

Abstract
Stereoselective syntheses of both functionalized tetrahydropyran subunits of (−)-lasonolide A are described. These tetrahydropyran rings were constructed using catalytic asymmetric hetero Diels-Alder reactions as the key steps. The C22 quaternary stereocenter present in the upper tetrahydropyran ring was constructed by a stereoselective alkylation and the C9 hydroxy stereochemistry of bottom tetrahydropyran was constructed by a stereoselective epoxidation followed by a regioselective epoxide opening reaction.
Lasonolide A was isolated from Caribbean marine sponge, Forcepia sp, by McConnell and co-workers in 1994.1 Lasonolide A exhibited potent antitumor activity against a range of cancer cell lines in the low nanomolar level. It has shown IC50 values of 8.6 nM and 89 nM against A-549 human lung carcinoma and Panc-1 human pancreatic carcinoma, respectively.1 Furthermore, it showed cell adhesion in the EL-4.IL-2 cell line, which detects signal-transduction agents. Interestingly, the mechanism of action of lasonolide A is still unknown. The natural abundance of lasonolide is very limited. The scarce supply and important biological properties of lasonolide A has attracted much interest in the chemistry and biology of lasonolide A. The structure of lasonolide A was initially determined by extensive NMR studies. Subsequently, its structure was revised through total synthesis2 and biological studies were reported by Lee and co-workers.3 Since then, two more total syntheses and numerous synthetic studies on both tetrahydropyran rings have been reported.4–6 We recently reported an asymmetric total synthesis of lasonolide A.5 Utilizing this synthetic lasonolide A, we have investigated the biological mechanism of action in collaboration with the National Cancer Institute.7 Our studies revealed that lasonolide A uniquely induces premature chromosome condensation which may lead to a new treatment of many disorders. In an effort to further elucidate lasonolide A’s structure-activity relationships as well as to identify its biological target, we sought to improve the synthesis of both tetrahydropyran subunits of lasonolide A. Herein, we report our studies leading to a stereoselective synthesis of both highly-substituted tetrahydropyran rings using asymmetric hetero Diels-Alder reactions as key steps.
Our previous route5 to the upper tetrahydopyran ring utilized a diastereoselective intramolecular 1,3-dipolar cycloaddition. The bicyclic isoxazoline led to the tetrahydropyran ring as well as the C22 quarternary stereocenter. However, the route was long with over 20 steps including a number of steps with low yields. The bottom tetrahydropyran ring was constructed using an asymmetric catalytic hetero Diels-Alder reaction developed by Jacobsen and co-workers.8 However, elaboration of the C9 hydroxy stereochemistry required ketone reduction producing a mixture of diastereoisomers (1:2 minor isomer being the desired isomer), separation, and then recycling of the major isomer through an oxidation/reduction sequence.5 Our retrosynthesis of lasonolide A is summarized in Figure 1. Key macrolactone 2 will be prepared from acyclic precursor 3, which we had previously constructed by a Julia-Kocienski reaction,5,9 using sulfone 4 and aldehyde 5.5 We further envisioned that both sulfone 4 and the aldehyde 5 could be stereoselectively constructed using Jacobsen’s chromium-catalyzed hetero Diels-Alder reactions as key steps.8
Figure 1.

Retrosynthetic analysis of lasonolide A
As shown in Scheme 1, the synthesis of silyloxy dienes for the hetero Diels-Alder reaction commenced with the known aldehyde 8.10 Methyl or ethylmagnesium bromide was added to aldehyde 8 to afford the respective alcohol in 97% and 95% yields, respectively. Parikh-Doering oxidation11 of the resulting alcohols provided the corresponding ketones. Reaction of these ketones with TMSOTf or TESOTf in the presence of triethylamine provided silyl enol ethers 912 and 10 in 99% and 91% yields, respectively. Asymmetric hetero Diels-Alder reactions of the silyl enol ethers 9 and 10 with benzyloxyacetaldehyde 1213 in the presence of chiral Cr(III) catalyst 118 (7.5 to 10 mol%) afforded cycloadducts 6 and 7. The enantiomeric excess of the cycloadducts 6 (91% ee) and 7 (93% ee) was determined by Chiral HPLC. These silyl enol ethers were then used as the key intermediates in the construction of the two tetrahydropyran rings.
Scheme 1.
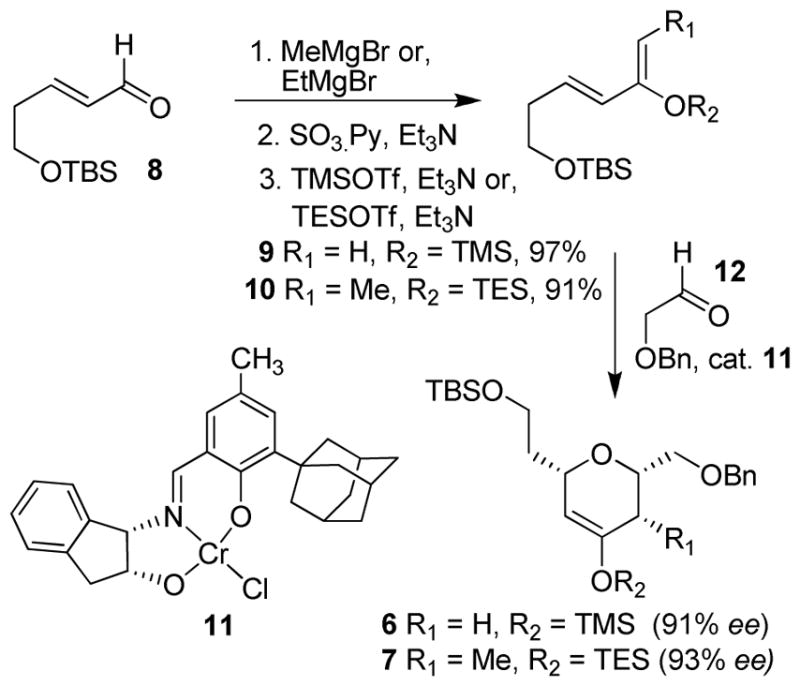
Synthesis of dihydropyrans 6 and 7
As shown in Scheme 2, for the construction of the top tetrahydropyran ring, silyl ether 6 was treated with MeLi at −78 °C for 1 h. The resulting lithium enolate was reacted with ethyl cyanoformate to provide the corresponding β-keto ester as a mixture in 82% yield. The mixture was subjected to alkylation with NaH and MeI to provide the desired methylation product 13 in 92% total yield (dr 6.6:1 by 1H NMR analysis). Exposure of β-keto ester 13 to L-selectride in THF provided the corresponding axial alcohol, which was subjected to LiAlH4 reduction in the same pot to afford diol 14 diastereoselectively (ratio 10:1) and diastereomers were separated to provide 14 in 82% yield. Protection of the diol as an acetonide followed by removal of the benzyl group using catalytic hydrogenation afforded the alcohol 15 in 84% yield over 2 steps. Parikh-Doering oxidation11 of 15 gave the precursor aldehyde which, was subjected to a Julia-Kocienski reaction.9 The requisite sulfone 17 was readily prepared by a Mitsunobu reaction with known alcohol 16,14 triphenyl phosphine, diisopropylazodicarboxylate (DIAD) and phenyl tetrazole thiol. Reaction of the sulfone 17 with KHMDS in a mixture (5:1) of DME and HMPA followed by addition of an aldehyde provided the corresponding trans-olefin as a major isomer (trans/cis = 10:1 by 1H NMR analysis) in 90% yield. Interestingly, trans/cis selectivity was reduced when THF was used as the solvent (75% yield, trans/cis = 5:1 by 1H NMR analysis). The resulting trans sulfide was oxidized to the corresponding sulfone using ammonium heptamolybdate15 as the catalyst in the presence of hydrogen peroxide. Subsequent protecting-group manipulation afforded the sulfone 4 in 10 steps from the hetero Diels-Alder product 6.5
Scheme 2.

Synthesis of the upper tetrahydropyran ring
The synthesis of the bottom tetrahydropyran ring is shown in Scheme 3. Hetero Diels-Alder product 7 was treated with TBAF in the presence of acetic acid in THF to provide ketone 19 in 72% yield over 2 steps. As described previously, selective reduction5 of this ketone to the desired axial alcohol 21 was very challenging. Direct reduction with a variety of hydride reagents provided undesired alcohol 20 as the major isomer (dr ≥ 2:1). Conversion of the undesired alcohol 20 back to alcohol 21 using Swern oxidation followed by reduction did improve the yield of 21. However, it required tedious separation, which was not very convenient even during large scale preparation. We therefore devised an alternative strategy to circumvent the poor diastereoselectivity issue. Thus, ketone 19 was converted to the corresponding enol triflate with NaHMDS and phenyl triflimide.16 Palladium-catalyzed reduction17 of the resulting enol triflate afforded alkene 22 in 75% yield over two steps. Epoxidation of the resulting olefin with dimethyldioxirane (DMDO) afforded the desired epoxide in 69% yield as a major isomer (5.3:1 ratio). Epoxidation presumably proceeded from the top-side as shown in the stereochemical model 23. The isomers were separated by silica gel chromatography. DIBAL-H reduction of the major epoxide provided alcohol 21 as a single isomer in excellent yield. The observed epoxide ring opening selectivity is consistent with the expected diaxial epoxide opening, in accordance with the Fürst-Plattner rule.18 Protection of the alcohol as a TBS ether followed by selective deprotection of the primary TBS group with CSA in MeOH provided the primary alcohol 24 in 89% yield. Parikh-Doering oxidation of alcohol 24 followed by Horner-Wadsworth-Emmons olefination of the resulting aldehyde furnished the α,β-unsaturated ester 25 in 91% yield. This was converted to alcohol 26 in a three step sequence involving, (1) DIBAL-H reduction; (2) protection of the resulting alcohol as a TBS ether, and (3) selective removal of the benzyl group19 with Li in liquid ammonia in 70% yield, over three steps. Alcohol 26 was converted to aldehyde 5 as described by us previously.5 Deprotonation of sulfone 4 using KHMDS in THF at −78 °C followed by addition of aldehyde 5 afforded trans-olefin 3 as a single isomer in 70% yield. This coupling product 3 was the key intermediate of our previous synthesis of lasonolide A.5
Scheme 3.

Synthesis of the bottom tetrahydropyran ring
In summary, we have accomplished a stereoselective synthesis of both tetrahydropyran subunits of lasonolide A using catalytic asymmetric hetero Diels-Alder reactions as key steps. The C22 quaternary carbon stereocenter present in the upper tetrahydropyran ring was installed using diastereoselective alkylation followed by diastereoselective reduction steps. The C9 hydroxy group in the bottom tetrahydropyran ring was introduced stereoselectively using selective epoxidation with DMDO and reduction with DIBAL-H. Julia-Kocienski reaction of the two subunits provided an advanced intermediate 3 of our previous synthesis of lasonolide A. The synthesis of 3 has been carried out in 12 linear steps (13.5% overall yield from 8), and 25 overall steps. In comparison, our previous synthesis of 3 was accomplished in 23 linear steps (2.78% overall yield) and 38 overall steps.5 The overall routes are amenable to the synthesis of structural analogs of lasonolide A.
EXPERIMENTAL SECTION
General experimental details are provided as Supporting Information.
(2-((2S,6S)-6-(benzyloxymethyl)-4-(trimethylsilyloxy)-5,6-dihydro-2H-pyran-2-yl)ethoxy)(tert-butyl)dimethylsilane (6)
A mixture of enol ether 912 (1.05 g, 3.51 mmol), 12 (1.58 g, 10.5 mmol), 4Å molecular sieves (1.2 g) was treated with the Jacobsen’s catalyst 11 (123 mg, 0.26 mmol) at 23 °C for 24 h. The reaction was diluted with EtOAc and filtered through a short pad of Celite, eluted with EtOAc. The filtrate was collected and concentrated in vacuo. The residue was purified quickly by a short pad of silica gel (1% NEt3/5% EtOAc/Hexanes) to afford enol ether 6 (1.12 g, 71%) as a yellow oil. [α]D25 −23.2(c 0.6, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.35-7.26 (m, 5H), 4.80 (s, 1H), 4.59 (ABq, JAB = 12.3 Hz, 2H), 4.32-4.27 (m, 1H), 3.82-3.71 (m, 3H), 3.55 (dd, J = 10.3, 6.2 Hz, 1H), 3.49 (dd, J = 10.3, 4.2 Hz, 1H), 2.15-2.06 (m, 1H), 1.91 (m, 1H), 1.80-1.68 (m, 2H), 0.89 (s, 9H), 0.20 (s, 2H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 148.0, 138.3, 128.3, 127.6, 127.5, 106.6, 73.6, 73.3, 72.7, 71.2, 59.6, 39.4, 32.8, 25.9, 18.3, 0.2, −5.4; FT-IR (film) 2955, 2928, 1668, 1098 cm−1. ESI-MS m/z 451.2 ([M+H]+), 473.4 ([M+Na]+). ESI-HRMS calcd for C24H43O4Si2 ([M+H]+) 451.2694; found: 451.2708.
(2S,3S,6S)-Ethyl-6-((benzyloxy)methyl)-2-(2-((tert-butyldimethylsilyl)oxy)ethyl)-3-methyl-4-oxotetrahydro-2H-pyran-3-carboxylate (13)
To a solution of 6 (1.12 g, 2.49 mmol) in dry THF (10 mL) was added MeLi (1.6 M in diethyl ether, 4.7 mL) at −78 °C. The reaction was then kept at −20 °C for 1 h and then cooled down to −78 °C. NCCO2Et (0.74 mL, 7.46 mmol) was added. The mixture was warmed to 23 °C and quenched with water. The aqueous layer was extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (10% EtOAc/Hexanes) of the residue provided alkylated product (922 mg, 82%) as a yellow oil.
To a solution of the former oil (392 mg, 0.87 mmol) in dry THF (5 mL) was added NaH (42 mg, 60% in oil, 1.04 mmol) at 0 °C. After 30 min, MeI (494 mg, 3.48 mmol) was added. The reaction was kept at 0 °C for 1 h and then at 23 °C overnight. The reaction was quenched with aq. NH4Cl. The aqueous layer was extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (10% EtOAc/Hexanes) of the residue afforded the desired isomer 13 (324 mg, 80%) as a colorless oil. [α]D25 45.2 (c 1.1, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.35-7.26 (m, 5H), 4.60 (ABq, JAB = 12.3 Hz, 2H), 4.27-4.23 (m, 1H), 4.20 (q, J = 6.8 Hz, 2H), 3.97-3.90 (m, 1H), 3.81-3.70 (m, 2H), 3.64-3.55 (m, 2H), 2.63 (dd, J = 12.1, 5.5 Hz, 1H), 2.32 (dd, J = 15.2, 2.8 Hz, 1H), 1.81-1.70 (m, 1H), 1.45-1.41 (m, 1H), 1.37 (s, 3H), 1.26 (t, J = 6.9 Hz, 3H), 0.89 (s, 9H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 206.4, 170.6, 137.9, 128.4, 127.7, 127.5, 76.6, 76.1, 73.4, 71.9, 61.8, 61.2, 59.1, 39.8, 33.7, 25.8, 18.2, 14.3, 14.0, −5.4, −5.6; FT-IR (film) 2955, 2930, 2858, 1738, 1715, 1254 1103 cm−1. ESI-MS m/z 465.1 ([M+H]+), 487.1 ([M+Na]+). ESI-HRMS calcd for C25H40O6SiNa ([M+Na]+) 487.2486; found: 487.2496.
(2S,3R,4R,6S)-6-((Benzyloxy)methyl)-2-(2-((tert-butyldimethylsilyl)oxy)ethyl)-3-(hydroxymethyl)-3-methyltetrahydro-2H-pyran-4-ol (14)
To a solution of 13 (320 mg, 0.69 mmol) in dry THF (5 mL) was added L-selectride (1 M in THF, 2.07 mL) at −78 °C. The reaction was kept at −78 °C for 3 h. LiAlH4 (26 mg, 0.69 mmol) was then added and the reaction was allowed to stir at 0 °C for 2 h. The reaction was quenched slowly with aq. NaOH (1 M, 2 mL) and aq. 30% H2O2 (1 mL). The mixture was stirred at 0 °C for another 1 h. The aqueous layers were extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (40% EtOAc/Hexanes to 50% EtOAc/Hexanes) of the residue afforded diol 14 (240 mg, 82%) as a colorless oil. [α]D25 34.6 (c 0.70, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.36-7.25 (m, 5H). 4.57 (s, 2H), 4.11 (d, J = 6.8 Hz, 1H), 4.03 (m, 1H), 3.90 (s, 1H), 3.82-3.78 (m, 2H), 3.67 (d, J = 11.6 Hz, 1H), 3.55-3.46 (m, 3H), 1.80-1.63 (m, 2H), 1.61-1.42 (m, 2H), 0.90 (s, 9H), 0.76 (s, 3H), 0.07 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 138.4, 128.3, 127.5, 127.4, 74.6, 73.21, 73.16, 71.6, 71.5, 69.7, 61.3, 39.9, 32.5, 26.0, 18.4, 15.1, −5.4; FT-IR (film) 3376, 2955, 2928, 1089 cm−1. ESI-MS m/z 425.2 ([M+H]+), 447.1 ([M+Na]+). ESI-HRMS calcd for C23H40O5SiNa ([M+Na]+) 447.2543; found: 447.2539.
((4aS,5S,7S,8aR)-5-(2-((tert-Butyldimethylsilyl)oxy)ethyl)-2,2,4a-trimethylhexahydropyrano[4,3-d][1,3]dioxin-7-yl)meth anol (15)
To a solution of 14 (276 mg, 0.651 mmol) in dry CH2Cl2 (5 mL) was added 2,2-dimethoxypropane (2 mL) followed by PPTS (16.4 mg, 0.065 mmol). The reaction was kept at 23 °C for 10 h. The reaction was quenched with aq. NaHCO3. The aqueous layers were extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (10% EtOAc/Hexanes) of the residue provided the acetonide derivative as a colorless oil (278 mg, 92%).
A mixture of the former colorless oil (295 mg, 0.64 mmol), Pd(OH)2-C (20% wt, 50% wet, 60 mg) in EtOAc (6 mL) was stirred at 23 °C under H2 atmosphere for 10 h. The solid was filtered off and washed with EtOAc twice. The combined organic phase was concentrated in vacuo and purified by flash chromatography to afford alcohol 15 (217 mg, 91%) as a colorless oil. [α]D25 36.0 (c 1.05, CHCl3). 1H NMR (400 MHz, CDCl3) δ 4.26 (d, J = 10.3 Hz, 1H), 3.89-3.80 (m, 2H), 3.77-3.70 (m, 2H), 3.65-3.54 (m, 1H), 3.54-3.45 (m, 2H), 3.46-3.37 (m, 1H), 2.10-2.01 (br, 1H), 1.84-1.72 (m, 1H), 1.70-1.64 (m, 1H), 1.52-1.32 (m, 2H), 1.43 (s, 3H), 1.39 (s, 3H), 0.89 (s, 9H), 0.73 (s, 3H), 0.06 (s, 3H), 0.05 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 98.4, 72.3, 71.7, 71.3, 66.0, 65.9, 60.1, 34.5, 32.3, 29.24, 29.17, 25.9, 18.8, 18.2, 14.7, −5.36, −5.42; FT-IR (film) 3460, 2955, 1089 cm−1. ESI-MS m/z 397.2 ([M+Na]+). ESI-HRMS calcd for C19H38O5SiNa ([M+Na]+) 397.2381; found: 397.2387.
1-Phenyl-5-((3-((1-phenyl-1H-tetrazol-5-yl)sulfonyl)prop yl)thio)-1H-tetrazole (17)
To a solution of 16 (4.34 g, 16.19 mmol), 1-phenyl-1H-tetrazole-5-thiol (3.33 g, 19 mmol) and Ph3P (7.86g, 30mmol) in dry CH2Cl2 (150 mL) was added DIAD (4.44 g, 22 mmol) dropwise. The reaction was kept at 23 °C overnight. The solvent was removed and the residue was purified by silica gel chromatography to give the desired sulfide 17 as a white solid (5.26 g, 76%). 1H NMR (400 MHz, CDCl3) δ 7.71-7.26 (m, 10H), 3.95 (t, J = 7.0 Hz, 2H), 3.60 (t, J = 6.9 Hz, 2H), 2.61 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 153.2, 153.1, 133.3, 132.8, 131.5, 130.3, 129.8, 129.7, 125.0, 123.8, 54.2, 31.0, 22.3; FT-IR (film) 1730, 1498, 1340 cm−1. ESI-MS m/z 429.1 ([M+H]+). ESI-HRMS calcd for C17H17O2N8S2 ([M+H]+) 429.0910; found: 429.0925.
2-((4aS,5S,7S,8aR)-2,2,4a-Trimethyl-7-((E)-4-((1-Phenyl-1H-tetrazol-5-yl)sulfonyl)but-1-en-1-yl)hexahydropyrano[4,3-d] [1,3]dioxin-5-yl)ethanol (18)
To a solution of 15 (210 mg, 0.56 mmol) in CH2Cl2 (5 mL) and DMSO (2 mL) was added NEt3 (0.63 mL, 4.49 mmol) followed by SO3.Py (358 mg, 2.25 mmol) at 0 °C. The reaction was kept at 0 °C for 2 h. The reaction was quenched with aq. NaHCO3. The aqueous layers were extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over Na2SO4 and concentrated. Purification of the residue by flash chromatography (30% EtOAc/Hexanes) afforded the desired aldehyde (206 mg, 99%) as a colorless oil.
To a solution of 17 (960 mg, 2.25 mmol) in DME/HMPA (v/v, 10 mL/2 mL) was added KHMDS (0.5 M in toluene, 4.5 mL, 2.25 mmol) at −78 °C. After 30 min, a solution of above aldehyde (206 mg, 0.56 mmol) in dry DME (1 mL) was added. The reaction was kept at −78 °C for 2 h and then quenched with aq. NH4Cl. The layers were separated and the aqueous layers were extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over Na2SO4 and concentrated. Purification of the residue by flash chromatography (15% EtOAc/Hexanes) afforded the corresponding olefin (290 mg, 91%) as a colorless oil. [α]D25 24.9 (c 0.95, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.60-7.51 (m, 5H), 5.70-5.61 (m, 1H), 5.59-5.48 (m, 1H), 4.27-4.12 (m, 2H), 3.85-3.80 (m, 1H), 3.76-3.63 (m, 2H), 3.53-3.44 (m, 2H), 3.43-3.36 (m, 2H), 2.58-2.49 (m, 2H), 1.73-1.62 (m, 2H), 1.52-1.46 (m, 2H), 1.42 (s, 3H), 1.39 (s, 3H), 0.90 (s, 9H), 0.72 (s, 3H), 0.06 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 154.2, 133.9, 133.6, 130.0, 129.7, 127.4, 123.8, 98.4, 77.2, 71.9, 71.1, 66.0, 59.9, 34.2, 33.5, 32.8, 32.2, 31.8, 29.2, 25.9, 18.8, 18.2, 14.8, −5.35, −5.42; FT-IR (film) 2928, 1384, 836 cm−1. ESI-MS m/z 575.2 ([M+H]+), 597.2 ([M+Na]+). ESI-HRMS calcd for C29H46N4O4SSiNa ([M+Na]+) 597.2907; found: 597.2904.
To a solution of above olefin (245 mg, 0.43 mmol) in EtOH (5 mL) and THF (2 mL) was added buffer (pH = 7.5, Na2HPO4-NaH2PO4, 0.5 mL) followed by (NH4)6Mo7O24.7H2O (132 mg, 0.107 mmol), aq. H2O2 (30%, 0.5 mL) at 0 °C. The reaction was allowed to stir at 23 °C for 2 h until another portion of (NH4)6Mo7O24.7H2O (132 mg, 0.11 mmol), aq. H2O2 (30%, 0.5 mL) was added. The mixture was stirred for 12 h and then carefully quenched with aq. Na2SO3. The mixture was extracted with ethyl acetate. The combined extracts was washed with water and brine, dried over Na2SO4 and concentrated.
Above residue was dissolved in THF (5 mL). HF.Py (0.16 mL, 4.27 mmol) was added. The reaction was allowed to stir at 23 °C for 4 h and then it was quenched with aq. NaHCO3. The mixture was extracted with ethyl acetate. The combined extracts was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (50% EtOAc/Hexanes) of the residue afforded the desired alcohol 185 (160 mg, 76% for 2 steps) as a colorless oil. 1H NMR: (400 MHz, CDCl3) δ 7.65-7.54 (m, 5H), 5.68-5.48 (m, 2H), 4.35 (t, J = 7.6 Hz, 1H), 4.27-4.24 (m, 1H), 3.84-3.69 (m, 5H), 3.50 (ABq, JAB = 12.5 Hz, 2H), 2.95 (br, 1H), 2.68-2.60 (m, 2H), 1.71-1.64 (m, 3H), 1.50 (m, 1H), 1.41 (s, 3H), 1.39 (s, 3H), 0.75 (s, 3H).
TES-enol ether (10)
To a solution of 8 (1.9 g, 8.88 mmol) in dry THF (40 mL) was added ethylmagnesium bromide (3 M in ether, 3.85 mL) at −20 °C. The reaction was kept at −20 °C for 2 h and then quenched with aq. NH4Cl. The mixture was extracted with ethyl acetate. The combined extracts was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (20% EtOAc/Hexanes) of the residue gave the desired alcohol (2.05 g, 95%) as a colorless oil.
To a solution of above alcohol (1.71 g, 7 mmol) in CH2Cl2/DMSO (2:1, 45 mL) was added Et3N (9.75 mL, 70 mmol) followed by SO3.Py (2.79 g, 17.5 mmol) at 0 °C. When the reaction was complete as shown by TLC, it was quenched with aq. NaHCO3. The mixture was extracted with ethyl acetate. The combined extracts was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (10% EtOAc/Hexanes) of the residue gave the desired ketone (1.61 g, 95%) as a yellow oil.
To a solution of the former ketone (2.63 g, 10.9 mmol) in diethyl ether (15 mL) was added NEt3 (3.8 mL, 27.2 mmol) followed by TESOTf (4.3 g, 16.3 mmol) at −78 °C. The reaction was kept at −78 °C for 30 min and at 0 °C for 1 h. The reaction was quenched with aq. NaHCO3. The mixture was extracted with ethyl acetate. The combined extracts was washed with water and brine, dried over Na2SO4 and concentrated. Purification by flash chromatography (1% NEt3/5% EtOAc/Hexane) afforded TES-enol ether 10 (3.53 g, 91%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 5.90 (d, J = 15.4 Hz, 1H), 5.82-5.70 (m, 1H), 4.73 (q, J = 7.0 Hz, 1H), 3.64 (t, J = 6.8 Hz, 2H), 2.29 (q, J = 6.9 Hz, 2H), 1.64 (d, J = 7.0 Hz, 3H), 1.03-0.91 (m, 12H), 0.89 (s, 9H), 0.70 (q, J = 8.1, 5.9 Hz, 6H), 0.05 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 149.5, 130.4, 124.3, 107.4, 62.9, 35.8, 25.8, 18.2, 11.3, 6.7, 6.3, 5.4, −5.4; FT-IR (film) 2954, 1657, 1627, 1097 cm−1. MS m/z 357.3 ([M+H]+). ESI-HRMS calcd for C19H41O2Si2 ([M+H]+) 357.2640; found: 357.2648.
(2-((2S,5S,6S)-6-(benzyloxymethyl)-5-methyl-4-(triethylsilyloxy)-5,6-dihydro-2H-pyran-2-yl)ethoxy)(tert-butyl)dimethylsilane (7)
A mixture of the TES-enol ether 10 (3.4 g, 9.55 mmol), 12 (4.3 g, 28.7 mmol), 4Å molecular sieves (3.2 g) was treated with the Jacobsen’s catalyst 11 (278 mg, 0.57 mmol) at 23 °C for 48 h. The reaction was diluted with EtOAc and filtered through a short pad of Celite, eluted with EtOAc. The combined filtrate was concentrated in vacuo. The residue was purified with flash chromatography (1% NEt3/5% EtOAc/Hexane) to afford the desired cycloadduct 7 as a colorless oil. [α]D25 + 46.3 (c 0.8, CHCl3). 1H NMR (300 MHz, CDCl3) δ 7.37-7.21 (m, 5H), 4.69 (m, 1H), 4.58 (ABq, JAB = 12.0 Hz, 2H), 3.23 (t, J = 5.1 Hz, 1H), 3.90-3.63 (m, 3H), 3.58 (dd, J = 9.8, 6.9 Hz, 1H), 3.47 (dd, J = 9.8, 6.1 Hz, 1H), 2.10-1.98 (m, 1H), 1.80-1.68 (m, 2H), 0.99 (t, J = 3.8 Hz, 9H), 0.98 (d, J = 7.5 Hz, 3H), 0.91 (s, 9H), 0.68 (q, J = 7.9 Hz, 6H), 0.06 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 153.4, 138.2, 128.2, 127.6, 127.4, 104.1, 75.5, 73.3, 71.4, 70.4, 59.6, 39.4, 36.1, 25.9, 18.3, 11.9, 6.6, 4.9, −5.4; FT-IR (film) 2935, 2955, 1664, 1089 cm−1. MS m/z 507.25 ([M+H]+). ESI-HRMS calcd for C28H51O4Si2 ([M+H]+) 507.3320; found: 507.3335.
(2S,3S,6S)-2-((Benzyloxy)methyl)-6-(2-((tert-butyldimethylsilyl)oxy)ethyl)-3-methyldihydro-2H-pyran-4(3H)-one (19)
To a solution of the former silyl enol ether 7 in dry THF (100 mL) was added HOAc (1.1 mL, 19.1 mmol) followed by TBAF (1 M in THF, 9.55 mL, 9.55 mmol) dropwise at 0 °C. The reaction was allowed to stir at 0 °C for 2 h. The reaction was quenched with aq. NaHCO3. The mixture was extracted with ethyl acetate. The combined extracts was washed with water and brine, dried over Na2SO4 and concentrated. Purification of the residue by flash chromatography (10% EtOAc/Hexanes) gave ketone 19 (2.69 g, 72% over 2 steps) as a colorless oil. [α]D25 + 14.6 (c 1.07, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.32-7.27 (m, 5H), 4.59 and 4.50 (ABq, JAB = 11.7 Hz, 2H), 3.90-3.85 (m, 1H), 3.85-3.70 (m, 3H), 3.65 (dd, J = 9.6, 6.9 Hz, 1H), 3.49 (dd, J = 9.6, 6.2 Hz, 1H), 2.58-2.52 (m, 1H), 2.46 (dd, J = 13.0, 11.6 Hz, 1H), 2.28 (d, J = 15.1 Hz, 1H), 1.90-1.82 (m, 1H), 1.78-1.68 (m, 1H), 1.09 (d, J = 7.5 Hz, 3H), 0.89 (s, 9H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 210.8, 137.9, 128.3, 127.7, 127.6, 74.1, 73.3, 69.4, 58.8, 46.8, 44.5, 39.2, 25.9, 18.2, 10.8, −5.4, −5.5; FT-IR (film) 2928, 1717, 1093 cm−1. ESI-MS m/z 393.2 ([M+H]+), 415.1 ([M+Na]+). ESI-HRMS calcd for C22H36O4SiNa ([M+Na]+) 415.2275; found: 415.2282.
(2S,3R,4R,6S)-2-((Benzyloxy)methyl)-6-(2-((tert-butyldimethylsilyl)oxy)ethyl)-3-methyltetrahydro-2H-pyran-4-ol(18) and (2S,3R,4S,6S)-2-((Benzyloxy)methyl)-6-(2-((tert-butyl dimethylsilyl)oxy)ethyl)-3-methyltetrahydro-2H-pyran-4-ol (20)
To a solution of 19 (2.7 g, 6.89 mmol) in dry CH2Cl2 (50 mL) was added DIBAL-H (1 M in CH2Cl2, 10.3 mL) dropwise at −78 °C. The reaction was kept at −78 °C for 2 h before it was quenched with MeOH (1 mL). EtOAc and aq. Rochelle salt were added. The reaction was stirred for 2 h and then extracted with ethyl acetate. The combined extracts was wash with water and brine, dried over Na2SO4 and concentrated. The residue was purified by flash chromatography (20% EtOAc/Hexanes to 30% EtOAc/Hexanes) afforded alcohol 21 (810 mg, 30%) as a colorless oil. [α]D25-12.6 (c 1.10, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.34-7.26 (m, 5H), 4.60 and 4.49 (ABq, JAB = 12.3 Hz, 2H), 4.13 (dt, J = 2.8, 6.9 Hz, 1H), 3.95-3.86 (m, 2H), 3.75-3.68 (m, 2H), 3.53 (dd, J = 9.6, 6.8 Hz, 1H), 3.39 (dd, J = 9.7, 6.1 Hz, 1H), 1.78-1.65 (m, 2H), 1.65-1.48 (m, 3H), 0.89 (s, 9H), 0.88 (d, J = 7.2 Hz, 3H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 138.4, 128.3, 127.6, 127.5, 73.2, 72.7, 71.0, 70.5, 69.0, 59.5, 39.2, 36.5, 34.4, 25.9, 18.3, 10.9, −5.4; FT-IR (film) 3443, 2927, 1096 cm−1. MS m/z 395.2 ([M+H]+). 417.2 ([M+Na]+). ESI-HRMS calcd for C22H38O4SiNa ([M+Na]+) 417.2437; found: 417.2440.
Undesired alcohol 20 (1.76 g, 65%) was obtained as a colorless oil. [α]D25-6.7 (c 1.04, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.30-7.26 (m, 5H), 4.59 and 4.48 (ABq, JAB = 12.0 Hz, 2H), 3.90 (m, 1H), 3.89-3.61 (m, 2H), 3.60-3.45 (m, 3H), 3.45 (m, 1H), 2.02-1.99 (m, 1H), 1.80-1.71 (m, 1H), 1.70-1.60 (m, 2H), 1.56 (br, 1H), 1.43-1.33 (m, 1H), 0.88 (s, 9H), 0.83 (d, J = 6.9 Hz, 3H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 138.2, 128.3, 127.6, 127.5, 77.4, 73.3, 72.9, 70.8, 70.7, 59.4, 38.9, 36.1, 35.4, 25.9, 18.3, 4.8, −5.4; FT-IR (film) 3411, 2927, 1093 cm−1. MS m/z 417.3 ([M+Na]+). ESI-HRMS calcd for C22H38O4SiNa ([M+Na]+) 417.2437; found: 417.2437.
(2-((2S,5R,6S)-6-((Benzyloxy)methyl)-5-methyl-5,6-dihydro-2H-pyran-2-yl)ethoxy)(tert-butyl) dimethylsilane (22)
To a solution of 19 (245 mg, 0.63 mmol) in dry THF (6.3 mL) was added NaHMDS (1 M in THF, 0.94 mL) at −78 °C. The reaction was kept at −78 °C for 1 h before a solution of PhNTf2 (290 mg, 0.81 mmol) in THF (1 mL) was added. The reaction was stirred at that temperature for 30 min and then at 23 °C overnight. The reaction was quenched with aq. NH4Cl. The mixture was extracted with ethyl acetate and the combined extracts was washed with water and brine, dried over Na2SO4 and concentrated. Purification of the residue by passing through a short pad of silica gel (20% EtOAc/Hexanes) gave the crude enol triflate as a colorless oil.
To a solution of the former enol triflate, Pd(OAc)2 (7 mg, 0.03 mmol), PPh3 (16.4 mg, 0.06 mmol) and DIPEA (404 mg, 3.13 mmol) in dry DMF (5 mL) was added HCO2H (86.3 mg, 1.88 mmol) at 23 °C. After addition, the reaction was allowed to stir at 65 °C for 1 h. Then cooled to 23 °C, diluted with ether and washed with water and brine and dried over Na2SO4. Evaporation of solvent gave a residue which was chromatographed (20% EtOAc/Hexanes) to afford alkene 22 (176 mg, 75% over 2 steps) as a colorless oil. [α]D25 −46.7 (c 1.2, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.35-7.26 (m, 5H), 5.82-5.77 (m, 1H), 5.58 (d, J = 10.0 Hz, 1H), 4.62 and 4.52 (ABq, JAB = 11.6 Hz, 2H), 4.29 (m, 1H), 3.89-3.82 (m, 1H), 3.80-3.71 (m, 2H), 3.55 (dd, J = 10.4, 6.8 Hz, 1H), 3.44 (dd, J = 10.1, 6.0 Hz, 1H), 2.12 (m, 1H), 1.80-1.69 (m, 2H), 0.90 (d, J = 7.2 Hz, 3H), 0.89 (s, 9H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 138.4, 131.0, 129.3, 128.3, 127.6, 127.5, 75.1, 73.3, 72.4, 70.9, 59.5, 38.6, 30.8, 25.9, 18.3, 13.9, −5.4; FT-IR (film) 2928, 1255, 1089 cm−1. MS m/z 377.3 ([M+H]+). ESI-HRMS calcd for C22H37O3Si ([M+H]+) 377.2512; found: 377.2509.
(2S,3R,4S,6S)-2-((Benzyloxy)methyl)-6-(2-((tert-butyldime thylsilyl)oxy)ethyl)-3-methyltetrahydro-2H-pyran-4-ol (21)
To a solution of 22 (40 mg, 0.11 mmol) in MeCN (1 mL) and buffer (pH = 7.5, Na2HPO4-NaH2PO4, 1 mL) was added a solution of DMDO in acetone (1.5 mL) at −15 °C slowly. The reaction was allowed to stir at −15 °C for 20 min and then at 23 °C for 4 h. The reaction was quenched with aq. Na2SO3. The mixture was extracted with ethyl acetate. The combined extracts was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (10% EtOAc/Hexanes) of the residue gave the epoxide (29 mg, 69%) as a colorless oil.
To a solution of the epoxide (26 mg 0.07 mmol) in dry THF (1 mL) was added DIBAL-H (1 M in CH2Cl2, 0.2 mL) slowly at −15 °C. The reaction was allowed to stir at 0 °C to 23 °C for 2 h before it was quenched with aq. Rochelle salt. EtOAc was added and the reaction was allowed to stir vigorously at 23 °C for another 2 h. The layers were separated and the aqueous phase was extracted with ethyl acetate. The combined extracts was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel afforded the desired alcohol 21 (26 mg, 99%) as a colorless oil.
2-((2S,4S,5S,6S)-6-((Benzyloxy)methyl)-4-((tert-butyldimethylsilyl)oxy)-5-methyltetrahydro-2H-pyran-2-yl)ethanol (24)
To a solution of 21 (1.38 g, 3.49 mmol) in dry CH2Cl2 (20 mL) was added 2,6-lutidine (747 mg, 6.98 mmol) followed by TBSOTf (1.2 g, 4.53 mmol) at 0 °C. The reaction was kept at 0 °C for 1 h before it was quenched with aq. NaHCO3. The layers were separated and the aqueous phase was extracted with CH2Cl2. The combined extracts was dried over Na2SO4 and concentrated. Purification of the residue by flash chromatography (5% EtOAc/Hexanes) afforded di-TBS-protected diol (1.72 g, 97%) as a colorless oil.
To a solution of the former di-TBS ether (1.72 g, 3.39 mmol) in MeOH (15 mL) and CH2Cl2 (15 mL) was added CSA (78.6 mg, 0.34 mmol) at 23 °C. After 2 h, the reaction was quenched with aq. NaHCO3. The aqueous phase was extracted with ethyl acetate. The combined organic layers was washed with water and brine, dried over Na2SO4 and concentrated. Purification of the residue by flash chromatography (20% EtOAc/Hexanes) gave the primary alcohol 24 (1.18 g, 89%) as a colorless oil. [α]D25 + 9.9 (c 1.05, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.34-7.26 (m, 5H), 4.57 and 4.48 (ABq, JAB = 12.3 Hz, 2H), 4.21 (m, 1H), 4.06 (m, 1H), 3.88-3.75 (m, 3H), 3.47 (dd, J = 9.6, 8.2 Hz, 1H), 3.36 (dd, J = 9.9, 3.5 Hz, 1H), 1.81-1.53 (m, 4H), 1.38-1.29 (m, 1H), 0.89 (s, 9H), 0.83 (d, J = 6.8 Hz, 3H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 138.2, 128.4, 127.7, 127.5, 73.8, 73.3, 73.1, 70.6, 62.2, 37.4, 36.8, 34.7, 25.7, 18.0, 11.0, −5.0; FT-IR (film) 3443, 2928, 1254, 1098 cm−1. ESI-MS m/z 395.1 ([M+H]+), 417.2 ([M+Na]+). ESI-HRMS calcd for C22H39O4Si ([M+H]+) 395.2612; found: 395.2620.
(E)-Ethyl-4-((2S,4S,5S,6S)-6-((benzyloxy)methyl)-4-((tert-butyldimethylsilyl)oxy)-5-methyltetrahydro-2H-pyran-2-yl) but-2-enoate (25)
To a solution 24 (1.15 g, 2.92 mmol) in CH2Cl2/DMSO (2:1, 20 mL) was added Et3N (4 mL, 29.2 mmol) followed by SO3.Py (1.16 g, 7.31 mmol) at 0 °C. When the reaction was complete as shown by TLC, it was quenched with aq. NaHCO3. The aqueous phase was extracted with ethyl acetate. The combined organic layers was washed with water and brine, dried over Na2SO4 and concentrated. Purification of the residue by silica gel chromatography (10% EtOAc/Hexanes) gave the desired aldehyde (1.14 g, 99%) as a colorless oil.
To a solution of triethyl phosphonoacetate (1.3 g, 5.82 mmol) in dry THF (15 mL) was added NaH (60% in oil, 209 mg, 5.23 mmol) portionwise at 0 °C. A solution of the aldehyde (1.14 g, 2.91 mmol) in dry THF (5 mL) was then added slowly. The reaction was then stirred at 0 °C for 2 h before it was quenched with aq. NH4Cl. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined extracts was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (5% EtOAc/Hexanes) afforded ester 25 (1.22 g, 91%) as a colorless oil. [α]D25 + 2.3(c 1.2, CHCl3). 1H NMR (400 MHz, CDCl3) δ 7.35-7.26 (m, 5H), 7.01 (dt, J = 15.8, 6.9 Hz, 1H), 5.88 (d, J = 15.7 Hz, 1H), 4.63 and 4.51 (ABq, JAB = 12.0 Hz, 2H), 4.21-4.14 (m, 1H), 4.16 (q, J = 6.8 Hz, 2H), 3.94 (m, 1H), 3.78 (m, 1H), 3.50 (dd, J = 10.3, 7.5 Hz, 1H), 3.38 (dd, J = 10.3, 4.1 Hz, 1H), 2.47-2.36 (m, 1H), 2.35-2.27 (m, 1H), 1.65-1.50 (m, 2H), 1.38-1.32 (m, 1H), 1.26 (t, J = 7.2 Hz, 3H), 0.89 (s, 9H), 0.82 (d, J = 6.8 Hz, 3H), 0.03 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 166.4, 145.4, 138.5, 128.3, 127.5, 127.4, 123.1, 73.3, 73.2, 71.5, 70.78, 70.75, 60.0, 38.6, 36.8, 34.4, 25.7, 18.0, 14.2, 10.9, −4.9; FT-IR (film) 2929, 1722, 1257, 1073 cm−1. ESI-MS m/z 485.2 ([M+Na]+). ESI-HRMS calcd for C26H42O5SiNa ([M+Na]+) 485.2699; found: 485.2709.
((2S,3S,4S,6S)-4-((tert-Butyldimethylsilyl)oxy)-6-((E)-4-((tert-butyldimethylsilyl)oxy)but-2-en-1-yl)-3-methyltetrahydro -2H-pyran-2-yl)methanol (26)
To a solution of 25 (1.22 g, 2.64 mmol) in dry CH2Cl2 (10 mL) was added DIBAL-H (1 M in CH2Cl2, 7.93 mL) dropwise at −78 °C. The reaction was kept at −78 °C for 2 h before it was quenched with MeOH (1 mL). EtOAc and aq. Rochelle salt were added. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic layers was washed with water and brine, dried over Na2SO4 and concentrated. Purification of the residue by flash chromatography (5% EtOAc/Hexanes) gave the desired alcohol (1 g, 90%) as a colorless oil.
To a solution of the former oil (1 g, 2.39 mmol) in dry CH2Cl2 (10 mL) was added Et3N (1 mL, 7.16 mmol), DMAP (29.3 mg, 0.24 mmol) followed by TBSCl (540 mg, 3.58 mmol). The reaction was kept at 23 °C overnight. The reaction was quenched with aq. NaHCO3. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over Na2SO4 and concentrated. Purification of the residue by flash chromatography (25% EtOAc/Hexanes) to afford the di-TBS ether (1.22 g, 96%) as a colorless oil.
A solution of Li (120 mg, 17.1 mmol) in liquid NH3 (60 mL) was transferred dropwise to a solution of the di-TBS ether (400 mg, 0.75 mmol), allyl ethylether (645 mg, 7.49 mmol) in dry THF (7 mL) at −78 °C. The reaction was quenched with solid NH4Cl (1 g) at −78 °C and then gradually raised to 23 °C. Water and EtOAc were added. The layers were separated and the aqueous layer was extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over Na2SO4 and concentrated. Silica gel chromatography (25% EtOAc/Hexanes) of the residue gave alcohol 265 (283 mg, 81%) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 5.54-5.66 (m, 2H), 4.12 (d, J = 4.4 Hz, 2H), 4.01 (dt, J = 9.0, 3.0 Hz, 1H), 3.74-3.82 (m, 2H), 3.64 and 3.41 (ABq, JAB = 11.0 Hz, 2H), 2.25 (m, 1H), 2.14 (m, 1H), 2.09 (br, 1H), 1.47-1.52 (m, 2H), 1.38 (m, 1H), 0.89 (s, 9 H), 0.88 (s, 9 H), 0.83 (d, J = 7.5 Hz, 3H), 0.05 (s, 6H), 0.02 (s, 6H); 13C NMR (125MHz, CDCl3) δ 135.2, 117.1, 75.4, 72.1, 71.4, 64.9, 40.8, 37.4, 34.7, 26.2, 18.4, 11.8, −4.5.
Supplementary Material
Acknowledgments
Financial support for this work was provided by the National Institutes of Health and Purdue University.
Footnotes
SUPPORTING INFORMATION: 1H NMR and 13C NMR of all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Horton PA, Koehn FE, Longley RE, McConnell OJ. J Am Chem Soc. 1994;116:6015–6016. [Google Scholar]
- 2.(a) Lee E, Song HY, Kang JW, Kim DS, Jung CK, Joo JM. J Am Chem Soc. 2002;124:384–385. doi: 10.1021/ja017265d. [DOI] [PubMed] [Google Scholar]; (b) Song HY, Joo JM, Kang JW, Kim DS, Jung CK, Kwak HS, Park JH, Lee E, Hong CY, Jeong S, Jeon K, Park JH. J Org Chem. 2003;68:8080–8087. doi: 10.1021/jo034930n. [DOI] [PubMed] [Google Scholar]
- 3.(a) Kang SH, Kang SY, Choi H, Kim CM, Jun H, Youn J. Synthesis. 2004;7:1102–1114. [Google Scholar]; (b) Kang SH, Kang SY, Kim CM, Choi H, Jun H, Lee BM, Park CM, Jeong JW. Angew Chem, Int Ed. 2003;42:4779–4782. doi: 10.1002/anie.200352016. [DOI] [PubMed] [Google Scholar]
- 4.Yoshimura T, Yakushiji F, Kondo S, Wu X, Shindo M, Shishido K. Org Lett. 2006;8:475–478. doi: 10.1021/ol0527678. [DOI] [PubMed] [Google Scholar]
- 5.(a) Ghosh AK, Gong GL. Org Lett. 2007;9:1437–1440. doi: 10.1021/ol0701013. [DOI] [PubMed] [Google Scholar]; (b) Ghosh AK, Gong GL. Chem Asian J. 2008;3:1811–1823. doi: 10.1002/asia.200800164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Gurjar MK, Kumar P, Rao BV. Tetrahedron Lett. 1996;37:8617–8620. [Google Scholar]; (b) Nowakowski M, Hoffmann HMR. Tetrahedron Lett. 1997;38:1001–1004. [Google Scholar]; (c) Gurjar MK, Chakrabarti A, Rao BV, Kumar P. Tetrahedron Lett. 1997;38:6885–6888. [Google Scholar]; (d) Misske AM, Hoffmann MRH. Chem-Eur J. 2000;6:3313–3320. doi: 10.1002/(sici)1521-3765(20000915)6:18<3313::aid-chem3313>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]; (e) Deba T, Yakushiji F, Shindo M, Shishido K. Synlett. 2003;10:1500–1502. [Google Scholar]; (f) Hart DJ, Patterson S, Unch JP. Synlett. 2003;9:1334–1338. [Google Scholar]; (g) Yoshimura T, Bando T, Shindo M, Shishido K. Tetrahedron Lett. 2004;45:9241–9244. [Google Scholar]; (h) Dalgard JE, Rychnovsky SD. Org Lett. 2005;7:1589–1591. doi: 10.1021/ol050270s. [DOI] [PubMed] [Google Scholar]; (i) Sawant KB, Ding F, Jennings MP. Tetrahedron Lett. 2006;47:939–942. [Google Scholar]; (j) Sawant KB, Ding F, Jennings MP. Tetrahedron Lett. 2007;48:5177–5180. [Google Scholar]
- 7.Pommier Y, Zhang Y, Ghosh AK. US 201000041619A12010218 US Pat Appl Publ. 2010
- 8.(a) Dossetter AG, Jamison TF, Jacobsen EN. Angew Chem, Int Ed. 1999;38:2398–2400. doi: 10.1002/(sici)1521-3773(19990816)38:16<2398::aid-anie2398>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]; (b) Gademann K, Chavez DE, Jacobsen EN. Angew Chem, Int Ed. 2002;41:3059–3061. doi: 10.1002/1521-3773(20020816)41:16<3059::AID-ANIE3059>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 9.(a) Baudin JB, Hareau G, Julia SA, Ruel O. Tetrahedron Lett. 1991;32:1175–1178. [Google Scholar]; (b) Blakemore PR, Cole WJ, Kocienski PJ, Morley A. Synlett. 1998;1:26–28. [Google Scholar]
- 10.Caron PY, Deslongchamps P. Org Lett. 2010;12:508–511. doi: 10.1021/ol902711b. [DOI] [PubMed] [Google Scholar]
- 11.Parikh JR, Doering WvE. J Am Chem Soc. 1967;89:5505–5507. [Google Scholar]
- 12.Vijn RJ, Hiemstra H, Kok JJ, Knotter M, Speckamp WN. Tetrahedron. 1987;43:5019–5030. [Google Scholar]
- 13.Denmark SE, Herbert B. J Org Chem. 2000;65:2887–2896. doi: 10.1021/jo991990d. [DOI] [PubMed] [Google Scholar]
- 14.Kang SH, Kang SY, Choi H, Kim CM, Jun H, Youn J. Synthesis. 2004:1102–1114. [Google Scholar]
- 15.Williams DR, Brooks DA, Berliner MA. J Am Chem Soc. 1999;121:4924–4925. [Google Scholar]
- 16.For a similar reaction sequence for 4-ketotetrahydropyran 19 to its endocyclic olefins as in 22, see: Liu P, Hong S, Weinreb SM. J Am Chem Soc. 2008;130:7562–7563. doi: 10.1021/ja802700z.
- 17.Cacchi S, Morera E, Ortar G. Tetrahedron Lett. 1984;25:4821–4824. [Google Scholar]; (b) Mori M, Nakanishi M, Kajishima D, Sato S. J Am Chem Soc. 2003;125:9801–9807. doi: 10.1021/ja029382u. [DOI] [PubMed] [Google Scholar]
- 18.(a) Fürst vA, Plattner PlA. Helv Chim Acta. 1949:275–283. doi: 10.1002/hlca.19490320139. [DOI] [PubMed] [Google Scholar]; (b) Alt GH, Barton DHR. J Chem Soc. 1954:4284–4294. [Google Scholar]
- 19.Zakarian A, Batch A, Holton RA. J Am Chem Soc. 2003;125:7822–7824. doi: 10.1021/ja029225v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.