

Abstract
Objective
Caspase Activated DNase (CAD) is an endonuclease that is activated by active caspase 3 during apoptosis and is responsible for degradation of chromatin into nucleosomal units. These nucleosomal units are then included in apoptotic bodies. The presence of apoptotic bodies is considered important for the generation of auto-antigens in autoimmune diseases such as lupus, which are characterized by the presence of anti-nuclear antibodies.
Methods
The present study was carried out to determine the role of CAD in Sle1, Sle123 and 3H9 spontaneous models of lupus, where autoimmunity is genetically pre-determined. We also determined the ability of lupus auto-antibodies to bind to CAD deficient or sufficient apoptotic cells.
Results
The deficiency of CAD resulted in higher anti-dsDNA antibody titers in lupus-prone mice. Surprisingly, the absence of CAD only exacerbated genetically pre-determined autoimmune responses. To further determine whether nuclear modifications are required to maintain tolerance to nuclear auto-antigens, we used the 3H9 mouse, an anti-DNA heavy chain knock-in. In this model, the autoreactive B cells are tolerized by anergy. In line with the Sle1 and Sle123 CAD mutant mice, CAD deficient 3H9 mice spontaneously generated anti-DNA antibodies. We finally show that auto-antibodies with specificities towards histone/DNA complexes bind more to CAD deficient apoptotic cells compared to CAD sufficient apoptotic cells.
Conclusions
We propose that in mice genetically predisposed to lupus, nuclear apoptotic modifications are required to maintain tolerance. In the absence of these modifications, apoptotic chromatin is abnormally exposed, facilitating the autoimmune response.
Introduction
Nuclear antigens generated during apoptosis have been proposed to be a source of auto-antigens (AutoAgs) in lupus (1). Impaired clearance of apoptotic cells leads to lupus-like disease (2, 3). During apoptosis, chromatin undergoes fragmentation leading to formation of micronuclei, blebs and apoptotic bodies (4); the latter are nuclear fragments enclosed in cytoplasmic membranes. We and others have shown that antigen presenting cells such as dendritic cells phagocytose blebs and cross-present nuclear Ags (5, 6). Apoptotic chromatin fragmentation also allows release in the extracellular milieu of nucleic acid/protein complexes, which can induce IFNα production through TLRs (7, 8).
Caspase Activated DNase (CAD) is an endonuclease responsible for cleaving chromatin into fragments (9, 10). CAD is bound to an inhibitor, iCAD, and is constitutively inactive. Activation of the effector caspase 3 leads to cleavage of iCAD, and releases the active CAD, which translocates to the nucleus and cleaves chromatin specifically at the inter-nucleosomal level (11). Using the pristane-induced model of lupus, we showed previously that absence of CAD inhibits the generation of anti-nuclear antibodies (ANA). Thus, in the pristane-induced lupus model, the nuclear modifications during apoptosis are required for presentation of nuclear autoAg (12)
The present study was carried out to determine the role of chromatin fragmentation in spontaneous mouse models of lupus where the autoimmune response is antigen-driven and B cells are prone to auto-reactivity toward nuclear components. We used the NZM2410-derived Sle1 and Sle123 models. Sle1 is characterized by loss of tolerance to chromatin, whereas Sle123 also develops proliferative glomerulonephritis (13, 14). To further determine the role of chromatin fragmentation in B cell tolerance, we used 3H9 mice (15). These knock-in mice carry a rearranged dsDNA-reactive heavy chain derived from MRL/lpr (16, 17). On a non-autoimmune background, 3H9 B cells undergo follicular exclusion and exhibit an anergic phenotype (18). On an autoimmune background the mice usually have a worse or accelerated course of disease (19, 20). Anergy in 3H9 mice has been shown to be antigen dependent making these mice suitable for our purpose because the absence of CAD alters the display of chromatin during apoptotic cell death.
Here we show that absence of CAD results in higher levels of anti-chromatin antibodies (Abs) in Sle1 and Sle123 mice and more severe nephritis in Sle123 mice. We also show that in 3H9CAD−/− B cells do not undergo follicular exclusion and spontaneously produce anti-dsDNA and anti-chromatin Abs. These results strongly suggest that in mice genetically predisposed to autoimmunity, the absence of chromatin fragmentation and the resulting abnormal display of apoptotic AutoAgs, result in failure to achieve anergy and instead promote B cell autoreactivity. We also show that lupus antibodies bind more to CAD deficient apoptotic cells, thereby likely facilitating or propagating auto-reactivity in the periphery.
Based upon our data, we propose that the absence of CAD during B-cell development results in the escape of auto-reactive B cells. The apoptotic CAD-deficient cells are unable to form fragmented chromatin and therefore the auto-reactive B cells may be exposed to an altered nuclear Ag, which leads to activation and differentiation of autoreactive B cells in the periphery.
Materials and Methods
Mice
CAD deficient mice generated on the 129sv background were obtained from Dr. S. Nagata (Osaka University, Japan), and were backcrossed to C57Bl/6J mice for 8 generations. Their normal immune phenotype has been described previously (12). The NZM2410-derived Sle1 and Sle123 on C57Bl/6 background were obtained from Dr. L. Morel (University of Florida, Gainesville). The Sle1CAD−/− and Sle123CAD−/− were obtained by crossing CAD−/− to Sle1 and Sle123 for 6 generations. 3H9 heavy chain knock-in mice on C57Bl/6 background, were made by Dr. Martin Weigert (University of Chicago) (21). The CAD−/− mice were backcrossed onto 3H9 mice, and the presence of the 3H9 gene was confirmed by PCR of tail DNA (16). Mice were bred and maintained in accordance with the guidelines of the University Laboratory Animal Resource Office of Temple University, an American Association for the Accreditation of Laboratory Animal Care accredited facility. All experimental procedures were conducted according to the guidelines of the Institutional Animal Care and Use Committee.
Auto-antibodies (autoAbs)
Anti-dsDNA and anti-chromatin Abs in the sera were determined by ELISA as described previously (22, 23). Briefly, the plates were coated with chicken erythrocyte-derived chromatin at 3 μg/ml; or with calf thymus-derived dsDNA at 2.5 μg/ml in borate-buffered saline (BBS). Following addition of blocking buffer (3% BSA and 1% Tween 80 in 1X BBS), serum samples diluted 1:250 in BBT (BBS, 0.4% Tween 80, 0.5% BSA) were added and incubated overnight at 4°C. For the anti-dsDNA ELISA, plates were coated with poly-L-lysine (1μg/ml) (Sigma-Aldrich) prior to coating with antigen. Alkaline phosphatase conjugated goat anti-mouse IgG (Fcγ specific, Jackson ImmunoResearch, West Grove, PA) or goat anti-mouse IgM (Southern Biotech, Birmingham, AL) were used as secondary antibodies. The plates were developed using 1mg/ml para-nitrophenyl phosphate substrate (Sigma-Aldrich) in 0.01M diethanolamine (pH 9.8). Serum from MRL/lpr mouse with high titer of AutoAbs was also assayed at serial 2-fold dilutions from 1/250 to 1/128,000 to generate a standard curve.
Cell death
Apoptosis in mouse thymocytes was spontaneous or induced. Briefly, spontaneous apoptosis was achieved by culturing thymocytes from CAD+/+ or CAD−/− mice, 12h at 37°C. Induced apoptosis was achieved by UVB irradiation (20 mJ/cm2) (12). Both methods yielded at least 50% apoptotic cells (data not shown). Subsequently, cells were incubated with one of the following monoclonals: anti-H2B, anti-H2A-H2B-DNA, anti-nucleosome, and anti-DNA antibodies for 20min on ice, followed by goat anti-mouse FITC (Fcγ specific) and Annexin V PE for 15min in binding buffer. The latter was used to quantify apoptotic cells. Alternatively, cells were incubated with 1:50 dilution of MRL/lpr, Sle1 or Sle1CAD−/− sera. AutoAbs bound to the cells were detected using anti-mouse FITC (Fcγ specific). Data were acquired using a FACS Canto or FACS Calibur. Results were analyzed using FlowJo (version 8.8, Treestar Inc. Ashland, OR).
ANA
Staining was performed on prefixed HEp-2 cells following the manufacturer’s instructions (Antibodies Incorporated, Davis, CA). Briefly, mouse sera were used at 1/40 and 1/160 dilution in PBS+1%BSA+0.02%Azide and incubated for 30min at RT in a humidified chamber. Sera from 3H9 and 3H9CAD−/− with similar levels of anti-dsDNA Abs (determined by ELISA) were used. ANA were detected with a FITC-conjugated goat anti-mouse IgG (Fcγ specific, Jackson ImmunoResearch, West Grove, PA) Ab. Images were taken using a Zeiss Meta laser scanning confocal microscope with Zen software (Carl Zeiss MicroImaging, LLC, Thornwood, NY).
Spleen Immunofluorescence
Spleens were frozen in OCT, and stored at −80°C. The sections were fixed with acetone and treated with blocking buffer (3%BSA, 0.1%Tween 20, 10% rat serum) and stained with biotin conjugated anti-λ1, FITC conjugated anti-CD4, AlexaFluor647 conjugated anti-IgD, followed by AlexaFluor555 conjugated streptavidin. Images were taken using Zeiss confocal microscope. B cell follicles were defined as dense clusters of IgD+ cells and area of the follicle and total area of the image was determined (ImageJ Freeware, NIH, Bethesda, MD). λ1+ single cells in the follicle area were counted and number of λ1+ cells per mm2 area of the follicle were determined (24).
Flow cytometry
Cell suspensions were prepared from mouse femurs, tibias, and spleens. RBCs were lysed using ACK reagent. The following Abs were used for staining: IgM-FITC (polyclonal, Southern Biotech, Birmingham, AL), sIgMa-FITC (clone DS-1), sIgMb-PE (clone AF6-78), CD43-PE (clone S7), CD21-PE, CD23-PECy7, AA4.1-APC (BD Biosciences, San Jose, CA), B220-APCCy7 (clone 6B2, eBiosciences Inc., San Diego, CA), lambda-biotin (RML-42, Biolegend, San Diego, CA), kappa-PE (Clone 187.1, BD Biosciences). Flow cytometry was performed on BD Canto (BD Biosciences) and the data were analyzed using FlowJo software (version 8.8, Treestar Inc).
Western blot
HeLa cell lysates were prepared and protein concentrations were determined by bicinchoninic acid assay (Pierce, Rockford, IL). 50μg protein/well were separated on a 15% SDS PAGE gel and transferred to nitrocellulose membrane. The membrane was cut into 5mm strips. After blocking, each strip was incubated with 1:100 sera dilution. Sera from B6 and MRL/lpr were used as negative and positive controls respectively. The strips were then incubated with alkaline phosphatase conjugated goat anti-mouse IgG (Fcγ-specific) secondary Ab, and were developed using BCIP/NBT (Sigma, St. Louis, MO).
Statistical Analysis
Mann-Whitney or One-sided Student’s t tests were performed where appropriate using GraphPad Prism 4.0c (GraphPad Software, Inc.). Statistical significance was defined as p < 0.05.
Results
Absence of CAD results in increased production of anti-chromatin antibodies in Sle1 and Sle123 mice
To analyze the role of CAD in the spontaneous generation of lupus Abs, we generated CAD deficient Sle1 and Sle123 mice. Figure 1A shows that in the absence of CAD, Sle1 mice produced higher titers of anti-chromatin Abs compared to CAD sufficient Sle1 mice. A significant difference was observed beginning at 5–6 months of age, and was maintained through out the life of the mice (Fig.1A). Similar to Sle1CAD−/− mice, Sle123CAD−/− mice showed increased production of anti-chromatin Abs (Figure 1B). Interestingly Sle1CAD−/− mice showed increased production of anti-dsDNA antibodies while Sle123CAD−/− did not. (Figure 1C and 1D). Moreover Sle123CAD−/−, but not Sle1CAD−/−, showed increased immune complex deposition in the kidney at 8 months of age (Supplementary Figure 2). The results show that absence of chromatin fragmentation leads to increased production of anti-chromatin Abs and more severe nephritis in spontaneous lupus models. However, the altered autoAg generated in the absence of CAD does not induce renal immune complex deposition in Sle1, despite generating both anti-chromatin and anti-dsDNA autoAbs, possibly because the Sle1 background lacks the intrinsic factors that convey renal susceptibility to glomerulonephritis (25). These data demonstrate in vivo that apoptotic nuclear debris in genetically predisposed lupus models act as autoAgs and that alteration of self-ligand during cell death leads to a breakdown of self-tolerance.
Figure 1. Sle1 CAD−/− and Sle123 CAD−/− mice show higher levels of anti-chromatin auto-antibodies.

Sle1 (gray circles) and Sle1 CAD−/− (black circles) mice (5–9 month old) were serially bled and the anti-chromatin (A) and anti-dsDNA (C) auto-antibodies were measured by ELISA. By 6 months of age, CAD−/− mice showed significant higher levels of anti-chromatin Abs on the Sle1 background (p<0.0001). B and D show autoantibody levels in 8 month old Sle123 (gray circles) and Sle123 CAD−/−(black circles) mice. Sle123 CAD−/− mice also showed significantly higher anti-chromatin antibody levels (p<0.05) (B). The Sle1 CAD−/− mice had modest but statistically significant increase in anti-dsDNA antibodies at ~9 month of age (p<0.0001) (C). However the levels of anti-dsDNA Abs in both Sle1 and Sle1CAD−/− were lower than those observed in Sle123 and Sle123CAD−/− mice. The anti-dsDNA levels were similar between Sle123 and Sle123CAD−/− mice (D). All serum samples were tested at a 1:250 dilution.
Absence of CAD breaks tolerance of auto-reactive B cells in the 3H9 model of B cell anergy
The 3H9 mouse is knock-in for the MRL/lpr-derived autoreactive VH chain (3H9) (15); this heavy chain is reactive against dsDNA, chromatin and phospholipids, when it pairs with certain endogenous light chains including most lambda light chains (16, 26, 27). The 3H9 mouse on a non-autoimmune background shows a developmental arrest of auto-reactive B cells, surface IgM downregulation and follicular exclusion of lambda-1 B cells that express the transgenic heavy chain allele (16, 18). It is, therefore, an excellent model of peripheral and central tolerance towards chromatin. To determine whether in this model nuclear modifications during apoptosis are relevant to the maintenance of B cell tolerance we generated CAD deficient 3H9 mice. Figure 2 shows that unlike 3H9 mice, 3H9CAD−/− mice spontaneously produced IgM and IgG anti-dsDNA and anti-chromatin antibodies, as early as 1 month of age. Since the absence of nuclear modifications could have altered the autoAg recognized by auto-reactive B cells in the 3H9 mouse, we determined ANA pattern in 3H9 and 3H9CAD−/− sera. The antibodies made in the 3H9CAD−/− did not differ in their ANA pattern from those made in 3H9CAD+/+, although the titers in 3H9 mice were much lower. To investigate the specificities of those autoAbs we performed western blots with HeLa cells extracts. As positive control we used sera from MRL/lpr mice because the 3H9-ki is derived from this strain (15). We found that only the 3H9CAD−/− generated antibodies similar to MRL/lpr mice. (Supplementary Fig 3). These results strongly suggest that the absence of CAD alters the autoAb profile and facilitates the break of tolerance toward the autoAgs originally developed in the MRL/lpr mice.
Figure 2. 3H9 CAD−/− mice generate auto-antibodies spontaneously.

3H9 (gray circles) and 3H9CAD−/− (black circles) mice were bled at different ages and anti-dsDNA and anti-chromatin antibodies were detected in the sera by ELISA as described in the Methods. 3H9CAD−/− mice showed significantly higher anti-dsDNA antibodies, compared to 3H9 (p<0.01) (B), and showed a slight increase in anti-chromatin Abs (A). Both 3H9 and 3H9CAD−/− mice generated anti-dsDNA IgM (D) and anti-chromatin IgM (C) Abs, and the 3H9CAD−/− mice had significantly higher levels of both IgM anti-dsDNA (p=0.0008) and anti-chromatin (p=0.013). Sera from 3–6.5 months old mice were used to determine IgM levels. The data show that similarly to Sle1 and Sle123, the absence of CAD allowed auto-reactive B cells to escape tolerance. All serum samples were tested at a 1:250 dilution.
In the 3H9 mice there are at least two B cell tolerance checkpoints in play: the first is in the bone marrow where auto-reactive B cells can undergo receptor editing or apoptosis (27). B cells that pass the bone marrow checkpoints are further evaluated for self-reactivity in the periphery and, if needed, tolerized by anergy, follicular exclusion or deletion (16, 18). To better understand the manner in which self-tolerance is breached in 3H9CAD−/− mice, we analyzed B cell development and maturation by flow cytometry. The developing B cells from bone marrow (B220+CD43−) were divided into Hardy fractions D (small pre-B cells; IgM-, undergoing L chain rearrangement), E (immature IgM+), and F (IgM+, re-circulating B cells) (28). Figure 3A and Figure 3B show that the Hardy fractions D-F from 3H9CAD−/− have percentages and absolute numbers comparable to those observed in 3H9 mice, suggesting that the absence of CAD does not alter bone marrow B cell maturation, including expansion of Fr. D which accompanies the increased level of L chain receptor editing in this model ((29) and our unpublished observations). For the analysis of spleen B cells we divided them into mature (B220+AA4.1−) and immature transitional B cells (B220+AA4.1+). The mature B cells were further divided into follicular (CD23hiCD21−) and marginal zone (CD23loCD21hi) B cells. 3H9 mice showed accumulation of marginal zone B cells as previously reported (29). Similarly, 3H9CAD−/− mice showed marginal zone B cell numbers comparable to those observed in 3H9 mice, and the marginal zone B cells were more abundant than those observed in either B6 or CAD−/− (Fig 3C). The immature transitional B cells were further divided into T1 (CD23−IgMhi), T2 (CD23+IgM+), and T3 (CD23+IgMlo) (30). Figure 3D shows that both 3H9 and 3H9CAD−/− have reduced numbers of T2 cells and increased T3 and IgM−CD23+ cells as reported previously in 3H9 mice (29). These data show that absence of CAD does not alter B cell development or maturation in 3H9 mice. Finally both 3H9 and 3H9CAD−/− showed increased “a” allotype B cells in both spleens and bone marrow, reflecting the allotype of the transgenic heavy chain (Figure 3E).
Figure 3. B cell development and maturation is similar in 3H9 and 3H9CAD−/− mice.

B cell development and maturation in 3H9 CAD−/− mice was analyzed by flow cytometry. A. The developing B cells from bone marrow (B220+CD43−) were divided into Hardy fractions D, E, and F. Hardy fractions D–F from 3H9CAD−/− and 3H9 had comparable numbers. B. Representative absolute numbers of Hardy fractions D,E and F are shown. C. Splenic B cells were divided into mature (B220+AA4.1−) and into immature transitional (B220+AA4.1+) B cells. The mature B cells were divided into follicular (CD23hiCD21−) and marginal zone (CD23loCD21hi) B cells. 3H9 and 3H9CAD−/− mice showed increased marginal zone B cells. D. The immature transitional B cells were further divided into T1 (CD23-IgMhi), T2 (CD23+IgM+), and T3 (CD23+IgMlo). Both 3H9 and 3H9CAD−/− have reduced numbers of T2 cells. E. Both 3H9 and 3H9CAD−/− showed increased “a” allotype B cells in both spleens and bone marrow. The data are representative from 9 mice.
The 3H9 heavy chain can pair with the endogenous λ1 light chain to form an anti-dsDNA antibody (15). In 3H9 mice, the λ1+ auto-reactive cells are excluded from B cell follicles and accumulate at the T cell-B cell zone interface in the spleen (18, 31). To determine if CAD deficiency altered the degree of follicular exclusion of autoreactive B cells, we stained frozen spleen sections with IgD to identify B cell follicles and with anti-λ1 to identify the auto-reactive B cells in 3H9 and 3H9 CAD−/− mice. Figure 4A and 4B show that in 3H9 mice the λ1+ cells did not enter the follicles and accumulated in the T cell zone, consistent with previous studies (16, 32)(white arrows left panel). In 3H9CAD−/− mice, however, λ1+ cells did not accumulate in the T cell zone and were able to enter B-cell follicles (white arrows right panel). These data show that auto-reactive B cells escape to the periphery and do not undergo follicular exclusion in absence of CAD.
Figure 4. Auto-reactive B cells overcome follicular exclusion in 3H9CAD−/− mice.
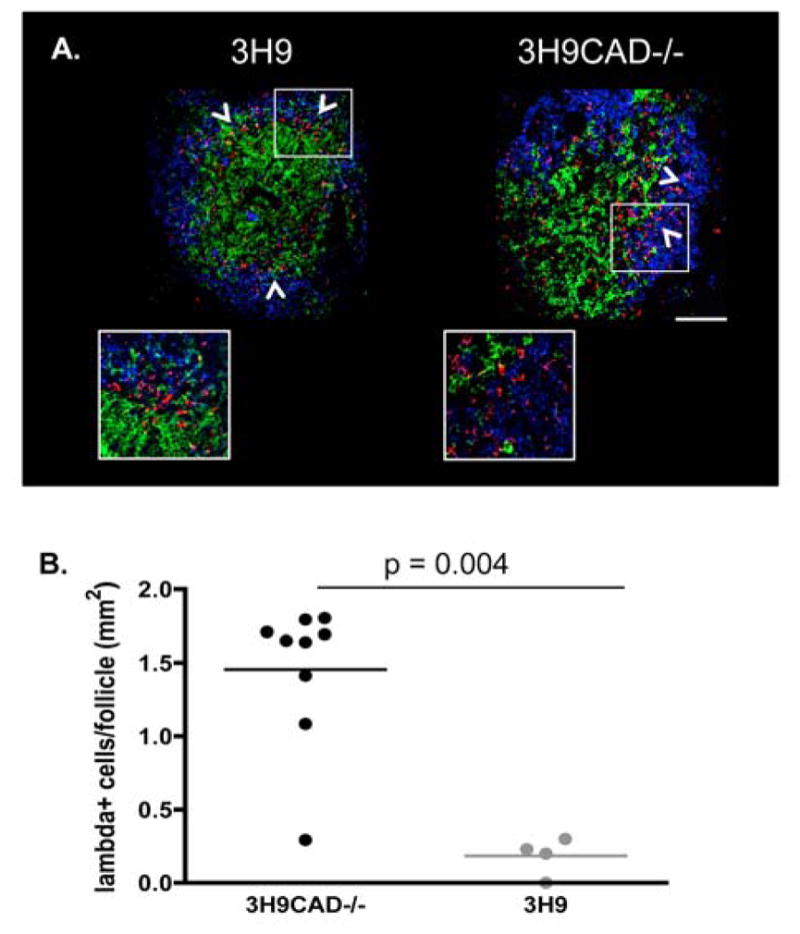
A. Frozen spleen sections from 3H9 and 3H9CAD−/− mice were fixed with acetone and stained using anti-IgD (Blue), anti-CD4 (Green), and anti-λ1 (Red). Images were acquired by Zeiss confocal microscope. Spleen sections from three mice/strain were stained. 2 to 4 follicles per mouse were used for quantitation. B. The number of λ1 positive cells in the B cell follicles were quantified using ImageJ. As previously reported, 3H9CAD+/+ mice showed follicular exclusion of autoreactive B cells (White arrows and inset left panel). In contrast 3H9CAD−/− mice did not (white arrows and inset, right panel). The results suggest that nuclear fragmentation during apoptosis is one of the mechanisms to maintain tolerance to chromatin.
Absence of CAD is relevant only in immune-competent cells
To determine whether absence of CAD was important in hematopoietic or stromal cells, we generated bone marrow chimeras, where CAD+/+ or CAD−/− mice were lethally irradiated and then reconstituted with Sle123 mice bone marrow. 2 months post reconstitution both CAD+/+ and CAD−/− mice produced similar levels of anti-chromatin Abs, which progressively increased to levels comparable to un-manipulated Sle123 mice (Supplementary Figure 4 and Figure 1).
Antibodies from lupus mice bind more to apoptotic CAD−/− cells
During apoptosis, the morphological changes observed in CAD+/+ and CAD−/− differ (12). Although CAD−/−cells undergo initial chromatin condensation during apoptosis, they do not fragment DNA or form nuclear blebs (11). The result is a dying cell that has lost most of its cytoplasm, but retains chromatin structure. This would potentially lead to an increased exposure of autoAgs or exposure of a modified autoAg. To determine whether these differences may affect the opsonization of apoptotic cells by auto-Abs, we determined the ability of serum from lupus prone mice to bind to apoptotic CAD+/+ or CAD−/− cells. Apoptotic cells were defined as Annexin V positive. Figure 5A shows the ability of sera from Sle1 or Sle1CAD−/− mice to bind to apoptotic cells. We used sera from mice that had either high or low titers of autoAbs by ELISA, and we used negative sera as controls. Figure 5A shows that sera from both Sle1 and Sle1CAD−/− with high titers of antibodies bound more to CAD−/− apoptotic cells, whereas sera negative for autoAbs did not bind apoptotic cells. These data show that CAD−/− cells are opsonized better by autoAbs. To demonstrate whether antigens exposed by CAD−/− and CAD+/+ apoptotic cells differ, we determined the ability of monoclonal antibodies to specific nuclear antigens to bind to CAD−/− or CAD+/+ apoptotic cells. Figure 5B shows that antibodies with specificities to histone H2B and to H2A-H2B-DNA complex bound more to CAD−/−apoptotic cells, whereas the antibodies with specificities towards DNA did not show any difference. Similar results were obtained with high titers anti-dsDNA sera from MRL/lpr mice (Supplementary Figure 5). The greater binding of autoAbs to CAD−/− apoptotic cells was not due to increased size or densities of apoptotic CAD−/− cells as both forward and size scatter by flow cytometry were similar (Figure 5C). The results suggest that in the absence of CAD, apoptotic chromatin is abnormally exposed, facilitating the autoimmune response by increasing auto-Ab binding.
Figure 5. AutoAbs from Sle1 and MRL/lpr mice preferentially bind CAD−/− apoptotic cells.

A. We determined whether the absence of nuclear modifications during apoptosis altered the ability of sera from autoimmune mice to bind apoptotic cells. Apoptosis was induced in CAD+/+ or CAD−/− cells by γ radiation. AutoAbs from both Sle1 and Sle1.CAD−/− mice bound more to apoptotic cells from CAD−/− mice than to apoptotic cells from CAD+/+ mice. B. To determine whether antigens exposed on CAD+/+ and CAD−/− apoptotic cells differ, apoptotic thymocytes from either CAD+/+ or CAD−/− mice were stained with Annexin V and monoclonal antibodies towards H2B, H2A-H2B-DNA complexes, nucleosomes, or DNA. The figure shows Annexin V+ gated cells. These results demonstrate that monoclonal antibodies with specificities towards histones and histones/DNA preferentially bind CAD−/− apoptotic cells. C. Apoptotic thymocytes from CAD+/+ and CAD−/− mice were analyzed by flow cytometry. Annexin V positive thymocytes from both strains showed similar forward scatter and side scatter, indicating that apoptotic CAD−/− and CAD+/+ have similar sizes.
Discussion
Our data show that the deficiency of CAD in lupus prone mice results in increased production of autoAbs and greater severity of renal disease. Our data also show that CAD deficiency leads to spontaneous production of autoAbs in the anergic 3H9 mouse model, suggesting that CAD may be responsible for maintaining tolerance to nuclear autoAgs.
Apoptotic cellular debris has been suggested to be a major source of nuclear autoAg (1, 33). Yet, under physiological conditions, the apoptotic blebs and bodies may also serve as a source of autoAgs that induce self-tolerance by anergy, receptor editing, or deletion of auto-reactive cells. Spontaneous lupus models and patients have intrinsic defects in one or more tolerance checkpoints that make them prone to autoimmunity (34). The presence of blebs and bodies limit but do not completely eliminate this escape. Therefore, we reasoned that the absence of these modifications during cell death (as observed in CAD deficiency) would allow the escape of auto-reactive B cells. Our data indeed show that in the Sle1 and Sle123 models where auto-reactive B cells are already known to escape, the absence of CAD results in increased production of anti-chromatin Abs. Furthermore, in the 3H9 model, tolerance is broken in the absence of CAD as there is de novo production of autoAbs. The absence of apoptotic modifications not only alters bone marrow B cell selection, but also results in defective peripheral tolerance. The end-result is activation of auto-reactive B cells that have escaped multiple tolerance checkpoints.
The congenic strains derived from the NZM2410 constitute an excellent model where the different manifestations of lupus can be dissected (35). In this study we used C57Bl/6 single congenic Sle1 and tricongenic Sle123 models of spontaneous lupus (36). The Sle1 locus mediates loss of tolerance to chromatin (37). Although the interval is required for the development of disease in NZM2410 mice, Sle1 alone leads only to serological autoreactivity (38). However in epistasis with Sle2 and Sle3, Sle1 leads to fatal glomerulonephritis. Interestingly, Sle1 single congenic mice develop accelerated lupus when backcrossed with other lupus susceptibility loci. For example animals with the Sle1 region and the Y autoimmune accelerator gene (Yaa gene), a single locus derived from BXSB lupus prone mouse, develop severe SLE with approximately 60% mortality at 9 months of age (39). The epistatic interaction of Sle1 with Fas mutation also leads to increased severity of disease, characterized by splenomegaly, ANA production, glomerulonephritis, and 80% mortality by 5–6 mo of age (40).
We show here that absence of CAD resulted in increased anti-chromatin Abs in Sle1 and Sle123 mice. The Sle123 mice also showed a greater severity of renal disease suggesting that absence of CAD, as the above susceptibility genes, acts as an accelerator of disease. However, the nephritis score was not statistically significant.
The low affinity 3H9 anti-DNA/chromatin heavy chain knock-in (3H9) mice have proved to be a very useful model to study B cell tolerance mechanisms. In this model, the rearranged V(D)J heavy chain, 3H9, replaces the chromosomal JH locus. The transgenic locus undergoes editing, isotype switching, and somatic mutation (21). The 3H9 mouse does not generate high levels of autoAbs, the auto-reactive B cells are anergic and excluded from the follicular zone (18), presumably because B cells have encountered the Ag (i.e. DNA/Chromatin) (32). This model therefore allowed us to investigate whether and at which stage apoptotic chromatin is required for the anergy or deletion of auto-reactive B cells. The auto-reactive 3H9 heavy chain pairs with certain endogenous light chains, such as λ1 that can be tracked in vivo, to generate anti-dsDNA specificity. Indeed we show here that in the CAD deficient 3H9 mice, the auto-reactive B cells λ1+ overcome follicular exclusion and the mice spontaneously generate autoAbs toward chromatin and dsDNA. These results might be partially explained by previous work in mice and humans in which apoptotic cell remnants lead to abnormal recognition of AutoAgs and to selection of autoAbs (41, 42). In the absence of CAD, autoreactive B cells in the germinal centers are exposed to abnormal chromatin complexes that could lead to accidental selection of autoAbs.
Overall the results in the 3H9CAD−/− mouse are in agreement with the Sle1CAD−/− and Sle123CAD−/− models and suggest that fragmented apoptotic chromatin is required for inducing tolerance to nuclear autoAgs. Similar to Sle1 single congenics, 3H9 mice can break tolerance to DNA in the presence of other autoimmune modifiers. NZB/W F1 mice with the 3H9 knock-in generated anti-DNA IgM and IgG with homogenous ANA (19). Moreover anti-DNA Abs were higher in MRL/lpr mice expressing 3H9 (43). Thereby CAD deficiency may act as an autoimmune modifier in Sle1, Sle123 and 3H9 models of lupus autoimmunity.
We have shown that lupus autoAbs bind more to CAD deficient apoptotic cells, Better opsonization of CAD−/− apoptotic cells by autoAbs may result in more efficient uptake and presentation of nuclear antigens, which may further activate peripheral auto-reactive B cells to differentiate into antibody secreting cells.
Moreover we have previously shown that antibody opsonization of autoAgs results in increased phagocytosis by dendritic cells. This increased phagocytosis significantly enhanced the ability of dendritic cells to activate T cells (5). FcγR-mediated phagocytosis may therefore result in vigorous immune responses and promote autoimmunity. Recently Munoz et al. showed that in SLE patients a defective clearance leads to generation of secondary necrotic cells (41). The opsonization of cell remnants with nuclear autoAbs drives FcγR-mediated phagocytosis, which is inflammatory and thus may contribute to chronic inflammation and tissue damage (3, 32, 44).
Our observation that autoAbs bind more to CAD deficient apoptotic cells suggests that the opsonization of cells lacking CAD increases the FcγR-mediated phagocytosis of these remnants and inflammation. However the non-fragmented chromatin could also increase the affinity of auto-reactive B cells towards chromatin (as suggested by Figure 5).
It has been shown that in 3H9 mice, the auto-reactive B cells express light chains that veto DNA binding (so-called “editor” light chains) and these edited B cells show a much-reduced affinity for DNA. We therefore propose that the absence of CAD biases B cell repertoire toward DNA binding and contributes to the loss of tolerance to DNA.
We have previously published that the lack of chromatin fragmentation in the pristane-induced lupus model resulted in the absence of ANA (12). Following pristane injection, the presence of local inflammation and increased apoptosis in the peritoneum along with the adjuvant properties of pristane lead to the generation of granulomas where lymphoid structures are formed. Those are then responsible for the generation of auto-reactive B cells and eventually autoAbs (45, 46). This model reflects an induction of autoimmunity, which “acutely” necessitates autoAgs to trigger and sustain the autoimmune response, such as during lupus flares. Therefore the lack of apoptotic blebs and nuclear Ag due to the absence of CAD may result in absence of ANA. Another difference may also explain the loss of tolerance to dsDNA in the 3H9 background and increased levels of autoantibodies in the Sle1 and Sle123 background. It is generally accepted that the anti-histone/DNA response is Toll-like Receptor (TLR) 9 dependent, while the anti-Protein/RNA response is TLR7 dependent (47). The absence of CAD could primarily trigger models in which the anti-histone/DNA response and possibly the TLR9 dependence are strong. Instead it would act the opposite in the Pristane model, which produces high levels of anti-ribonucleoprotein Abs and has been recently demonstrated to be dependent on activation through TLR7 (13).
We therefore infer that CAD is necessary to maintain tolerance toward nuclear antigens in lupus-prone genetic backgrounds; once tolerance is broken CAD deficiency increases the provision of autoAgs to fuel the autoimmune response.
In this study we demonstrate that apoptotic morphological differences in the nuclear Ags allow auto-reactive anergic B cells to break tolerance and produce autoAbs. Interestingly the absence of nuclear fragmentation increased the binding of autoAbs directed toward histones or histones/DNA complexes but not DNA, suggesting that 1) DNA exposure by apoptotic cells is CAD independent and that 2) nuclear fragmentation is mainly required to maintain tolerance toward chromatin.
In conclusion our work demonstrates that nuclear modifications during cell death are an important mechanism for maintaining tolerance to chromatin and other lupus autoAgs. Therefore apoptotic nuclear material is required to delete or render potential auto-reactive cells anergic. Pharmacological manipulation of specific steps during apoptosis might lead to a better control of autoimmunity in humans.
Supplementary Material
Supplementary Figure 1. Sle1 CAD−/− mice do not have defects in T and B cell development compared to B6 and Sle1 mice. A. The frequencies of double negative (DN, CD4−CD8−) T cells, double positive (DP CD4+CD8+), and single positive (SP) T cells (CD4−CD8+ and CD4+CD8−) were determined. 3–5 month-old B6, Sle1 and Sle1 CAD−/− were studied (N=3). B. Peripheral B cell development and maturation were evaluated by flow cytometry using the following markers- Transitional B cells in the spleen: T1 (CD19+B220+AA4.1+IgMhiCD23−), T2 CD19+B220+AA4.1+IgMhiCD23+ and T3 CD19+B220+AA4.1+IgMloCD23+ and Marginal Zone (MZ) B cells (CD19+B220+IgMhiCD21hi). 3–5 month-old mice were investigated (N=3). These results demonstrate that the T and B cell development and maturation in the absence of CAD is normal.
Supplementary Figure 2. Sle123 CAD−/− had more severe nephritis compared to Sle123 mice. A. Sle123 CAD−/− showed greater immune complex deposition. 7–8 month old mice were evaluated. Frozen kidney sections were stained with FITC conjugated goat anti-mouse IgG (Fcγ-specific). Images were taken with a Nikon TE300 scanning confocal microscope, equipped with Nomarski differential interphase contrast optics, coupled to the Bio-Rad Radiance 2000 laser. B. H&E staining was performed and a previously described scoring method was used to assess the disease severity. In particular, the severity of nephritis was evaluated based on the presence of hyperthrophy, proliferative changes, hyaline deposits, crescents, necrosis/fibrosis/sclerosis and basement membrane thickening. Overall severity was graded from 0–4. Specifically 0–1+ is normal or occasional changes. 2+ is definite disease with less than 30% involvement of glomeruli, no tubular necrosis and less than 30% of vessels involved. 3+ is more than 30–50% glomeruli involved, with crescents formation, large interstitial infiltrate with necrosis and more than 30% of vessels involved. 4+ diffuse proliferative changes with crescents and sclerosis in more than 50% of glomeruli, extensive interstitial infiltration with necrosis, extensive fibrosis and more than 50% of vessels involvement with necrosis and fibrosis. A grade higher than 2 was considered pathologic (12, 48). The data show that CAD deficient Sle123 develop more severe nephritis with greater immune-complex deposition in the kidneys.
Supplementary Figure 3. Antibodies from 3H9CAD−/− and MRL/lpr, but not from 3H9 mice have similar antigen specificities. Whole cell lysates from HeLa cells were separated on a 15% SDS PAGE gel and transferred to nitrocellulose membrane. The membrane was cut into 5mm strips. After blocking, each strip was incubated with 1:100 dilution of the sera. Sera from B6 and MRL/lpr were used as negative and positive controls respectively. The strips were then incubated with alkaline phosphatase conjugated goat anti-mouse IgG (Fcγ-specific) secondary Ab, and were developed using BCIP/NBT (Sigma). M: protein standards. Black arrow indicates Abs found only in 3H9CAD−/− and control MRL/lpr.
Supplementary Figure 4. Absence of CAD is relevant only in immuno-competent cells. Bone marrow chimeras were generated by reconstituting CAD+/+ and CAD−/− mice with bone marrow from Sle123 mice as described previously (23). Briefly, recipient mice were irradiated with a lethal dose of 900 rads (450 rads twice, with 4h interval), and reconstituted with 3×107 unsorted bone marrow cells from Sle123 mice. Mice were bled monthly, and anti-chromatin antibodies were determined by ELISA. There was no statistical difference between the two strains. Data are represented as Mean ± SEM (N= 5 mice in each group).
Supplementary Figure 5. AutoAbs from MRL/lpr mice preferentially bind CAD−/− apoptotic cells. We determined whether the absence of nuclear modifications during apoptosis altered the ability of sera from autoimmune mice to bind apoptotic cells. Apoptosis was induced in CAD+/+ or CAD−/− cells by γ radiation. Sera from old MRL/lpr mice, but not B6 mice, stained higher numbers of apoptotic CAD−/− cells.
Table 1.
ANA patterns in 3H9 and 3H9CAD−/− sera
| 3H9 | 3H9CAD−/− | |
|---|---|---|
| Nuclear+Cytoplasmic | 3/4 | 3/6 |
| Nuclear speckled | 4/4 | 5/6 |
| Nuclear homogenous with fine speckled | 1/4 | 1/6 |
Sera from 3H9 and 3H9CAD−/− mice with similar levels of autoAbs as determined by ELISA were used to determine ANA pattern. The table shows the number of sera showing a particular pattern out the total sera tested.
Acknowledgments
The authors thank Drs. P.L. Cohen, R.A. Eisenberg and M.F. Denny for critically reading the manuscript and Drs S. Gallucci and A. Marshak-Rothstein for their excellent suggestions. The authors also thank Dr. Laurence Morel for providing the Sle1 and Sle123 mice and for helping generating the mutant mice.
This work was supported by the Lupus Research Institute and NIH-NIAMS R03-R03-AR-51387 to R.C; NIH R01DE017590 to ELP; NIH AR40072, 44076, and support from Rheuminantions, Inc to J.C
References
- 1.Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med. 1994;179(4):1317–30. doi: 10.1084/jem.179.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140(5):619–30. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 3.Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6(5):280–9. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 4.Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16(8):1093–107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frisoni L, McPhie L, Colonna L, Sriram U, Monestier M, Gallucci S, et al. Nuclear autoantigen translocation and autoantibody opsonization lead to increased dendritic cell phagocytosis and presentation of nuclear antigens: a novel pathogenic pathway for autoimmunity? J Immunol. 2005;175(4):2692–701. doi: 10.4049/jimmunol.175.4.2692. [DOI] [PubMed] [Google Scholar]
- 6.Fransen JH, van der Vlag J, Ruben J, Adema GJ, Berden JH, Hilbrands LB. The role of dendritic cells in the pathogenesis of systemic lupus erythematosus. Arthritis Res Ther. 2010;12(2):207. doi: 10.1186/ar2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amoura Z, Koutouzov S, Piette JC. The role of nucleosomes in lupus. Curr Opin Rheumatol. 2000;12(5):369–73. doi: 10.1097/00002281-200009000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes. J Exp Med. 2004;199(12):1631–40. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature. 1998;391(6662):43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 10.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391(6662):96–9. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 11.Kawane K, Nagata S. Nucleases in programmed cell death. Methods Enzymol. 2008;442:271–87. doi: 10.1016/S0076-6879(08)01414-6. [DOI] [PubMed] [Google Scholar]
- 12.Frisoni L, McPhie L, Kang SA, Monestier M, Madaio M, Satoh M, et al. Lack of chromatin and nuclear fragmentation in vivo impairs the production of lupus anti-nuclear antibodies. J Immunol. 2007;179(11):7959–66. doi: 10.4049/jimmunol.179.11.7959. [DOI] [PubMed] [Google Scholar]
- 13.Fairhurst AM, Mathian A, Connolly JE, Wang A, Gray HF, George TA, et al. Systemic IFN-alpha drives kidney nephritis in B6. Sle123 mice. Eur J Immunol. 2008;38(7):1948–60. doi: 10.1002/eji.200837925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morel L. Genetics of SLE: evidence from mouse models. Nat Rev Rheumatol. 2010;6(6):348–57. doi: 10.1038/nrrheum.2010.63. [DOI] [PubMed] [Google Scholar]
- 15.Luning Prak ET, Monestier M, Eisenberg RA. B cell receptor editing in tolerance and autoimmunity. Ann N Y Acad Sci. 2011;1217:96–121. doi: 10.1111/j.1749-6632.2010.05877.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erikson J, Radic MZ, Camper SA, Hardy RR, Carmack C, Weigert M. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature. 1991;349(6307):331–4. doi: 10.1038/349331a0. [DOI] [PubMed] [Google Scholar]
- 17.Ota M, Duong BH, Torkamani A, Doyle CM, Gavin AL, Ota T, et al. Regulation of the B cell receptor repertoire and self-reactivity by BAFF. J Immunol. 2010;185(7):4128–36. doi: 10.4049/jimmunol.1002176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mandik-Nayak L, Bui A, Noorchashm H, Eaton A, Erikson J. Regulation of anti-double-stranded DNA B cells in nonautoimmune mice: localization to the T-B interface of the splenic follicle. J Exp Med. 1997;186(8):1257–67. doi: 10.1084/jem.186.8.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steeves MA, Marion TN. Tolerance to DNA in (NZB × NZW)F1 mice that inherit an anti-DNA V(H) as a conventional micro H chain transgene but not as a V(H) knock-in transgene. J Immunol. 2004;172(11):6568–77. doi: 10.4049/jimmunol.172.11.6568. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Li L, Kumar KR, Xie C, Lightfoot S, Zhou XJ, et al. Lupus susceptibility genes may breach tolerance to DNA by impairing receptor editing of nuclear antigen-reactive B cells. J Immunol. 2007;179(2):1340–52. doi: 10.4049/jimmunol.179.2.1340. [DOI] [PubMed] [Google Scholar]
- 21.Chen C, Nagy Z, Prak EL, Weigert M. Immunoglobulin heavy chain gene replacement: a mechanism of receptor editing. Immunity. 1995;3(6):747–55. doi: 10.1016/1074-7613(95)90064-0. [DOI] [PubMed] [Google Scholar]
- 22.Eisenberg R, Choudhury A. The anti-DNA knock-in model of systemic autoimmunity induced by the chronic graft-vs-host reaction. Methods Mol Med. 2004;102:273–84. doi: 10.1385/1-59259-805-6:273. [DOI] [PubMed] [Google Scholar]
- 23.Jog NR, Dinnall JA, Gallucci S, Madaio MP, Caricchio R. Poly(ADP-ribose) polymerase-1 regulates the progression of autoimmune nephritis in males by inducing necrotic cell death and modulating inflammation. J Immunol. 2009;182(11):7297–306. doi: 10.4049/jimmunol.0803565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, et al. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. The Journal of Experimental Medicine. 2008;205(12):2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi X, Xie C, Chang S, Zhou XJ, Tedder T, Mohan C. CD19 hyperexpression augments Sle1-induced humoral autoimmunity but not clinical nephritis. Arthritis Rheum. 2007;56(9):3057–69. doi: 10.1002/art.22825. [DOI] [PubMed] [Google Scholar]
- 26.Radic MZ, Mascelli MA, Erikson J, Shan H, Weigert M. Ig H and L chain contributions to autoimmune specificities. J Immunol. 1991;146(1):176–82. [PubMed] [Google Scholar]
- 27.Roark JH, Kuntz CL, Nguyen KA, Caton AJ, Erikson J. Breakdown of B cell tolerance in a mouse model of systemic lupus erythematosus. J Exp Med. 1995;181(3):1157–67. doi: 10.1084/jem.181.3.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hardy RR, Hayakawa K. B cell development pathways. Annu Rev Immunol. 2001;19:595–621. doi: 10.1146/annurev.immunol.19.1.595. [DOI] [PubMed] [Google Scholar]
- 29.Sekiguchi DR, Yunk L, Gary D, Charan D, Srivastava B, Allman D, et al. Development and selection of edited B cells in B6.56R mice. J Immunol. 2006;176(11):6879–87. doi: 10.4049/jimmunol.176.11.6879. [DOI] [PubMed] [Google Scholar]
- 30.Allman D, Lindsley RC, DeMuth W, Rudd K, Shinton SA, Hardy RR. Resolution of three nonproliferative immature splenic B cell subsets reveals multiple selection points during peripheral B cell maturation. J Immunol. 2001;167(12):6834–40. doi: 10.4049/jimmunol.167.12.6834. [DOI] [PubMed] [Google Scholar]
- 31.Shao WH, Kuan AP, Wang C, Abraham V, Waldman MA, Vogelgesang A, et al. Disrupted Mer receptor tyrosine kinase expression leads to enhanced MZ B-cell responses. J Autoimmun. 2010;35(4):368–74. doi: 10.1016/j.jaut.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paul E, Nelde A, Verschoor A, Carroll MC. Follicular exclusion of autoreactive B cells requires FcgammaRIIb. Int Immunol. 2007;19(4):365–73. doi: 10.1093/intimm/dxm002. [DOI] [PubMed] [Google Scholar]
- 33.Caricchio R, McPhie L, Cohen PL. Ultraviolet B radiation-induced cell death: critical role of ultraviolet dose in inflammation and lupus autoantigen redistribution. J Immunol. 2003;171(11):5778–86. doi: 10.4049/jimmunol.171.11.5778. [DOI] [PubMed] [Google Scholar]
- 34.Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol. 2008;20(6):632–8. doi: 10.1016/j.coi.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 35.Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Adv Immunol. 2006;92:1–69. doi: 10.1016/S0065-2776(06)92001-X. [DOI] [PubMed] [Google Scholar]
- 36.Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, et al. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci U S A. 2000;97(12):6670–5. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohan C, Alas E, Morel L, Yang P, Wakeland EK. Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J Clin Invest. 1998;101(6):1362–72. doi: 10.1172/JCI728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mohan C, Morel L, Yang P, Watanabe H, Croker B, Gilkeson G, et al. Genetic dissection of lupus pathogenesis: a recipe for nephrophilic autoantibodies. J Clin Invest. 1999;103(12):1685–95. doi: 10.1172/JCI5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103(26):9970–5. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shi X, Xie C, Kreska D, Richardson JA, Mohan C. Genetic dissection of SLE: SLE1 and FAS impact alternate pathways leading to lymphoproliferative autoimmunity. J Exp Med. 2002;196(3):281–92. doi: 10.1084/jem.20010955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Munoz LE, Janko C, Grossmayer GE, Frey B, Voll RE, Kern P, et al. Remnants of secondarily necrotic cells fuel inflammation in systemic lupus erythematosus. Arthritis Rheum. 2009;60(6):1733–42. doi: 10.1002/art.24535. [DOI] [PubMed] [Google Scholar]
- 42.Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46(1):191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Li H, Ni D, Weigert M. Anti-DNA B cells in MRL/lpr mice show altered differentiation and editing pattern. J Exp Med. 2002;196(12):1543–52. doi: 10.1084/jem.20021560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fukuyama H, Nimmerjahn F, Ravetch JV. The inhibitory Fcgamma receptor modulates autoimmunity by limiting the accumulation of immunoglobulin G+ anti-DNA plasma cells. Nat Immunol. 2005;6(1):99–106. doi: 10.1038/ni1151. [DOI] [PubMed] [Google Scholar]
- 45.Kuroda Y, Nacionales DC, Akaogi J, Reeves WH, Satoh M. Autoimmunity induced by adjuvant hydrocarbon oil components of vaccine. Biomed Pharmacother. 2004;58(5):325–37. doi: 10.1016/j.biopha.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 46.Calvani N, Caricchio R, Tucci M, Sobel ES, Silvestris F, Tartaglia P, et al. Induction of apoptosis by the hydrocarbon oil pristane: implications for pristane-induced lupus. J Immunol. 2005;175(7):4777–82. doi: 10.4049/jimmunol.175.7.4777. [DOI] [PubMed] [Google Scholar]
- 47.Avalos AM, Uccellini MB, Lenert P, Viglianti GA, Marshak-Rothstein A. FcgammaRIIB regulation of BCR/TLR-dependent autoreactive B-cell responses. Eur J Immunol. 2010;40(10):2692–8. doi: 10.1002/eji.200940184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chan O, Madaio MP, Shlomchik MJ. The roles of B cells in MRL/lpr murine lupus. Ann N Y Acad Sci. 1997;815:75–87. doi: 10.1111/j.1749-6632.1997.tb52046.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Sle1 CAD−/− mice do not have defects in T and B cell development compared to B6 and Sle1 mice. A. The frequencies of double negative (DN, CD4−CD8−) T cells, double positive (DP CD4+CD8+), and single positive (SP) T cells (CD4−CD8+ and CD4+CD8−) were determined. 3–5 month-old B6, Sle1 and Sle1 CAD−/− were studied (N=3). B. Peripheral B cell development and maturation were evaluated by flow cytometry using the following markers- Transitional B cells in the spleen: T1 (CD19+B220+AA4.1+IgMhiCD23−), T2 CD19+B220+AA4.1+IgMhiCD23+ and T3 CD19+B220+AA4.1+IgMloCD23+ and Marginal Zone (MZ) B cells (CD19+B220+IgMhiCD21hi). 3–5 month-old mice were investigated (N=3). These results demonstrate that the T and B cell development and maturation in the absence of CAD is normal.
Supplementary Figure 2. Sle123 CAD−/− had more severe nephritis compared to Sle123 mice. A. Sle123 CAD−/− showed greater immune complex deposition. 7–8 month old mice were evaluated. Frozen kidney sections were stained with FITC conjugated goat anti-mouse IgG (Fcγ-specific). Images were taken with a Nikon TE300 scanning confocal microscope, equipped with Nomarski differential interphase contrast optics, coupled to the Bio-Rad Radiance 2000 laser. B. H&E staining was performed and a previously described scoring method was used to assess the disease severity. In particular, the severity of nephritis was evaluated based on the presence of hyperthrophy, proliferative changes, hyaline deposits, crescents, necrosis/fibrosis/sclerosis and basement membrane thickening. Overall severity was graded from 0–4. Specifically 0–1+ is normal or occasional changes. 2+ is definite disease with less than 30% involvement of glomeruli, no tubular necrosis and less than 30% of vessels involved. 3+ is more than 30–50% glomeruli involved, with crescents formation, large interstitial infiltrate with necrosis and more than 30% of vessels involved. 4+ diffuse proliferative changes with crescents and sclerosis in more than 50% of glomeruli, extensive interstitial infiltration with necrosis, extensive fibrosis and more than 50% of vessels involvement with necrosis and fibrosis. A grade higher than 2 was considered pathologic (12, 48). The data show that CAD deficient Sle123 develop more severe nephritis with greater immune-complex deposition in the kidneys.
Supplementary Figure 3. Antibodies from 3H9CAD−/− and MRL/lpr, but not from 3H9 mice have similar antigen specificities. Whole cell lysates from HeLa cells were separated on a 15% SDS PAGE gel and transferred to nitrocellulose membrane. The membrane was cut into 5mm strips. After blocking, each strip was incubated with 1:100 dilution of the sera. Sera from B6 and MRL/lpr were used as negative and positive controls respectively. The strips were then incubated with alkaline phosphatase conjugated goat anti-mouse IgG (Fcγ-specific) secondary Ab, and were developed using BCIP/NBT (Sigma). M: protein standards. Black arrow indicates Abs found only in 3H9CAD−/− and control MRL/lpr.
Supplementary Figure 4. Absence of CAD is relevant only in immuno-competent cells. Bone marrow chimeras were generated by reconstituting CAD+/+ and CAD−/− mice with bone marrow from Sle123 mice as described previously (23). Briefly, recipient mice were irradiated with a lethal dose of 900 rads (450 rads twice, with 4h interval), and reconstituted with 3×107 unsorted bone marrow cells from Sle123 mice. Mice were bled monthly, and anti-chromatin antibodies were determined by ELISA. There was no statistical difference between the two strains. Data are represented as Mean ± SEM (N= 5 mice in each group).
Supplementary Figure 5. AutoAbs from MRL/lpr mice preferentially bind CAD−/− apoptotic cells. We determined whether the absence of nuclear modifications during apoptosis altered the ability of sera from autoimmune mice to bind apoptotic cells. Apoptosis was induced in CAD+/+ or CAD−/− cells by γ radiation. Sera from old MRL/lpr mice, but not B6 mice, stained higher numbers of apoptotic CAD−/− cells.