).
Because BK channel currents are time dependent and IK1 channel currents are not, it is fairly easy to separate these, and get an accurate estimate of each component (see Materials and Methods). The main part of Figure 1A shows the relative magnitudes of BK (■) and IK1 (◯) currents (at +50 mV) obtained every 6 seconds during application and every 10 seconds during washout of DCEBIO. The reciprocal nature of the IK1/BK relationship is apparent: as IK1 currents gradually increased as a result of DCEBIO-induced activation, BK currents became inhibited. Washout of DCEBIO removed IK1 activation and the maxi-K currents recovered with a time course similar to the removal of IK1 activation.
The very tight link between BK current and IK1 activation can be seen in Figure 1B where we show the amount of BK current as a function of the IK1 current level recorded during washout of DCEBIO. There was a strong, inverse linear relationship between these two channel currents. The correlation coefficient for the linear fit was 0.9957. A similar analysis was made in a total of nine experiments and the mean correlation coefficient was 0.9953 ± 0.0014 of which the two smallest values were 0.9856 and 0.9910.
Since DCEBIO and its parent compound 1-EBIO increase IK1 current by increasing the channel open probability31–33 and not by increasing the single channel current or the number of channels, the inverse linear relationship seen in Figure 1B means that the BK current is inversely proportional to the IK1 channel open probability, PIK1. One simple way that such a relationship could occur is if each BK channel is paired with one IK1 channel and the probability that IK1 activation inhibits the BK channel is simply proportional to the IK1 channel open probability. Thus, the BK current will be proportional to the probability that the IK1 channel is NOT activated or 1 – αPIK1, where a represents the likelihood of BK inhibition for a given amount of IK1 activation, which predicts the simple, linear inverse relationship that is observed.
Another example of the tight link between IK1 channels and the inhibition of BK current is illustrated in Figure 2. In part A of this figure we show the relative magnitudes of IK1 and BK channel currents as a function of time during application of, first, 10 μM DCEBIO and then, in the continued presence of DCEBIO, 1 μM of the IK1 inhibitor TRAM-34.34 As in the experiment of Figure 1, activation of IK1 channels by DCEBIO inhibited BK channel current—in this case by 60%. Subsequent block of IK1 channels by TRAM-34 reversed the BK current inhibition (in this case to 95% of the control value). In Figure 2B we again show the inverse linear relationship between BK current and IK1 current during onset of DCEBIO (■). The tight link between these two channels is emphasized by the fact that exactly the same relationship between the currents was observed during onset and block of IK1 channel by TRAM-34 (○). Similar results were obtained in a total of 5 such experiments with TRAM-34 levels of 1 or 10 μM: the mean block of BK current by IK1 activation with DCEBIO was 66 ± 6% (N = 5) and the mean BK level after IK1 block by TRAM-34 was 88 ± 3% of control.
Figure 2.
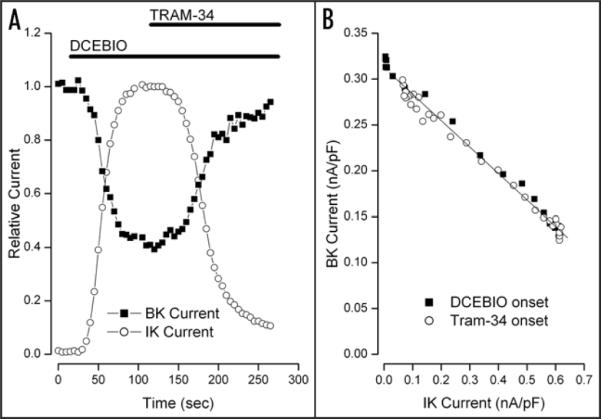
TRAM-34 reversal of BK current inhibition. (A) Time course of relative BK (■) and IK1 (○) current components at +50 mV recorded every 5 sec during application of, first, 10 μM DCEBIO and then 10 μM DCEBIO + 1 μM TRAM-34 as indicated by the solid bars. (B) The amount of BK current at the test voltage of +50 mV as a function of the IK1 current activated during application of DCEBIO (■) and during inhibition of IK1 current by TRAM-34 (○). The solid line is a linear fit to the data.
IK1-mediated inhibition of BK channels has little voltage dependence
As a step toward understanding the mechanism underlying the IK1-induced inhibition of BK channels, we examined this process over a range of membrane potentials. Since IK1 channel activation has little or no voltage dependence, it might be expected that its inhibition of BK channels might also have little or no voltage dependence. The results in Figure 3 show that was, indeed, the case. Part A of this figure illustrates the IK1-induced inhibition of BK channels in a parotid acinar cell at several test voltages. The inset shows raw data at several test voltages in the absence (Control) and presence of IK1 activation by DCEBIO. As in the data of Figure 1, large time and voltage dependent BK channel currents were recorded in the absence of IK1 activation. DCEBIO activated the time and voltage independent IK1 currents, which caused a decrease in the time dependent BK currents. The main part of the figure shows the voltage dependence of current recorded at the end of the test pulses. In the absence of IK1 activation, the current (■) was strongly outward rectifying reflecting the voltage dependence of BK channel gating. Upon IK1 activation, the current at the end of the pulse (○) reflected the presence of IK1 currents at all voltages and an apparent reduction in rectifying BK channel current.
Figure 3.
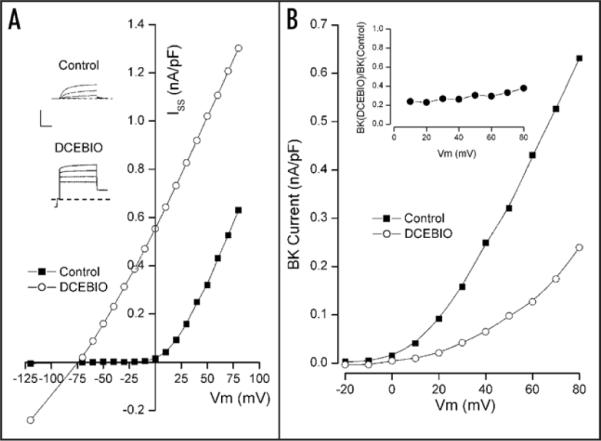
Test for voltage-dependence of IK1-induced inhibition of BK current. Shown in the inset are sample patch clamp current records in the absence (Control) and presence (DCEBIO) of 10 μM DCEBIO. Calib: 0.5 nA/pF, 20 msec. Main: total current measured at the end of the pulses to the indicated voltages in the absence (■) and presence (○) of 10 μM DCEBIO. (B) BK current component at the indicated potentials in the absence (■) and presence (○) of 10 μM DCEBIO. Inset: ratio of BK current in the presence of 10 μM DCEBIO to the control BK level at the indicated test voltages. All lines serve only to connect the data points.
The main part of Figure 3B shows the voltage dependence of just the time-dependent, BK current components recorded before (■) and during (○) activation of IK1 channels by DCEBIO. It is apparent that, as usual, the activation of IK1 channels caused a significant reduction in BK current. The inset in Figure 3B shows the ratio of BK current with IK1 activated to that in control conditions. It can be seen that the IK1-induced inhibition of BK channels occurred with very little voltage dependence.
We did a similar analysis in a total of 19 cells and in 15 of these cells the activation of IK1 produced a large reduction (58 to 92%) in BK current. The other four cells showed only modest reductions and were omitted from the analysis. We do not know the reason for this variability but we have recently found that the IK1-induced inhibition of BK is a steep function of cell cholesterol35 and we speculate that cell-to-cell variations in cholesterol level may underlie the above variability. The pooled results from the 15 cells with large reductions in BK current are illustrated in Figure 4. Part A of this figure shows the mean BK current (normalized to the value at +50 mV) recorded in the absence (■) and presence of 10 μM DCEBIO (○). In part B of Figure 4 we show the mean ratio of BK current values with IK1 activated to phase of control levels. As with the particular example shown in the inset of Figure 3B, there was relatively little and no consistent voltage dependence to the action of IK1 activation.
Figure 4.
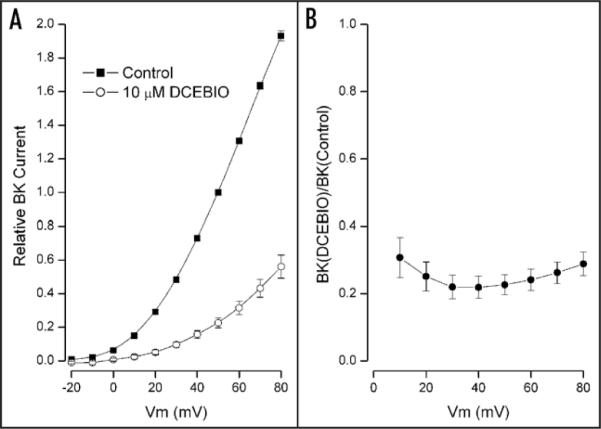
Pooled data testing the voltage-dependence of IK1-induced BK inhibition. (A) Time dependent BK current component at the indicated potentials in the absence (■) and presence (○) of 10 μM DCEBIO. Mean values from 15 cells with standard error limits. The data have been normalized to the control BK current value at +50 mV. (B) Ratio of BK current in the presence of 10 μM DCEBIO to the control BK level at the indicated test voltages. Mean values with standard error limits are shown. All lines serve only to connect the data points.
IK1-mediated inhibition of BK channels does not affect BK gating kinetics
The voltage dependence of BK channel activity is also reflected in the kinetics of channel gating. We examined the effects of IK1 activation on the time dependence of BK activation by fitting a single exponential time function to the BK currents. This was done using 2 μM DCEBIO to produce a smaller amount of IK1 activation because use of the higher, 10 μM concentration often produced so much inhibition of BK current that it was difficult to get a reliable fit over a broad voltage range. Figure 5A shows BK current-voltage relations (pooled from 5 cells) before (■) and during IK1 activation with 2 μM DCEBIO (○). As with the larger amount of IK1 activation (Figs. 3 and 4), the lower degree of IK1 activation produced a simple, voltage-independent reduction of BK current—in this case a 50% reduction (dashed line).
Figure 5.
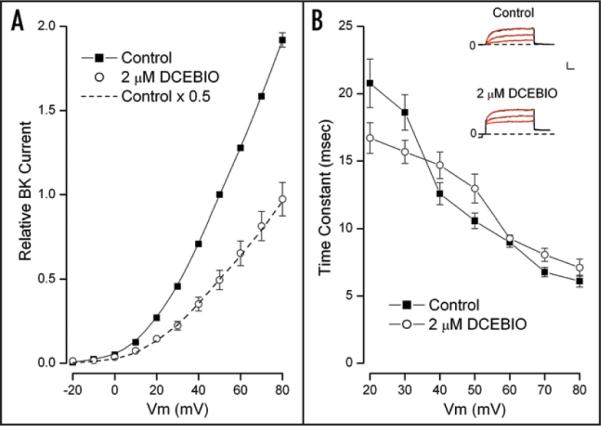
BK current-voltage relation and gating kinetics in 2 μM DCEBIO. (A) Time dependent BK current component at the indicated potentials in the absence (■) and presence (○) of 2 μM DCEBIO. Mean values from 5 cells with standard error limits. The data have been normalized to the control BK current value at +50 mV. The dashed line is the control data scaled by a constant factor of 0.5. (B) Inset: raw current data in response to voltage steps to 30, 50 and 70 mV in the absence (Control) and presence of 2 μM DCEBIO. The red lines are single exponential time function fits to the data. Calib: 0.25 nA/pF, 10 msec. Main: mean values of the fitted time constants with standard error limits at the indicated membrane potentials in the absence (■) and presence (○) of 2 μM DCEBIO.
The inset of Figure 5B contains currents (black traces) recorded in the absence (Control) and presence of 2 μM DCEBIO. Shown also (red) are fits of single exponential time functions to the BK currents which are clearly accurate representations of the data. The main part of Figure 5B shows the voltage dependence of the time constants from these fits in control conditions (■) and during IK1 activation with 2 μM DCEBIO (○). It is apparent from these data that the BK current inhibition was not accompanied by any large effect on the BK kinetics. Indeed, use of the Student's paired t-test revealed no statistically significant difference at any voltage. Thus, even though IK1 activation caused a large change in BK channel current, there was no associated effect on BK channel gating kinetics.
The IK1 N-terminus
As noted in Introduction, the IK1-induced inhibition of BK channels occurs in small, cytoplasm-free inside-out patches.22 This observation and the above data showing that the interaction was essentially voltage-independent led us to hypothesize that the inhibition mechanism may be a direct interaction between the two channel proteins. The voltage independent inhibition of BK current by IK1 channel activation and the lack of any alteration in BK gating kinetics is suggestive of a simple block mechanism—a block in the cytoplasmic end of the BK pore away from the membrane electric field. As noted in Introduction, the N-terminal region of IK1 is similar to the N-terminal region of BK channel β2 and β3 subunits in that it contains a high density of charged residues:

It has been suggested36,37 that the N-terminus of these inactivating BK channel β subunits inserts through large cytoplasmic side portals in the BK protein in such a way as to inhibit ion flow through the pore. Internal application of peptides made from these BK β subunits produce a time dependent block of BK channel currents.27,38 If the IK1 N-terminal acts in a similar way to these BK β subunits to inhibit BK channels, then a peptide derived from this region should also produce a time dependent block of BK channels. The data presented in Figure 6 shows that this expectation was met.
Figure 6.
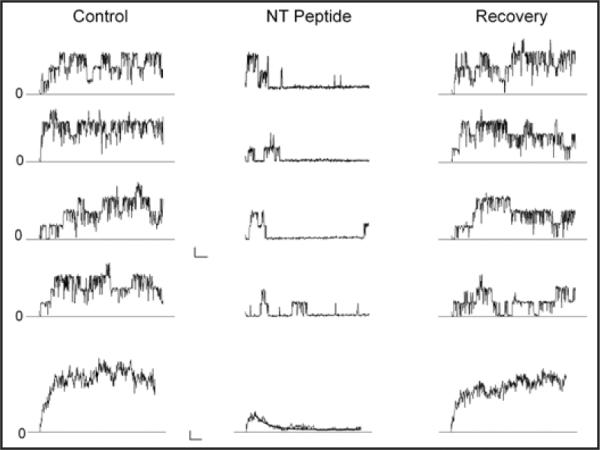
Time dependent block by the IK1 N-terminal peptide. Currents in response to 250 msec voltage clamp pulses to +50 mV from inside/out patches containing a few BK channels. The vertical panels (left to right) contain current data before (Control), during (NT Peptide), and after removal (Recovery) of 25 μM of the IK1 N-terminal peptide. The first 4 records in each panel are representative examples of current from a single voltage application; the bottom record is the average of several individual records: 7, 16 and 25 for the Control, NT peptide, and Recovery conditions, respectively. The black line in the ensemble average trace with the NT peptide is from the fit of an exponential function to the data-see text for details. Calib: 10 pA/25 msec for singles and 5 pA/25 msec for ensemble averages.
The IK1 N-terminal peptide and block of BK channels
Shown in Figure 6 are BK currents recorded from an inside/out patch in the absence (Control), presence (NT peptide), and after removal (Recovery) of 25 μM of a peptide consisting of the first 24 amino acids of the IK1 N-terminus. For each condition, four example records in response to a 250 msec pulse to +50 mV are illustrated along with an average of several such records (bottom traces). It is apparent from these data that there were only a few BK channels in such membrane patches. In the absence of peptide, BK channels activated quickly after the depolarization and current persisted for the duration of the pulse. The bottom, average record confirms this observation. With the peptide present BK channels still activated rather quickly but their activity was reduced at later times during the pulse; again, this conclusion is substantiated by the mean behavior illustrated in the lower record. That is, the IK1 N-terminal peptide produced a time dependent block of BK channels just as do peptides made from BK β subunit N-termini27,38 (reviewed in ref. 28). The black line in the ensemble average with the peptide is a single exponential function with a time constant of 27 msec. In 5 similar, inside/out experiments like that illustrated in Figure 6 with 25 μM peptide, we obtained an average blocking time constant of 21 ± 3 msec. While we did not specifically determine the peptide dissociation constant, the 25 μM concentration used blocked a very large fraction of the current so the apparent Kd for block will be much lower than this concentration. The steady- state block was in the range of 85%–90% with 25 μM peptide so the Kd can be estimated to be near 3 μM.
Competition between IK1 activation and block by the N-terminal peptide
The data of Figure 6 show that a peptide of the N-terminus of IK1 inhibited BK channels. This result is consistent with this structure being the molecular element responsible for the IK1-induced inhibition of BK but is certainly not sufficient to confirm the hypothesis. If, as suggested, the N-terminus of IK1 physically inhibits BK channels, then the activation of IK1 should compete with block by the exogenous peptide and slow the peptide on-rate. A quantitative prediction of this competition can be made by considering that the BK channel can be inhibited by IK1 activation or by the peptide in a mutually exclusive manor. Previous work with peptides derived from the BK β2 subunit showed that the peptides block BK channels in a bi-molecular fashion.27,38 The monoexponential nature of the time dependence of block by the IK1 N-terminal peptide (see Figs. 6 and 7 inset) suggests a similar process and so can be described by:
where (NP) represents the N-terminal peptide with intrinsic on and off-rates given by kon and koff, respectively. BK and NP•BK represent the unblocked and blocked channel, respectively.
Figure 7.

Peptide block rate and IK1 activation. Inset: Sample, whole-cell current records in response to a voltage pulse to 50 mV from an experiment with 250 μM of the IK1 N-terminal peptide in the patch pipette. Data are shown before (Control), during (DCEBIO), after (Washout) application of 10 μM DCEBIO. The red lines are the results of single exponential time function fits to the data with the indicated time constant values. Calib: 0.2 nA/pF, 100 msec. Main: The peptide block rate (1/τ) as a function of the IK1 level during washout of DCEBIO. The dashed line is a linear fit to the data.
The time dependent peptide blocking action will be described by:
| (1) |
where P NP•BK and PBK represent the probability that the BK channel is blocked and not blocked by the N-terminal peptide, respectively. The relationship among the probabilities of finding the BK channel in one of its three states is:
where PIK•BK is the probability that the BK channel is inhibited by IK1. As noted above in the analysis associated with Figure 1B, in the absence of the peptide, PIK•BK is proportional to the IK1 open probability so PIK•BK = αPIK. However, with the peptide present in a mutually exclusive way,
so the relationship among the probabilities becomes:
| (2) |
For high peptide conditions, the koff term in Eq. 1 can be ignored and Eqs. 1 and 2 can be solved for the time course of the peptide blocking event and this will have an apparent block rate given by:
| (3) |
which predicts a linear relationship between the block rate and IK1 open probability (and IK1 current).
A sufficiently accurate, quantitative test of this prediction is difficult to make with the few channels present with inside/out patches. Therefore, we used a whole-cell approach with the peptide in the pipette. Preliminary experiments with even 75 μM peptide produced little time dependent block even several minutes after achieving whole-cell mode-suggesting limited cell dialysis of the peptide. Thus, we used 250 μM of the IK1 N-terminal peptide and the experiment described in Figure 7 illustrates the results obtained.
As can be seen in the inset of Figure 7 (Control), the peptide produced a clear, time-dependent inhibition of the BK current. The time dependent block was well described by a single exponential function (red trace) with a time constant of 37 msec. After activation of IK1 with DCEBIO (middle trace), the rate of peptide block was considerably slowed and the blocking time constant was 68 msec. After removal of DCEBIO and the consequent reversal of IK1 activation, the rate of peptide block was faster again—a 28 msec time blocking time constant. The faster block rate after DCEBIO may reflect continued cell dialysis with the peptide. We computed the mean peptide block rate in the absence of IK1 activation from the Control and Washout time constants as 30 sec−1. The block rate with IK1 activated was 15 sec−1. That is, IK1 activation decreased the peptide block rate by a factor of two. Nine of ten experiments showed a similar result with IK1 activation slowing the rate of N-terminal peptide block by a mean factor of 2.8 ± 0.59.
The close association between IK1 activation and the peptide block rate can be seen in the main part of Figure 7. The reduction in IK1 activation following DCEBIO removal is sufficiently slow to determine the peptide block rate at several levels of IK1 activation. It is clear that increased IK1 activation slowed the apparent rate of block by the N-terminal peptide. The dashed line in the figure, which appears to be a reasonably accurate description of the data, is reminiscent of the linear relationship between IK1 activation and BK channel inhibition shown in Figure 1B and is what was predicted in Eq. 3 for the relationship between the peptide block rate and the level of IK1 activation. The correlation coefficient for this inverse linear fit was 0.946. Four of 9 such analyses yielded R values greater than 0.925 with an overall mean of 0.726 ± 0.087; all but one had p-values ≤0.064.
The results of Figure 7 show that there was clear competition between BK inhibition produced by IK1 activation and block by the IK1 N-terminal peptide. The results of the simple competition model account for both the linear relationship between IK1 activation and BK inhibition (Fig. 1B) and for the linear relationship between the apparent block rate of the exogenous N-terminal peptide and IK1 activation (Fig. 7). These results also demonstrate that the IK1-induced inhibition of BK channels involves events in the cytoplasmic part of the inner pore otherwise the peptide would not be able to compete with the IK1-induced inhibition process.
Discussion
The results presented above showed a very tight link between IK1 channel activity and BK channel current inhibition: IK1 activation with DCEBIO inhibited BK current and IK1 channel block by TRAM-34 exactly restored the inhibited BK current. Analogous to the fact that IK1 channel activation is voltage independent the IK1-mediated inhibition of BK channels had little or no voltage dependence. In addition IK1 activation inhibited BK channels with no alteration in gating kinetics. We have previously shown that the BK single channel current level was not affected.22 The moderately rapid, reversible inhibition of BK (see Introduction) argues against a change in the number of membrane channels. Thus, we conclude that IK1 activation produced a mostly voltage independent reduction of the BK channel open probability.
We also found that a peptide derived from the N-terminus of the IK1 channel was a potent, open channel blocker of BK channels similar to the action of N-terminus peptides from BK β2 and β3 subunits-subunits that produce an inactivation of BK channels by inhibiting ion flux through the channel pore. Importantly, the activation of IK1 channels interfered with BK channel block by the IK1 N-terminus peptide. This result demonstrates that the mechanism of the IK1-induced inhibition of BK channels involves events in the cytoplasmic part of the BK channel. The simplest model to account for all these observations is based on what is known about BK channel inhibition by the BK β subunits (reviewed in ref. 29). In our simple model we consider IK1 and BK channels to exist in close proximity and the activation of IK1 channels causes its N-terminus to insert into a cytoplasmic side portal and inhibit ion flow through the BK pore.
For this model to be viable, the IK1 N-terminal must be long enough to pass through side portals in the large cytosolic domains that hang from the membrane embedded part of the BK channel.39–43 The β2 and β3 amino terminal regions up to the first membrane spanning domain are predicted to be about 46 and 35 amino acids, respectively; the IK1 N-terminus about 25 amino acids. While the IK1 N-terminus is considerably shorter than its counterparts in the β2 and β3 subunits, it is likely quite long enough to achieve inhibition of the BK channel. Studies by Xia et al.44 showed that an artificial poly-Q linker of 12 glutamine residues connecting the terminal three amino acids of the β2 N-terminus to the first membrane-spanning domain was capable of producing BK inactivation. In addition, shortening the β2 N-terminus to only 26 amino acids was also effective. Thus, the IK1 N-terminus appears long enough to reach sites in the BK channel capable of inhibiting current flow.
It makes sense that a peptide of the BK β2 N-terminus inhibits BK channels since these two proteins are physiologically relevant partners but these channels are also blocked by a peptide made from the Shaker channel N-terminus45,46—a region, like the β subunits and IK1 N-termini, that contains a high concentration of charged amino acids. Since maxi-K and Shaker channels are entirely unrelated, this result suggests a certain promiscuity with regard to maxi-K channels and strongly charged peptides. However, if the properties of block by the IK1 peptide have more in common with those of the β2 than the Shaker peptide, that would suggest a more specific relationship between the IK1 peptide and the maxi-K channel. As described above, we estimated an approximate 3 μM Kd value for the IK1 N-terminal peptide and a blocking time constant of 21 msec at 25 μM, which indicates on- and off-rates near 2 × 106 M−1sec−1, and 6 sec−1, respectively. The estimated on-rate for the IK1 N-terminal peptide is comparable to the on-rate for block by both the β2 and the Shaker peptide, which are roughly 3 × 106 M−1sec−1 and 6 × 106 M−1sec−1, respectively.27,38,45,46 The biggest difference (more than 100-fold) between the β2 and the Shaker peptides is in their off rates which are roughly 0.8 sec−1 and 110 sec−1, respectively.27,38,45,46 While our estimated off-rate for the IK1 N-terminal peptide is about 7-fold faster than the β2 peptide, it is almost 20-fold slower than the Shaker peptide. The very slow off-rate of the β2 and IK1 peptides indicates a highly favorable binding to the BK channel—a useful prerequisite for a physiological inhibition mechanism and consistent with the idea that the N-terminal region of the IK1 channel that, upon activation of IK1, inhibits BK.
As shown in Results, BK channel current was a strictly inverse linear function of IK1 current activation. Likewise, the apparent on-rate for BK block by the IK1 N-terminal peptide was inversely linearly related to IK1 activation. A simple model that included a single IK1 channel associated with each BK channel was able to account for how BK current and the apparent peptide on-rate can both be an inverse linear function of IK1 activation. In our previous work22 we reported a non-linear relationship between BK and IK1 current and suggested that more than one IK1 channel was associated with each BK channel. We now believe that result was contaminated by incomplete series resistance compensation and that the most likely arrangement is for one BK channel to be inhibited by only one IK1 channel. Each tetrameric IK1 channel will have four amino terminal regions but the distance constraints discussed above would seem to make it unlikely that all four N-termini could be positioned close enough to the central axis of the BK channel to produce the observed inhibition. The three dimensional orientation of the associated IK1 and BK channels is unknown but it seems reasonable to expect that only one of the four N-terminal regions is the active one.
Our simple model is able to account for both the inverse linear relationship between IK1 channel activation and BK current inhibition and the inverse linear relationship between the IK1 N-terminal peptide block rate and IK1 channel activation. The overarching assumption in this model is that the ability of the IK1 N-terminus to inhibit the BK channel is directly related to the IK1 channel open probability. That is, whatever conformational change occurs when the IK1 channel opens is also accompanied by a change in its N-terminus that puts it in a position to inhibit the BK channel. Recent work strongly47,48 suggests that gating of the IK1 (and SK) channels occurs at the level of the selectivity filter which might be seen to question an N-terminus movement. However, it should be noted that even if IK1 gating does indeed occur at the selectivity filter, this does not rule out additional conformational changes in the channel protein. There is no direct data on the position of the IK1 N-terminus and very little in other channels. However, the crystal structures of both an open and closed conformation of the bacterial inward rectifier K channel, KirBac3.1 have been solved49 and these reveal a movement of the N-terminal domain outward, away from the main channel and a conformational change at its extreme terminus. This observation in no way confirms our hypothesis but neither is there direct evidence against it—more work is necessary.
An underlying assumption in our derivation of the inverse relationship between the N-terminal peptide block rate and IK1 activation is that the IK1 openings and closings are much faster than the peptide block kinetics. The effective peptide blocking time constant (from the type of experiments illustrated in Fig. 7) was about 30 msec and, as discussed above, we estimate the off-rate to be near 6 sec−1 or an off time constant of about 160 msec. Thus, our assumption is valid if the IK1 open and closed times with DCEBIO activation are much faster than these values. We did not measure these kinetics in our experiments and we are unaware of published data with DCEBIO. However, these measurements have been made with 1-EBIO-stimulated IK1 channels.50 This work showed that IK1 channels have two open states and channels activated with 1-EBIO have dwell times in these two states of 1 and 7 msec. Three closed times have been observed but in channels activated by 1-EBIO, 97% of the events are accounted for by two closed times of about 1.6 msec (66%) and 5 msec (31%). These values are all much faster than the peptide on- and off-rates and so validate the assumption behind our mathematical model.
A potential argument against the idea that it is the IK1 N-terminus that physically inhibits BK channels is the extensive work done44 on the structure-activity relationship of the BK β2 subunit. This work demonstrated a critical role for three hydro-phobic residues immediately adjacent to the initial N-terminus methionine. This and previous work with inactivating peptides on Shaker K channels (reviewed in ref. 51) have lead to the general conclusion that such a hydrophobic “tail” is a requirement for any ball-and-chain inhibition mechanism. However, a detailed comparison (Fig. 8) of many inactivating K channels and β subunits reveals several N-termini without a strong hydrophobic end. For this analysis we used a 5-point, color-coded hydrophobic scale employing the experimentally determined, whole-residue interface approach described in ref. 52. As can be seen in the figure, the BK β2 N-terminus has, perhaps, the strongest hydrophobic character and so may not be representative of all inactivating N-termini. The IK1 N-terminus is not alone in having a strongly hydrophilic residue near the terminal methionine: the KV1.4 terminus, like the IK1 protein, has a glutamate and the ShakerC and KVβ1.1 structures contain a glutamine. Significantly, the BKβ3 protein has two neutral amino acids (threonine and alanine) and the hydrophilic proline surrounding the very hydrophobic phenylalanine. This analysis suggests that the rules for optimal design of inactivating/inhibiting proteins may be more complex and that the IK1 N-terminus cannot be ruled out a priori.
Figure 8.
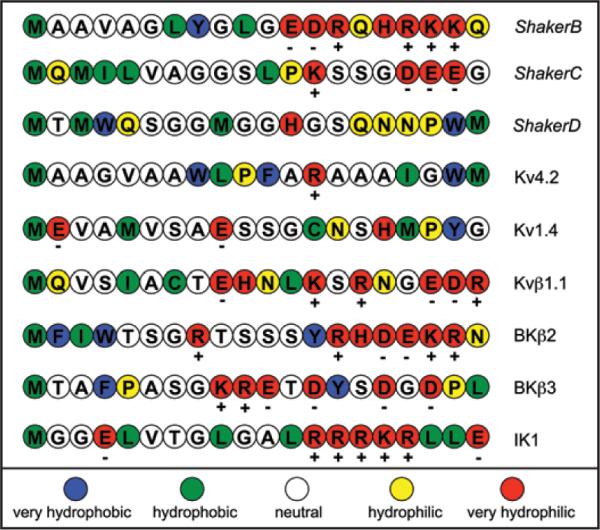
Hydropathy analysis of the N-terminus region of several inactivating K channels and accessory, β, peptides. The single letter amino acid code for the various N-termini are shown in colors representing the relative hydrophobic/hydrophilic character of these amino acids according to the scale at the bottom of the figure. This 5-point relative scale was derived from the experimentally determined, whole-residue interface approach described in reference 52. The figure was designed after a similar presentation in reference 51 and includes sequence data from a similar figure in reference 44.
While there are similarities between the inactivation of BK channels by their β subunits and the IK1-induced BK channel inhibition, there are at least a couple of differences. Exogenous β subunit N-terminus peptides compete with cytoplasmic blockers like TEA,27,28 but inactivation via the native proteins does not— even with such large cytosolic blockers as decamethonium and a 22 amino acid peptide derived from the Shaker inactivating N-terminus.53 In contrast we showed here that IK1 activation clearly competed with block by the IK1 N-terminus peptide. This difference could be due to different kinetics between the various competing structures or perhaps by slightly different blocking locations of the N-terminal regions.
Another apparent difference between the actions of the β subunits and IK1 activation is that the β subunits produce a time dependent block of BK channel current but no such time dependence is apparent upon IK1 activation. That is, either the N-terminal region of the intact β subunits cannot gain final access to the BK side portals until the BK channel protein achieves its open conformation or it is the opening of the BK channel that triggers the N-terminal inactivation. In contrast, it appears that it is the IK1 activation that induces the inhibition of BK channels not the opening of the BK channels. The lack of an apparent time dependence to the IK1-induced inhibition of BK channels could be that the IK1 inhibitory apparatus (whether the N-terminal or not) has access to its inhibitory location independent of the state of the BK channel. Alternatively, but perhaps less likely, the kinetics of IK1-induced BK inhibition may be so fast that no clear time dependence can be seen—analogous to how open channel block by internal TEA has no apparent time dependence. Such rapid block kinetics might be expected to produce a reduction in the BK single channel current level (as does TEA) but no such reduction is seen.22 Note that one of the BK β3 isoforms produces a very rapid BK inactivation and very flickery single channel openings.54
In summary, the results of our study have begun to reveal some of the functional molecular properties of the BK channel inhibition produced when IK1 channels are activated. Unlike many other modulators of BK activity that occurs through a shift of the BK voltage dependence, the IK1-induced inhibition appeared to be an essentially voltage independent reduction in BK channel open probability-reminiscent of a simple pore-block mechanism. We showed that a peptide derived from the N-terminus of the IK1 channel efficiently inhibited the current through BK channels and activation of IK1 channels competed with block by this peptide. The similarities between the N-termini of IK1 channels and BK channel β subunits and the similarities between our results here and the body of work on BK channel inactivation by their β subunits lead us to suggest that activation of an IK1 channel induces its N-terminal to enter a side portal in the cytoplasmic part of an associated BK channel and block current flow through the central pore. Since the IK1/BK interaction can be reconstituted in expression systems,22 a direct way to test this idea would be to truncate or otherwise mutate the IK1 N-terminus. Unfortunately, this domain is critical for the assembly and trafficking of IK1 channels to the surface membrane55 limiting this approach. In the absence of such a direct approach, the specific role of the IK1 N-terminus cannot be ascertained with certainty. Nevertheless, we have shown that the IK1-induced inhibition of BK channels occurs at a cytoplasmically accessible site and has little or no voltage dependence. The ability of the IK1 peptide to efficiently block BK channels with a slow off-rate makes its N-terminus a good candidate for the inhibitory structure.
Materials and Methods
Ethical approval
All animals were housed in a pathogen-free area at the University of Rochester and all experiments were carried out in accordance with procedures reviewed and approved by the local University Committee on Animal Resources. Mice were sacrificed by exsanguination following exposure to CO2.
Parotid acinar cell preparations
Detailed descriptions of the procedures for producing single parotid acinar cells have been previously described.22 Briefly, glands from mice (BlackSwiss × 129/SvJ hybrid) were finely minced and digested with trypsin after which the cells were dissociated with Liberase and, finally, plated onto 5 mm diameter glass coverslips. All dissociation solutions were gassed continuously with 95% O2 + 5% CO2 and maintained at 37°C. The procedures for animal handling, maintenance, and surgery were approved by the University of Rochester Committee on Animal Resources.
Electrophysiology
Whole-cell patch clamp recordings were done at room temperature (20–22°C) with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Currents were filtered (at 5 kHz) by a 4-pole Bessel low-pass filter prior to being sampled by a 12 bit analog/digital converter controlled by a personal computer. Patch pipettes were constructed from quartz (Garner Glass Co.,). Compensation for access (series) resistance was made with the patch clamp circuitry. This compensation is especially important in testing if the mechanism for the reduction of BK current by IK1 channel activity is voltage-dependent (Figs. 3–5). In these tests the mean measured access resistance (and SEM value) was 5.8 ± 0.28 MΩ (N = 35). The mean series resistance compensation was 88 ± 0.81% and the mean uncompensated resistance was 0.69 ± 0.07 MΩ. Unless otherwise indicated, results are reported as mean ± SEM values.
As described in the text, the total current measured is composed of time-independent current from IK1 channels and time-dependent current from BK channels. In addition to simply determining the total steady-state current (e.g., the current-voltage relations in Fig. 3A), we also measured the individual IK1 and BK components. The IK1 current was measured immediately after the settling of the capacity transient at a given test potential before BK channels are significantly activated. The BK current was obtained as the difference between the current at the end of the pulse and the current immediately after the capacity transient. We have previously shown, through the use of the specific BK channel blocker, paxilline, and with gene-ablation studies8,22 that all the time dependent current is through BK channels.
The external solution for both whole-cell and inside/out patch recordings consisted of (in mM): 135 Na-glutamate, 5 K-glutamate, 2 CaCl2, 2 MgCl2, 10 HEPES (pH 7.2). For whole-cell experiments we used an internal solution that consisted of 135 mM K-glutamate, 10 mM HEPES (pH 7.2), 10 mM BAPTA, and with CaCl2 added to establish an 80 nM Ca2+ concentration;56 see also http://www.stanford.edu/~cpatton/maxc.html). For inside/out patches the bath (internal) solution was the same as for whole-cell experiments except that 5 mM EGTA replaced the BAPTA and, again, CaCl2 added to establish 80 nM free Ca2+. The peptide derived from the first 24 amino acids of the IK1 N-terminus (mggelvtglgalrrrkrlleqekr) was provided by Alpha Diagnostics International, San Antonio, Texas. This peptide was acetylated at its amino terminal and amidated at its carboxy-terminus and was provided at 92% purity. DCEBIO (5,6-Dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one) and TRAM-34 ([1-(2-chlorophenyl)diphenyl)methyl]-1H-pryazol) were obtained from Sigma Chemical Co., (St. Louis, MO).
Acknowledgements
We are grateful to James E. Melvin for discussions throughout the course of this work and for critically reading the manuscript. We thank the following members of Dr. Melvin's lab: Laurie Koek for maintaining the mouse colonies and Mark Wagner, Victor Romanenko, and Margarit Sievert for preparing the parotid acinar cells. This work was supported by National Institutes of Health Grant DE016960.
Abbreviations
- BK
a big conductance, Ca2+-activated K channel
- IK1
an intermediate conductance, Ca2+-activated K channel
- DCEBIO
5,6-dichloro-1-ethyl-1,3-dihydro-2H-benzimidazol-2-one
- TRAM-34
[1-(2-chlorophenyl)diphenyl)methyl]-1H-pryazol
References
- 1.Hille B. Ion Channels of Excitable Membranes. Sinauer Associates, Inc; Sunderland, Mass: 2001. [Google Scholar]
- 2.Jensen BS, Odum N, Jorgensen NK, Christophersen P, Olesen SP. Inhibition of T cell proliferation by selective block of Ca2+-activated K+ channels. Proc Natl Acad Sci USA. 1999;96:10917–21. doi: 10.1073/pnas.96.19.10917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khanna R, Chang MC, Joiner WJ, Kaczmarek LK, Schlichter LC. hSK4/hIK1, a calmodulin-binding KCa channel in human T lymphocytes. Roles in proliferation and volume regulation. J Biol Chem. 1999;274:14838–49. doi: 10.1074/jbc.274.21.14838. [DOI] [PubMed] [Google Scholar]
- 4.Vandorpe DH, Shmukler BE, Jiang L, Lim B, Maylie J, Adelman JP, et al. cDNA cloning and functional characterization of the mouse Ca2+-gated K+ channel, mIK1. Roles in regulatory volume decrease and erythroid differentiation. J Biol Chem. 1998;273:21542–53. doi: 10.1074/jbc.273.34.21542. [DOI] [PubMed] [Google Scholar]
- 5.Begenisich T, Nakamoto T, Ovitt CE, Nehrke K, Brugnara C, Alper SL, et al. Physiological roles of the intermediate conductance, Ca2+-activated potassium channel Kcnn4. J Biol Chem. 2004;279:47681–7. doi: 10.1074/jbc.M409627200. [DOI] [PubMed] [Google Scholar]
- 6.Petersen OH, Maruyama Y. Calcium-activated potassium channels and their role in secretion. Nature. 1984;307:693–6. doi: 10.1038/307693a0. [DOI] [PubMed] [Google Scholar]
- 7.Melvin JE, Yule D, Shuttleworth T, Begenisich T. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol. 2005;67:445–69. doi: 10.1146/annurev.physiol.67.041703.084745. [DOI] [PubMed] [Google Scholar]
- 8.Romanenko V, Nakamoto T, Srivastava A, Melvin JE, Begenisich T. Molecular identification and physiological roles of parotid acinar cell maxi-K channels. J Biol Chem. 2006;281:27964–72. doi: 10.1074/jbc.M603871200. [DOI] [PubMed] [Google Scholar]
- 9.Ishii TM, Silvia C, Hirschberg B, Bond CT, Adelman JP, Maylie J. A human intermediate conductance calcium-activated potassium channel. Proc Natl Acad Sci USA. 1997;94:11651–6. doi: 10.1073/pnas.94.21.11651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Logsdon NJ, Kang J, Togo JA, Christian EP, Aiyar J. A novel gene, hKCa4, encodes the calcium-activated potassium channel in human T lymphocytes. J Biol Chem. 1997;272:32723–6. doi: 10.1074/jbc.272.52.32723. [DOI] [PubMed] [Google Scholar]
- 11.Maruyama Y, Gallacher DV, Petersen OH. Voltage and Ca2+-activated K+ channel in baso-lateral acinar cell membranes of mammalian salivary glands. Nature. 1983;302:827–9. doi: 10.1038/302827a0. [DOI] [PubMed] [Google Scholar]
- 12.Nehrke K, Quinn CC, Begenisich T. Molecular identification of Ca2+-activated K+ channels in parotid acinar cells. Am J Physiol Cell Physiol. 2003;284:535–46. doi: 10.1152/ajpcell.00044.2002. [DOI] [PubMed] [Google Scholar]
- 13.Takahata T, Hayashi M, Ishikawa T. SK4/IK1-like channels mediate TEA-insensitive, Ca2+-activated K+ currents in bovine parotid acinar cells. Am J Physiol Cell Physiol. 2003;284:127–44. doi: 10.1152/ajpcell.00250.2002. [DOI] [PubMed] [Google Scholar]
- 14.Warth R, Hamm K, Bleich M, Kunzelmann K, von Hahn T, Schreiber R, et al. Molecular and functional characterization of the small Ca2+-regulated K+ channel (rSK4) of colonic crypts. Pflugers Arch. 1999;438:437–44. doi: 10.1007/s004249900059. [DOI] [PubMed] [Google Scholar]
- 15.Joiner WJ, Basavappa S, Vidyasagar S, Nehrke K, Krishnan S, Binder HJ, et al. Active K+ secretion through multiple KCa-type channels and regulation by IKCa channels in rat proximal colon. Am J Physiol Gastrointest Liver Physiol. 2003;285:185–96. doi: 10.1152/ajpgi.00337.2002. [DOI] [PubMed] [Google Scholar]
- 16.Sausbier M, Matos JE, Sausbier U, Beranek G, Arntz C, Neuhuber W, et al. Distal Colonic K+ Secretion Occurs via BK Channels. J Am Soc Nephrol. 2006;17:1275–82. doi: 10.1681/ASN.2005101111. [DOI] [PubMed] [Google Scholar]
- 17.Hanley PJ, Musset B, Renigunta V, Limberg SH, Dalpke AH, Sus R, et al. Extracellular ATP induces oscillations of intracellular Ca2+ and membrane potential and promotes transcription of IL-6 in macrophages. Proc Natl Acad Sci USA. 2004;101:9479–84. doi: 10.1073/pnas.0400733101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grgic I, Eichler I, Heinau P, Si H, Brakemeier S, Hoyer J, et al. Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler Thromb Vasc Biol. 2005;25:704–9. doi: 10.1161/01.ATV.0000156399.12787.5c. [DOI] [PubMed] [Google Scholar]
- 19.Neylon CB, Lang RJ, Fu Y, Bobik A, Reinhart PH. Molecular cloning and characterization of the intermediate-conductance Ca2+-activated K+ channel in vascular smooth muscle: relationship between KCa channel diversity and smooth muscle cell function. Circ Res. 1999;85:33–43. doi: 10.1161/01.res.85.9.e33. [DOI] [PubMed] [Google Scholar]
- 20.Kohler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, et al. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. 2003;108:1119–25. doi: 10.1161/01.CIR.0000086464.04719.DD. [DOI] [PubMed] [Google Scholar]
- 21.Si H, Grgic I, Heyken WT, Maier T, Hoyer J, Reusch HP, et al. Mitogenic modulation of Ca2+-activated K+ channels in proliferating A7r5 vascular smooth muscle cells. Br J Pharmacol. 2006;148:909–17. doi: 10.1038/sj.bjp.0706793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson J, Begenisich T. Membrane-delimited inhibition of maxi-K channel activity by the intermediate conductance Ca2+-activated K channel. J Gen Physiol. 2006;127:159–69. doi: 10.1085/jgp.200509457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakamoto T, Srivastava A, Romanenko VG, Ovitt CE, Perez-Cornejo P, Arreola J, et al. Functional and molecular characterization of the fluid secretion mechanism in human parotid acinar cells. Am J Physiol Regul Integr Comp Physiol. 2007;292:2380–90. doi: 10.1152/ajpregu.00591.2006. [DOI] [PubMed] [Google Scholar]
- 24.Romanenko VG, Nakamoto T, Srivastava A, Begenisich T, Melvin JE. Regulation of membrane potential and fluid secretion by Ca2+-activated K+ channels in mouse submandibular glands. J Physiol. 2007;581:801–17. doi: 10.1113/jphysiol.2006.127498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gray PT. Oscillations of free cytosolic calcium evoked by cholinergic and catecholaminergic agonists in rat parotid acinar cells. J Physiol. 1988;406:35–53. doi: 10.1113/jphysiol.1988.sp017367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bruce JI, Shuttleworth TJ, Giovannucci DR, Yule DI. Phosphorylation of inositol 1,4,5-trisphosphate receptors in parotid acinar cells. A mechanism for the synergistic effects of cAMP on Ca2+ signaling. J Biol Chem. 2002;277:1340–8. doi: 10.1074/jbc.M106609200. [DOI] [PubMed] [Google Scholar]
- 27.Wallner M, Meera P, Toro L. Molecular basis of fast inactivation in voltage and Ca2+-activated K+ channels: a transmembrane beta-subunit homolog. Proc Natl Acad Sci USA. 1999;96:4137–42. doi: 10.1073/pnas.96.7.4137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xia XM, Ding JP, Lingle CJ. Molecular basis for the inactivation of Ca2+- and voltage-dependent BK channels in adrenal chromaffin cells and rat insulinoma tumor cells. J Neurosci. 1999;19:5255–64. doi: 10.1523/JNEUROSCI.19-13-05255.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Z, Zhou Y, Ding JP, Xia XM, Lingle CJ. A limited access compartment between the pore domain and cytosolic domain of the BK channel. J Neurosci. 2006;26:11833–43. doi: 10.1523/JNEUROSCI.3812-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Yao J, Tong X, Guo Z, Wu Y, Sun L, et al. Interaction sites between the Slo1 pore and the NH2 terminus of the beta2 subunit, probed with a three-residue sensor. J Biol Chem. 2007;282:17720–8. doi: 10.1074/jbc.M607063200. [DOI] [PubMed] [Google Scholar]
- 31.Singh S, Syme CA, Singh AK, Devor DC, Bridges RJ. Benzimidazolone activators of chloride secretion: potential therapeutics for cystic fibrosis and chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2001;296:600–11. [PubMed] [Google Scholar]
- 32.Pedarzani P, Mosbacher J, Rivard A, Cingolani LA, Oliver D, Stocker M, et al. Control of electrical activity in central neurons by modulating the gating of small conductance Ca2+-activated K+ channels. J Biol Chem. 2001;276:9762–9. doi: 10.1074/jbc.M010001200. [DOI] [PubMed] [Google Scholar]
- 33.Pedersen KA, Schroder RL, Skaaning-Jensen B, Strobaek D, Olesen SP, Christophersen P. Activation of the human intermediate-conductance Ca2+-activated K+ channel by 1-ethyl-2-benzimidazolinone is strongly Ca2+-dependent. Biochim Biophys Acta. 1999;1420:231–40. doi: 10.1016/s0005-2736(99)00110-8. [DOI] [PubMed] [Google Scholar]
- 34.Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci USA. 2000;97:8151–6. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Romanenko VG, Roser K, Melvin JE, Begenisich T. The Role of Cell Cholesterol and the Cytoskeleton in the Interaction between IK1 and Maxi-K Channels. Am J Physiol Cell Physiol. 2009;296:878–88. doi: 10.1152/ajpcell.00438.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benzinger GR, Xia XM, Lingle CJ. Direct observation of a preinactivated, open state in BK channels with beta2 subunits. J Gen Physiol. 2006;127:119–31. doi: 10.1085/jgp.200509425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z, Zeng XH, Xia XM, Lingle CJ. N-terminal inactivation domains of beta subunits are protected from trypsin digestion by binding within the antechamber of BK channels. J Gen Physiol. 2009;133:263–82. doi: 10.1085/jgp.200810079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bentrop D, Beyermann M, Wissmann R, Fakler B. NMR structure of the “ball-and-chain” domain of KCNMB2, the beta 2-subunit of large conductance Ca2+- and voltage-activated potassium channels. J Biol Chem. 2001;276:42116–21. doi: 10.1074/jbc.M107118200. [DOI] [PubMed] [Google Scholar]
- 39.Gulbis JM, Zhou M, Mann S, MacKinnon R. Structure of the cytoplasmic beta subunit-T1 assembly of voltage-dependent K+ channels. Science. 2000;289:123–7. doi: 10.1126/science.289.5476.123. [DOI] [PubMed] [Google Scholar]
- 40.Jiang Y, Pico A, Cadene M, Chait BT, MacKinnon R. Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in the human BK channel. Neuron. 2001;29:593–601. doi: 10.1016/s0896-6273(01)00236-7. [DOI] [PubMed] [Google Scholar]
- 41.Sokolova O, Kolmakova-Partensky L, Grigorieff N. Three-dimensional structure of a voltage-gated potassium channel at 2.5 nm resolution. Structure. 2001;9:215–20. doi: 10.1016/s0969-2126(01)00578-0. [DOI] [PubMed] [Google Scholar]
- 42.Jiang Y, Lee A, Chen J, Cadene M, Chait BT, MacKinnon R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 2002;417:515–22. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 43.Xia XM, Zhang X, Lingle CJ. Ligand-dependent activation of Slo family channels is defined by interchangeable cytosolic domains. J Neurosci. 2004;24:5585–91. doi: 10.1523/JNEUROSCI.1296-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia XM, Ding JP, Lingle CJ. Inactivation of BK channels by the NH2 terminus of the beta2 auxiliary subunit: an essential role of a terminal peptide segment of three hydrophobic residues. J Gen Physiol. 2003;121:125–48. doi: 10.1085/jgp.20028667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Foster CD, Chung S, Zagotta WN, Aldrich RW, Levitan IB. A peptide derived from the Shaker B K+ channel produces short and long blocks of reconstituted Ca2+-dependent K+ channels. Neuron. 1992;9:229–36. doi: 10.1016/0896-6273(92)90162-7. [DOI] [PubMed] [Google Scholar]
- 46.Toro L, Stefani E, Latorre R. Internal blockade of a Ca2+-activated K+ channel by Shaker B inactivating “ball” peptide. Neuron. 1992;9:237–45. doi: 10.1016/0896-6273(92)90163-8. [DOI] [PubMed] [Google Scholar]
- 47.Garneau L, Klein H, Banderali U, Longpre-Lauzon A, Parent L, Sauve R. Hydrophobic interactions as key determinants to the KCa3.1 channel closed configuration. An analysis of KCa3.1 mutants constitutively active in zero Ca2+ J Biol Chem. 2009;284:389–403. doi: 10.1074/jbc.M805700200. [DOI] [PubMed] [Google Scholar]
- 48.Bruening-Wright A, Lee WS, Adelman JP, Maylie J. Evidence for a deep pore activation gate in small conductance Ca2+-activated K+ channels. J Gen Physiol. 2007;130:601–10. doi: 10.1085/jgp.200709828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuo A, Domene C, Johnson LN, Doyle DA, Venien-Bryan C. Two different conformational states of the KirBac3.1 potassium channel revealed by electron crystallography. Structure. 2005;13:1463–72. doi: 10.1016/j.str.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 50.Syme CA, Gerlach AC, Singh AK, Devor DC. Pharmacological activation of cloned intermediate- and small-conductance Ca2+-activated K+ channels. Am J Physiol Cell Physiol. 2000;278:570–81. doi: 10.1152/ajpcell.2000.278.3.C570. [DOI] [PubMed] [Google Scholar]
- 51.Kukuljan M, Labarca P, Latorre R. Molecular determinants of ion conduction and inactivation in K+ channels. Am J Physiol. 1995;268:535–56. doi: 10.1152/ajpcell.1995.268.3.C535. [DOI] [PubMed] [Google Scholar]
- 52.Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3:842–8. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- 53.Solaro CR, Ding JP, Li ZW, Lingle CJ. The cytosolic inactivation domains of BKi channels in rat chromaffin cells do not behave like simple, open-channel blockers. Biophys J. 1997;73:819–30. doi: 10.1016/S0006-3495(97)78114-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xia XM, Ding JP, Zeng XH, Duan KL, Lingle CJ. Rectification and rapid activation at low Ca2+ of Ca2+-activated, voltage-dependent BK currents: consequences of rapid inactivation by a novel beta subunit. J Neurosci. 2000;20:4890–903. doi: 10.1523/JNEUROSCI.20-13-04890.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jones HM, Hamilton KL, Papworth GD, Syme CA, Watkins SC, Bradbury NA, et al. Role of the NH2 terminus in the assembly and trafficking of the intermediate conductance Ca2+-activated K+ channel hIK1. J Biol Chem. 2004;279:15531–40. doi: 10.1074/jbc.M400069200. [DOI] [PubMed] [Google Scholar]
- 56.Bers DM, Patton CW, Nuccitelli R. A practical guide to the preparation of Ca2+ buffers. Methods Cell Biol. 1994;40:3–29. doi: 10.1016/s0091-679x(08)61108-5. [DOI] [PubMed] [Google Scholar]