

Abstract
Objective
To determine the role for APRIL in the development of SLE.
Methods
Wild-type (WT) NZM 2328, NZM.April-/-, NZM.Baff-/-, and NZM.Baff-/-.April-/- mice were evaluated for lymphocyte phenotype by flow cytometry, for serum total IgG and IgG autoantibody levels by ELISA, for glomerular deposition of IgG and C3 by immunofluorescence, for renal histopathology, and for clinical disease (severe proteinuria).
Results
In comparison to WT mice, NZM.April-/- mice harbored increased spleen B cells, T cells, and plasma cells (PC); increased serum levels of IgG anti-chromatin antibodies; and decreased numbers of bone marrow (BM) PC. In addition, glomerular deposition of IgG and C3 was similar in NZM.April-/- and WT mice; renal histopathology tended to be more severe in NZM.April-/- mice than in WT mice; and development of clinical disease was identical in NZM.April-/- and WT mice. BM (but not spleen) PC and serum IgG anti-chromatin and anti-dsDNA antibody levels were lower in NZM.Baff-/-.April-/- mice than in NZM.Baff-/- mice, whereas renal immunopathology in each cohort was equally mild.
Conclusions
APRIL is dispensable for development of full-blown SLE in NZM mice. Moreover, the elimination of both APRIL and BAFF has no discernable effect on development of renal immunopathology or clinical disease beyond that of elimination of BAFF alone. The reduction in BM PC in hosts doubly-deficient in APRIL and BAFF beyond that in hosts deficient only in BAFF raises concern that combined antagonism of APRIL and BAFF may lead to greater immunosuppression without concomitant increase in therapeutic efficacy.
Introduction
The clinical diagnosis of systemic lupus erythematosus (SLE) is based on a constellation of signs, symptoms, and clinical laboratory-based abnormalities. This inherent heterogeneity notwithstanding, certain common threads tend to run across all “varieties” of SLE. One of these common threads is B cell hyperactivity. A priori, any factor that enhances B cell survival and/or function may play a pathogenetic role in SLE.
One such factor is BAFF (commonly also known as BLyS), a 285-amino acid type-II transmembrane protein member of the TNF ligand superfamily (1, 2). Cleavage of surface BAFF by a furin protease results in release of a soluble, biologically active 17-kDa molecule (2, 3) which binds to three receptors (BCMA, TACI, and BR3 [also known as BAFFR]) on the surface of B cells (4-7).
The connection between BAFF and development of SLE is very strong. In mice that otherwise are not autoimmune-prone, constitutive overexpression of BAFF consequent to introduction of a BAFF transgene (Tg) leads to SLE-like features, including elevated circulating titers of multiple autoantibodies and immune-complex glomerulonephritis (8-10). Moreover, introduction of a BAFF Tg into mice that otherwise have an incomplete diathesis to SLE (i.e., mice that develop SLE-associated autoantibodies but rarely develop renal disease) results in precocious development of glomerular pathology (11). Indeed, SLE-associated autoantibodies develop in BAFF-Tg mice even in the absence of class II major histocompatibility complex expression (12), and SLE-like features develop in otherwise non-autoimmune-prone BAFF-Tg mice even in the complete absence of T cells as long as MyD88-mediated signaling is intact (13).
Observations in humans are consonant with those in mice. Circulating BAFF levels are elevated in as many as 50% of SLE patients (14-16), and a large 2-year longitudinal study and three independent smaller studies documented a significant correlation between BAFF expression and clinical disease activity (17-20).
Importantly, prevention/amelioration of SLE is associated with elimination/neutralization of BAFF as well. Treatment of SLE-prone (NZBxNZW)F1 (BWF1) mice or MRL-lpr mice with a BAFF antagonist retards disease progression and improves survival (10, 21-23). Furthermore, very limited clinical disease develops in SLE-prone NZM 2328 (NZM) mice (an inbred recombinant strain derived from BWF1 mice which closely mirrors the parental SLE phenotype and shares many features with human SLE [24]) in which the Baff gene has been disrupted (25). Indeed, NZM.Baff-/- mice are completed protected from the virulent SLE disease that rapidly develops in young (pre-autoimmune) wild-type (WT) NZM mice upon overexpression of interferon-α (26). Finally, the anti-BAFF monoclonal antibody (mAb) belimumab has been shown to be efficacious in two separate human SLE phase-III trials (27, 28).
In contrast to the large body of evidence that links BAFF with SLE, the evidence linking the closely related APRIL to SLE is limited at most. Although APRIL does not bind to BR3 (6), its three-dimensional structure is sufficiently similar to that of BAFF to permit APRIL to bind to the other two BAFF receptors (BCMA and TACI) (29-32). Of potential importance, BAFF and APRIL naturally form heterotrimers that are biologically active in in vitro assays and which circulate at elevated concentrations in patients with SLE (33, 34). What regulates formation of heterotrimers rather than BAFF or APRIL homotrimers and whether the potency of the heterotrimers under in vivo conditions is greater than, equal to, or less than those of the BAFF or APRIL homotrimers remain unknown. Nevertheless, given that APRIL can co-stimulate B cells, induce Ig class switching, and promote plasma cell (PC) survival (29, 30, 35-38), APRIL may be an important (co-)contributor to SLE pathogenesis and, thereby, represent an appropriate therapeutic target in SLE.
To date, there have been no studies in either murine or human SLE that have solely targeted APRIL. That is, the benefit garnered from antagonizing APRIL per se remains uncertain. This point is not merely academic, since the combined antagonism of BAFF and APRIL may be more immunosuppressive than the antagonism of BAFF alone. Indeed, BR3-Ig (which antagonizes BAFF but not APRIL) and TACI-Ig (which antagonizes both BAFF and APRIL) displayed identical clinical efficacy in a head-to-head study despite TACI-Ig having a greater inhibitory effect on humoral immunity than did BR3-Ig (23).
To directly assess the contribution of APRIL to the development SLE, we generated NZM.April-/- mice. Counterintuitively, T cells and B cells were globally expanded, and serological autoimmunity and renal immunopathology tended to be greater, in NZM.April-/- mice than in WT mice. Moreover, development of renal immunopathology was identically mild in NZM.Baff-/- .April-/- mice as in NZM.Baff-/- mice despite the greater reduction in the former of bone marrow (BM) PC. Collectively, these findings demonstrate that APRIL is dispensable for full-blown SLE in NZM mice, and they raise the concern that the combined antagonism of APRIL and BAFF may lead to greater immunosuppression beyond that of antagonism of BAFF alone but without a concomitant gain in therapeutic efficacy.
Materials and methods
General
All mice were maintained in specific pathogen-free quarters at USC, and the experiments were approved by the Institutional Animal Care and Use Committee.
Mice
Female mice from four congenic NZM strains were studied: NZM WT, NZM.Baff-/-, NZM.April-/-, and NZM.Baff-/-.April-/-. The generation of NZM.Baff-/- mice has been previously described (25). To generate NZM.April-/- mice, the April-/- genotype from April-/- mice (mixed B6/129 background) (39) was introgressed into NZM mice using a marker-assisted selection protocol, using markers to include those regions identified as susceptibility loci in NZM mice (25). To detect the disrupted April gene fragment (containing a neo insert), genomic DNA extracted from mouse tail clippings was PCR-amplified for 30 cycles at 95°C for 60 sec, 60°C for 30 sec, and 72°C for 90 sec. The primer sequences were:
April primer 1:5'-CAG TCC TGC ATC TTG TTC CA-3'
April primer 2:5'-GCA GAT AAA TTC CAG TGT CCC-3'
neo: 5'-CTC CCA CTC ATG ATC TAT AAG ATCC-3'.
All three primers were added to a single reaction mix. Band size for the intact April gene fragment is 712 bp, and band size for the disrupted April gene fragment (neo) is 464 bp. Thus, mice bearing the April+/+ genotype would yield only a 712-bp band; mice bearing the April-/- genotype would yield only a 464-bp band; and mice bearing the April+/- genotype would yield both 712-bp and 464-bp bands.
NZM.Baff-/-.April-/- mice were generated by intercrossing NZM.Baff-/- and NZM.April-/- mice and screening by PCR for the Baff-/-.April-/- genotype.
Cell surface staining
For determination of T cell and B cell subsets, murine spleen mononuclear cells were stained with combinations of fluorochrome-conjugated mAb specific for murine CD3, CD4, CD8, CD44, CD62L, CD45R (B220), CD19, CD21, CD23, or CD69 (BD PharMingen, San Diego, CA, or eBioscience, San Diego, CA) and analyzed by flow cytometry (40). For PC determination, spleen or BM cells were surface-stained with a combination of fluorochromeconjugated mAb specific for murine CD4, CD8, Gr1, F4/80, and B220 and intracellular-stained with fluorochrome-conjuated mAb specific for murine Igκ and Igλ. PC were taken as the CD4-CD8-Gr1-F4/80-B220-Igκ/λ+ cells.
Serum total IgG and IgG autoantibody determinations
Serum levels of total IgG, IgG anti-chromatin, and anti-dsDNA antibodies were determined by ELISA (26). Autoantibody OD values were normalized to the mean OD of serum from 5-month-old MRL-lpr mice, the latter being arbitrarily assigned a value of 100 U/ml.
Serum BAFF determination
Serum BAFF was measured by ELISA (41). Assay plates were coated with a mouse BR3:human Fc fusion protein (Alexis Biochemicals, Plymouth Meeting, PA) as the capture reagent, and biotinylated anti-mouse BAFF mAb 16D7 (Human Genome Sciences) followed by streptavidin-horseradish peroxidase was used as the detector. Results were interpolated from a mouse BAFF standard curve that was run on each plate.
Kidney histology
Sections of formalin-fixed kidneys from WT or NZM.April-/- mice were stained with hematoxylin and eosin (H&E), periodic acid-Schiff (PAS), Masson's trichrome, and Jones’ silver methenamine and were assessed by light microscopy for glomerular activity (hypercellularity, necrotizing lesions, karyorrhexis, cellular crescents, hyaline deposits), tubulointerstitial activity (interstitial cellular infiltration, tubular cell necrosis), chronic glomerular pathology (glomerulosclerosis, fibrous crescents), and chronic tubulointerstitial pathology (tubular atrophy, interstitial fibrosis). Each category was subjectively scored on a 0-3 scale, for a maximum composite score of 12. Kidneys from NZM.Baff-/- or NZM.Baff-/-.April-/- mice were evaluated by H&E.
Kidney immunofluorescence
Five-micron sections of snap-frozen kidneys were stained for total IgG deposition using FITC-conjugated goat F(ab’)2 fragment anti-mouse IgG or C3 antibodies (MP Biomedicals, Solon, OH) (26). Staining intensity was subjectively scored on a 0-4 scale.
Assessment of proteinuria
Reagent strips for urinary protein (Albustix, Bayer, Elkhart, IN) were dipped in mouse urine and were assigned a score (0-4) by visual color comparison to the supplied standard color key. Severe proteinuria was defined as ≥3 on two consecutive examinations.
Statistical analysis
All analyses were performed using SigmaStat software (SPSS, Chicago, IL). Parametric testing between two matched or unmatched groups was performed by the paired or unpaired t test, respectively. Parametric testing among three or more groups was performed by one-way ANOVA. When the data were not normally distributed or the equal variance test was not satisfied, non-parametric testing was performed by the Mann-Whitney rank sum test between two groups and by Kruskal-Wallis one-way ANOVA on ranks among three or more groups.
Results
Expansion of B cells and T cells in NZM.April-/- mice
In non-autoimmune-prone (mixed B6/129 background) mice, the numbers and percentages of total spleen B and T cells in April-/- mice are identical to those in WT mice (39, 42), although an increase in the percentage of memory (CD44hiCD62Llo) CD4+ cells in the spleens of April-/- mice was reported by one group (39). Unexpectedly, NZM.April-/- mice underwent a global expansion of mononuclear cells in their spleens, affecting total B (CD19+) cells, follicular (CD23hiCD21int) B cells, marginal zone (CD23loCD21+) B cells, activated (CD69+) B cells, total T (CD3+) cells, total CD4+ cells, naive (CD44loCD62Lhi) CD4+ cells, memory (CD44hiCD62Llo) CD4+ cells, and CD8+ cells (Figures 1A-J). Percentages of these individual cell populations were not significantly different between WT and NZM.April-/- mice (data not shown), with the exception that the mean percentage of CD8+ cells was modestly lower in NZM.April-/- mice than in WT mice (19.7% vs 23.7%, p = 0.043).
Figure 1. Spleen T and B cells in WT and NZM.April-/- mice.
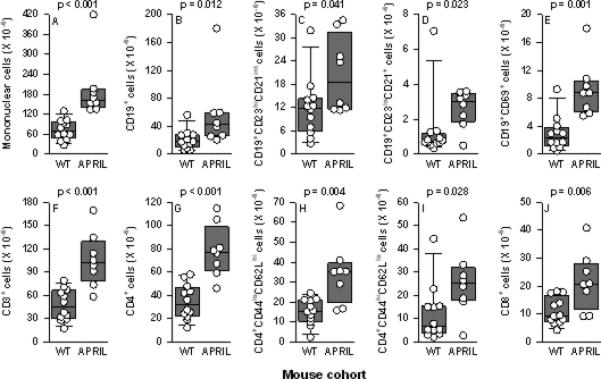
Spleen total mononuclear cells (panel A), total B (CD19+) cells (panel B), follicular (CD23hiCD21int) B cells (panel C), marginal zone (CD23loCD21+) B cells (panel D), activated (CD69+) B cells (panel E), total T (CD3+) cells (panel F), total CD4+ cells (panel G), naive (CD44loCD62Lhi) CD4+ cells (panel H), memory (CD44hiCD62Llo) CD4+ cells (panel I), and CD8+ cells (panel J) from NZM WT mice (n = 12) and NZM.April-/- mice (APRIL; n = 8) (all mice 7-9 months old) are plotted. Each symbol represents an individual mouse. The composite results are plotted as box plots. The lines inside the boxes indicate the medians; the outer borders of the boxes indicate the 25th and 75th percentiles; and the bars extending from the boxes indicate the 10th and 90th percentiles. The p values are shown above each panel for the comparisons between WT and NZM.April-/- mice.
Serologic autoimmunity in NZM.April-/- mice despite marked reduction in BM PC
Consistent with their global B (and T) cell expansion, NZM.April-/- mice harbored increased numbers of PC in their spleens. In sharp contrast, BM PC were markedly reduced in number (Figures 2A-B). This paucity of PC in the BM of NZM.April-/- mice likely reflects the vital role for APRIL produced by BM stromal cells in plasmablast survival (38).
Figure 2. Spleen and BM PC and serum total IgG and IgG autoantibody levels in WT and NZM.April-/- mice.
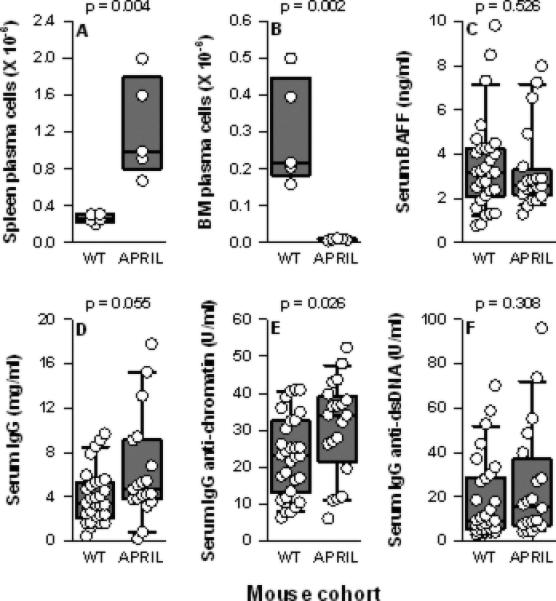
Spleen PC (panel A) and BM PC (panel B) from NZM WT (n = 5) and NZM.April-/- mice (APRIL; n = 5) (all mice 2-3 months old) are plotted as in Figure 1. Serum levels of BAFF (panel C) total IgG (panel D), IgG anti-chromatin (panel E), and IgG anti-dsDNA (panel F) from NZM WT (n = 30) and NZM.April-/- mice (n = 20) (all mice 7-12 months old) are plotted as in Figure 1.
The dearth of BM PC notwithstanding, serum IgG levels in NZM.April-/- mice tended to be higher, not lower, than those in WT mice. Similarly, serum IgG anti-chromatin, but not IgG anti-dsDNA, autoantibody levels were greater in NZM.April-/- mice than in WT mice (Figure 2D-F). This disparity in serum IgG and IgG anti-chromatin levels between the two mouse cohorts cannot be attributed to a difference in circulating BAFF levels per se, since their serum BAFF levels were identical (Figure 2C).
Renal immunopathology and clinical disease in NZM.April-/- mice
At 3 months of age, the kidneys of NZM.April-/- mice and WT mice were histologically normal, with either no or minimal deposition of IgG or C3 in their glomeruli (data not shown).
By 5 months of age, similar moderate degrees of glomerular deposition of IgG and C3 deposition had developed in WT and NZM.April-/- mice (Figures 3Aa-d). The deposits were predominantly in the mesangial areas, with minimal deposition along the glomerular capillary walls. Histologically, glomerular hypercellularity was readily appreciated in both mouse cohorts, with the hypercellularity actually being somewhat greater in NZM.April-/- mice than in WT mice. Glomerular size was increased in both, and cellular crescents were present. Moreover, mesangial matrix deposition (as detected by PAS staining) and collagen deposition in the glomeruli (as detected by silver staining) were readily detectable in both. In addition, some degree of interstitial inflammation with perivascular leukocyte infiltration was also observed in both (Figures 3Ae-l). Overall, the kidney scores tended to be higher in NZM.April-/- mice than in WT mice, although this difference did not achieve statistical significance (Figure 3B).
Figure 3. Renal immunopathology and clinical disease in WT and NZM.April-/- mice.
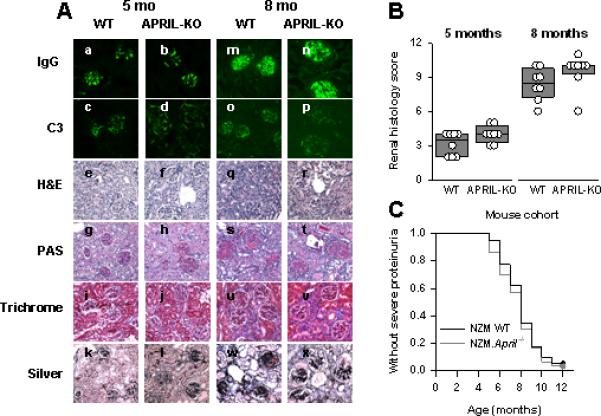
Panel A: Kidney sections from 5-month-old (a-l) or 8-month-old (m-x) WT (a, c, e, g, i, k, m, o, q, s, u, w) or NZM.April-/- (APRIL-KO; b, d, f, h, j, l, n, p, r, t, v, x) mice were stained for IgG (a, b, m, n) or C3 (c, d, o, p) immunofluorescence or were stained with H&E (e, f, q, r), PAS (g, h, s, t), Masson's trichrome (Trichrome; i, j, u, v), or Jones’ silver methenamine (Silver; k, l, w, x) for histological evaluation. Representative sections are illustrated. Panel B: The renal histology scores for the 8 mice in each cohort are plotted as in Figure 1. Panel C: WT (n = 40) and NZM.April-/- (n = 30) mice were monitored for 12 months for development of severe proteinuria. Data are plotted as the fraction of mice over time that did not develop severe proteinuria.
By 8 months of age, glomerular deposition of IgG, but not of C3, had greatly increased in both mouse cohorts (Figures 3Am-p). This was accompanied by marked renal histopathology, including the development of glomerular fibrotic crescents, glomerulosclerosis, tubular atrophy, increased interstitial inflammation and fibrosis, and striking degrees of perivascular leukocyte infiltration around both arterioles and venules (Figures 3Aq-x). As at 5 months of age, the kidney scores tended to be higher in NZM.April-/- mice than in WT mice, although this difference again did not achieve statistical significance (Figure 3B).
In both NZM.April-/- and WT mice, clinical disease (severe proteinuria) started to develop at 5 months of age, with 50% of the mice in each cohort being affected between 7 and 8 months of age (Figure 3C). Overall, the onset and incidence of clinical disease in these two cohorts were identical (Figure 3C). (Mortality could not be accurately assessed, since clinically sick mice were routinely sacrificed upon development of ascites and lethargy.)
Serologic autoimmunity, renal immunopathology, and clinical disease in NZM.Baff-/-.April-/- mice
Although development of clinical disease in NZM.April-/- mice was identical to that in WT mice, the increased numbers of T and B cells, the increased circulating levels of IgG anti-chromatin antibodies, and the trend to more severe renal immunopathology collectively suggested that the life-long absence of APRIL might promote or accelerate, rather than inhibit or delay, the development of SLE. Since TACI-Ig (atacicept), which antagonizes both BAFF and APRIL, has been evaluated in murine SLE (10, 22, 23) and is presently undergoing clinical evaluation in human SLE (43), we asked whether elimination of both BAFF and APRIL might lead to a more severe SLE phenotype than that resulting from elimination of BAFF only.
Comparison of NZM.Baff-/-.April-/- mice to NZM.Baff-/- mice revealed that this was not the case. Both spleen and BM PC populations were reduced in NZM.Baff-/- mice relative to those in WT mice (p = 0.016 and p = 0.007, respectively; Figures 4A-B). Of note, BM, but not spleen, PC in NZM.Baff-/-.April-/- mice were further reduced relative to those in NZM.Baff-/- mice (p = 0.016 for BM PC, p = 0.222 for spleen PC; Figures 4A-B), again consistent with an essential role in plasmablast survival for BM stromal cell-produced APRIL (38). Moreover, serum total IgG levels tended to be lower in NZM.Baff-/-.April-/- mice than in NZM.Baff-/- mice (p = 0.089; Figure 4C), and serum IgG anti-chromatin and anti-dsDNA antibody levels were unmistakably lower in NZM.Baff-/-.April-/- mice than in NZM.Baff-/- mice (p = 0.005 and p < 0.001, respectively; Figures 4D-E). Of note, serum IgG anti-dsDNA antibody levels were very high in some NZM.Baff-/- mice, consistent with our previous report of elevated IgG anti-dsDNA antibody titers in NZM.Baff-/- mice as they age (25).
Figure 4. Spleen and BM PC and serum total IgG and IgG autoantibody levels in NZM WT, NZM.Baff-/-, and NZM.Baff-/-.April-/- mice.
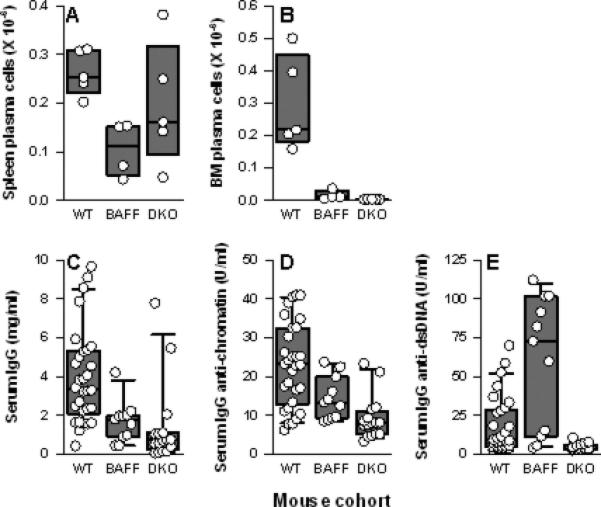
Spleen PC (panel A) and BM PC (panel B) from NZM WT (WT; n = 5), NZM.Baff-/- (BAFF; n = 4), and NZM.Baff-/-.April-/- mice (DKO; n = 5) are plotted as in Figure 1. Serum levels of total IgG (panel C), IgG anti-chromatin (panel D), and IgG anti-dsDNA (panel E) from WT (n = 30), NZM.Baff-/- (n = 11), and NZM.Baff-/-.April-/- mice (n = 16) are plotted as in Figure 1. (WT data are identical to those presented in Figure 2 and are included for clarity.)
In any case, glomerular IgG deposition was very limited in both NZM.Baff-/- and NZM.Baff-/- .April-/- mice, and no glomerular C3 deposition could be detected. This is in striking contrast to the copious deposition of both IgG and C3 in the glomeruli of age-matched WT mice (Figure 5). Moreover, histological evaluation demonstrated only occasional increases in glomerular cellularity in either NZM.Baff-/- or NZM.Baff-/-.April-/- mice without changes in glomerular size or increase in mesangial matrix. Perivascular leukocyte aggregates were rarely observed, and tubular atrophy or lesions were absent. Again, this is in striking contrast to the widespread pathological changes in WT mice. Importantly, none of the NZM.Baff-/- or NZM.Baff-/-.April-/- mice developed clinical disease by the time of sacrifice (8 months of age).
Figure 5. Renal immunopathology in NZM WT, NZM.Baff-/-, and NZM.Baff-/-.April-/- mice.
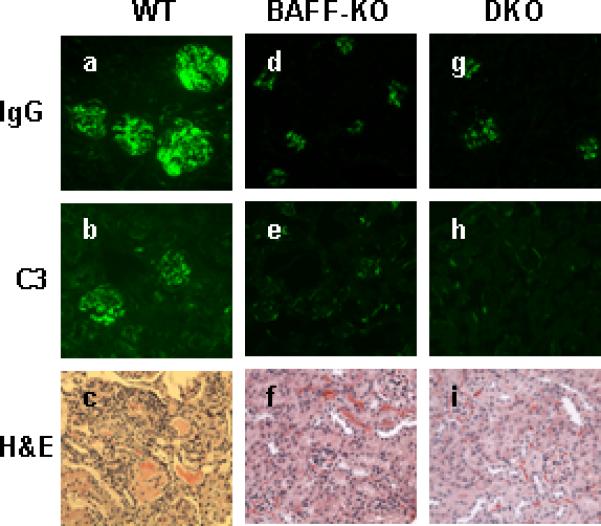
Kidney sections from 8-month-old NZM.Baff-/- (a-c) or NZM.Baff-/-.April-/- (d-f) mice were stained for IgG (a, d) or C3 (b,e ) immunofluorescence or were stained with H&E (c, f) for histological evaluation. Representative sections are illustrated.
Discussion
The contribution of BAFF to SLE has been firmly established. In mice, constitutive overexpression of BAFF in hosts that are otherwise not autoimmune-prone leads to SLE-like features (8-10), and treatment of SLE mice with a BAFF-specific antagonist ameliorates disease (21, 23). In humans, circulating BAFF levels are elevated in SLE patients and correlate with disease activity (14, 15, 17), and treatment with belimumab, a BAFF-specific antagonist, is efficacious in many SLE patients (27, 28).
In contrast, the connection between APRIL and SLE is tenuous. Unlike the SLE-like features that develop in BAFF-Tg mice, no such features develop in APRIL-Tg mice (44). Moreover, unlike the repeatedly observed increased circulating BAFF levels in human SLE (14, 15, 19, 45, 46), normal circulating APRIL levels in human SLE have been found in at least two independent studies (34, 47). Finally, no studies prior to the present one had addressed the effect on SLE of selective APRIL elimination. Although the combined antagonism of APRIL and BAFF is therapeutically successful in murine SLE (10, 22, 23) and is being evaluated in human SLE (43), the role for antagonism of APRIL per se cannot be unequivocally determined from such studies.
To directly assess the contribution of APRIL to SLE, we generated SLE-prone NZM mice that are deficient only in APRIL. Since in non-autoimmune-prone (B6/129 mixed background) April-/- mice, B cells (and T cells) are, for the most part, identical to those in WT mice, as are serum IgG levels (39, 42), we anticipated that these parameters in NZM.April-/- mice would be identical to those in their WT counterparts. Given the ability of APRIL to co-stimulate B cells, induce Ig class switching, and promote PC survival (29, 30, 35-38), we predicted that if there were any effects of APRIL deficiency on B cells and/or IgG levels in NZM.April-/- mice, those effects would be inhibitory. Unexpectedly, B cells (and T cells) were globally increased, rather than decreased, in NZM.April-/- mice, as were circulating levels of at least one IgG autoantibody (anti-chromatin).
The factors that promote expansion of B cells and augment serological autoimmunity in NZM.April-/- mice remain to be formally identified, but the absence of BAFF/APRIL heterotrimers in these mice may be highly relevant. Although BAFF/APRIL heterotrimers are biologically active in in vitro assays and circulate at elevated concentrations in SLE patients (33, 34), the in vivo activity of such heterotrimers is not known. Since BAFF/APRIL heterotrimers are less potent in vitro than are BAFF homotrimers (34), it is likely that BAFF/APRIL heterotrimers are also less potent in vivo. These heterotrimers can compete with BAFF homotrimers for the BAFF receptors, leading to functional down-modulation of BAFF activity. In the absence of APRIL, there would be no BAFF/APRIL heterotrimers. Thus, the natural heterotrimer-mediated down-modulation would be absent, leading to “uncontested” BAFF homotrimers and increased BAFF activity. That is, despite serum BAFF levels being identical in NZM.April-/- and WT mice, the net biological activity of BAFF in the former might be greater than in the latter. Such greater BAFF activity could well lead to increased numbers of B cells and circulating levels of autoantibodies in an APRIL-deficient SLE-prone host.
Moreover, BAFF has direct effects on T cells in addition to its direct effects on B cells. BAFF can co-stimulate in vitro proliferation of, and cytokine production by, T cells by directly engaging them (48, 49). Thus, loss of BAFF/APRIL heterotrimers in NZM.April-/- mice with the (presumed) attendant increase in BAFF potency may have contributed to the expansion of T cells. Unfortunately, reagents that specifically neutralize BAFF/APRIL heterotrimers without also neutralizing BAFF and/or APRIL homotrimers do not yet exist, so our hypothesis cannot yet be formally tested, and the role of BAFF/APRIL heterotrimers per se in SLE remains enigmatic.
In any case, NZM.April-/- mice developed renal immunopathology and clinical disease at least to the same degree and with the same time course as did WT NZM mice, demonstrating unequivocally that APRIL per se is dispensable to the development of full-blown SLE. This is in sharp contrast to the considerable protection from renal immunopathology and near-total protection from clinical disease in NZM.Baff-/- mice (25). Of note, the development of WT-like clinical disease in NZM.April-/- mice despite the T and B cell expansions and the increase in circulating IgG anti-chromatin antibody levels strongly suggests that cell number or serum autoantibody titers per se may offer only limited insights into development of clinically relevant parameters.
The dispensability of APRIL to development of full-blown SLE did not exclude a possible contributory role for APRIL in the development of SLE. That is, the role of APRIL in the development of SLE might only be appreciated when a more dominant contributor is lacking. Given the biologic relatedness of APRIL to BAFF, we reasoned that APRIL might play a more discernable role under BAFF-deficient conditions.
To test this, we compared NZM.Baff-/-.April-/- mice to NZM.Baff-/- mice. Rather than their levels being increased as might have been predicted from observations in NZM.April-/- mice, serum IgG anti-chromatin and IgG anti-dsDNA antibody levels were lower in NZM.Baff-/-.April-/- mice than in NZM.Baff-/- mice. Importantly, the superimposition of APRIL deficiency in a BAFF- deficient environment has no effect on BAFF/APRIL heterotrimers, since such heterotrimers already are not extant. Thus, the absence of APRIL in NZM.Baff-/-.April-/- mice aggravates, rather than ameliorates, the absence of BAFF and results in fewer BM PC and reduced circulating autoantibody levels. This preservation of spleen PC in NZM.Baff-/-.April-/- mice is reminiscent of the preservation of spleen PC in BWF1 mice following neutralization of both BAFF and APRIL with TACI-Ig (22). Of note, spleen PC were not preserved in NZM 2410 mice following treatment with TACI-Ig (23), indicating that the combined inhibition/elimination of BAFF and APRIL may have differential effects in different SLE models. In any case, renal immunopathology was identically minimal in NZM.Baff-/-.April-/- and NZM.Baff-/- mice.
With the caveats that findings in mice may not fully translate to humans and that studies in genetically-deficient hosts may not fully reflect outcomes in hosts treated with pharmacologic antagonists, our findings question the wisdom of antagonizing both BAFF and APRIL rather than antagonizing BAFF alone. Our cellular and serological findings in NZM.Baff-/-.April-/- mice are remarkably similar to those previously reported in NZM 2410 mice treated with TACI-Ig to antagonize both BAFF and APRIL (23). Importantly, NZM 2410 mice treated in that study with BR3-Ig (to antagonize BAFF only) had the same favorable clinical and pathological outcomes as did mice treated with TACI-Ig. That is, whether BAFF and APRIL were antagonized genetically or pharmacologically, clinical and pathological disease were ameliorated to the same degree as when only BAFF was antagonized. Thus, antagonism of both BAFF and APRIL may simply increase the risk of toxicity (especially infections) without adding a clinical benefit. One clinical trial of atacicept (in combination with mycophenolate mofetil) in SLE was prematurely terminated due to an increase in serious infections (ClinicalTrial.gov identifier NCT00573157), so circumspection is certainly warranted.
Acknowledgment
The authors thank Dr. Raif Geha, Children's Hospital of Boston, for his kind gift of April-/- mice (B6/129 mixed background).
This work was supported in part by NIH grants R01 AR050193 (WS), R01 AR048692 (CP), R01 AI073939 (MPC), T32 AI055428 (WJQ), a grant from the Arthritis Foundation Southern California Chapter (WS), an Arthritis Foundation Post-doctoral fellowship (RP), an American College of Rheumatology Research and Education Foundation Abbott Medical Student Clinical Preceptorship Award (NJ), and a sponsored research agreement from Human Genome Sciences (MPC).
References
- 1.Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, et al. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–263. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- 2.Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer J-L, Holler N, et al. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189:1747–1756. doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, et al. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204. doi: 10.1182/blood.v97.1.198. [DOI] [PubMed] [Google Scholar]
- 4.Laabi Y, Gras M-P, Brouet J-C, Berger R, Larsen C-J, Tsapis A. The BCMA gene, preferentially expressed during B lymphoid maturation, is bidirectionally transcribed. Nucleic Acids Res. 1994;22:1147–1154. doi: 10.1093/nar/22.7.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.von Bülow G-U, Bram RJ. NF-AT activation induced by a CAML-interacting member of the tumor necrosis factor receptor superfamily. Science. 1997;278:138–141. doi: 10.1126/science.278.5335.138. [DOI] [PubMed] [Google Scholar]
- 6.Thompson JS, Bixler SA, Qian F, Vora K, Scott ML, Cachero TG, et al. BAFF-R, a novel TNF receptor that specifically interacts with BAFF. Science. 2001;293:2108–2111. doi: 10.1126/science.1061965. [DOI] [PubMed] [Google Scholar]
- 7.Yan M, Brady JR, Chan B, Lee WP, Hsu B, Harless S, et al. Identification of a novel receptor for B lymphocyte stimulator that is mutated in a mouse strain with severe B cell deficiency. Curr Biol. 2001;11:1547–1552. doi: 10.1016/s0960-9822(01)00481-x. [DOI] [PubMed] [Google Scholar]
- 8.Mackay F, Woodcock SA, Lawton P, Ambrose C, Baetscher M, Schneider P, et al. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J Exp Med. 1999;190:1697–1710. doi: 10.1084/jem.190.11.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khare SD, Sarosi I, Xia X-Z, McCabe S, Miner K, Solovyev I, et al. Severe B cell hyperplasia and autoimmune disease in TALL-1 transgenic mice. Proc Natl Acad Sci U S A. 2000;97:3370–3375. doi: 10.1073/pnas.050580697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 11.Stohl W, Xu D, Kim KS, Koss MN, Jorgensen TN, Deocharan B, et al. BAFF overexpression and accelerated glomerular disease in mice with an incomplete genetic predisposition to systemic lupus erythematosus. Arthritis Rheum. 2005;52:2080–2091. doi: 10.1002/art.21138. [DOI] [PubMed] [Google Scholar]
- 12.Stohl W, Jacob N, Guo S, Morel L. Constitutive overexpression of BAFF in autoimmune-resistant mice drives only some aspects of systemic lupus erythematosus-like autoimmunity. Arthritis Rheum. 2010;62:2432–2442. doi: 10.1002/art.27502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groom JR, Fletcher CA, Walters SN, Grey ST, Watt SV, Sweet MJ, et al. BAFF and MyD88 signals promote a lupuslike disease independent of T cells. J Exp Med. 2007;204:1959–1971. doi: 10.1084/jem.20062567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Roschke V, Baker KP, Wang Z, Alarcón GS, Fessler BJ, et al. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001;166:6–10. doi: 10.4049/jimmunol.166.1.6. [DOI] [PubMed] [Google Scholar]
- 15.Cheema GS, Roschke V, Hilbert DM, Stohl W. Elevated serum B lymphocyte stimulator levels in patients with systemic immune-based rheumatic diseases. Arthritis Rheum. 2001;44:1313–1319. doi: 10.1002/1529-0131(200106)44:6<1313::AID-ART223>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 16.Stohl W, Metyas S, Tan S-M, Cheema GS, Oamar B, Xu D, et al. B lymphocyte stimulator overexpression in patients with systemic lupus erythematosus: longitudinal observations. Arthritis Rheum. 2003;48:3475–3486. doi: 10.1002/art.11354. [DOI] [PubMed] [Google Scholar]
- 17.Petri M, Stohl W, Chatham W, McCune WJ, Chevrier M, Ryel J, et al. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008;58:2453–2459. doi: 10.1002/art.23678. [DOI] [PubMed] [Google Scholar]
- 18.Collins CE, Gavin AL, Migone T-S, Hilbert DM, Nemazee D, Stohl W. B lymphocyte stimulator (BLyS) isoforms in systemic lupus erythematosus: disease activity correlates better with blood leukocyte BLyS mRNA levels than with plasma BLyS protein levels. Arthritis Res Ther. 2006;8:R6. doi: 10.1186/ar1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Becker-Merok A, Nikolaisen C, Nossent HC. B-lymphocyte activating factor in systemic lupus erythematosus and rheumatoid arthritis in relation to autoantibody levels, disease measures and time. Lupus. 2006;15:570–576. doi: 10.1177/0961203306071871. [DOI] [PubMed] [Google Scholar]
- 20.Ju S, Zhang D, Wang Y, Ni H, Kong X, Zhong R. Correlation of the expression levels of BLyS and its receptors mRNA in patients with systemic lupus erythematosus. Clin Biochem. 2006;39:1131–1137. doi: 10.1016/j.clinbiochem.2006.09.010. [DOI] [PubMed] [Google Scholar]
- 21.Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-κB2. Immunity. 2002;17:515–524. doi: 10.1016/s1074-7613(02)00425-9. [DOI] [PubMed] [Google Scholar]
- 22.Ramanujam M, Wang X, Huang W, Schiffer L, Grimaldi C, Akkerman A, et al. Mechanism of action of transmembrane activator and calcium modulator ligand interactor-Ig in murine systemic lupus erythematosus. J Immunol. 2004;173:3524–3534. doi: 10.4049/jimmunol.173.5.3524. [DOI] [PubMed] [Google Scholar]
- 23.Ramanujam M, Wang X, Huang W, Liu Z, Schiffer L, Tao H, et al. Similarities and differences between selective and nonselective BAFF blockade in murine SLE. J Clin Invest. 2006;116:724–734. doi: 10.1172/JCI26385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudofsky UH, Evans BD, Balaban SL, Mottironi VD, Gabrielsen AE. Differences in expression of lupus nephritis in New Zealand Mixed H-2z homozygous inbred strains of mice derived from New Zealand Black and New Zealand White mice: origins and initial characterization. Lab Invest. 1993;68:419–426. [PubMed] [Google Scholar]
- 25.Jacob CO, Pricop L, Putterman C, Koss MN, Liu Y, Kollaros M, et al. Paucity of clinical disease despite serological autoimmunity and kidney pathology in lupus-prone New Zealand Mixed 2328 mice deficient in BAFF. J Immunol. 2006;177:2671–2680. doi: 10.4049/jimmunol.177.4.2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jacob N, Guo S, Mathian A, Koss MN, Gindea S, Putterman C, et al. B cell and BAFF dependence of IFN-α-exaggerated disease in systemic lupus erythematosus-prone NZM 2328 mice. J Immunol. 2011;186:4984–4993. doi: 10.4049/jimmunol.1000466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Navarra SV, Guzmán RM, Gallacher AE, Hall S, Levy RA, Jimenez RE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial. Lancet. 2011;377:721–731. doi: 10.1016/S0140-6736(10)61354-2. [DOI] [PubMed] [Google Scholar]
- 28.Furie R, Petri M, Zamani O, Cervera R, Wallace DJ, Tegzová D, et al. A phase 3, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits BLyS, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011 doi: 10.1002/art.30613. DOI: 10.1002/art.30613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marsters SA, Yan M, Pitti RM, Haas PE, Dixit VM, Ashkenazi A. Interaction of the TNF homologues BLyS and APRIL with the receptor homologues BCMA and TACI. Curr Biol. 2000;10:785–788. doi: 10.1016/s0960-9822(00)00566-2. [DOI] [PubMed] [Google Scholar]
- 30.Yu G, Boone T, Delaney J, Hawkins N, Kelley M, Ramakrishnan M, et al. APRIL and TALL-1 and receptors BCMA and TACI: system for regulating humoral immunity. Nat Immunol. 2000;1:252–256. doi: 10.1038/79802. [DOI] [PubMed] [Google Scholar]
- 31.Wu Y, Bressette D, Carrell JA, Kaufman T, Feng P, Taylor K, et al. Tumor necrosis factor (TNF) receptor superfamily member TACI is a high affinity receptor for TNF family members APRIL and BLyS. J Biol Chem. 2000;275:35478–35485. doi: 10.1074/jbc.M005224200. [DOI] [PubMed] [Google Scholar]
- 32.Rennert P, Schneider P, Cachero TG, Thompson J, Trabach L, Hertig S, et al. A soluble form of B cell maturation antigen, a receptor for the tumor necrosis factor family member APRIL, inhibits tumor cell growth. J Exp Med. 2000;192:1677–1683. doi: 10.1084/jem.192.11.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roschke V, Sosnovtseva S, Ward CD, Hong JS, Smith R, Albert V, et al. BLyS and APRIL form biologically active heterotrimers that are expressed in patients with systemic immune-based rheumatic diseases. J Immunol. 2002;169:4314–4321. doi: 10.4049/jimmunol.169.8.4314. [DOI] [PubMed] [Google Scholar]
- 34.Dillon SR, Harder B, Lewis KB, Moore MD, Liu H, Bukowski TR, et al. B-lymphocyte stimulator/a proliferation-inducing ligand heterotrimers are elevated in the sera of patients with autoimmune disease and are neutralized by atacicept and B cell maturation antigen-immunoglobulin. Arthritis Res Ther. 2010;12:R48. doi: 10.1186/ar2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam K-P, et al. TACI and BAFF-R mediate isotype switching in B cells. J Exp Med. 2005;201:35–39. doi: 10.1084/jem.20032000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakurai D, Hase H, Kanno Y, Kojima H, Okumura K, Kobata T. TACI regulates IgA production by APRIL in collaboration with HSPG. Blood. 2007;109:2961–2967. doi: 10.1182/blood-2006-08-041772. [DOI] [PubMed] [Google Scholar]
- 37.He B, Xu W, Santini PA, Polydorides AD, Chiu A, Estrella J, et al. Intestinal bacteria trigger T cell-independent immunoglobulin A2 class switching by inducing eipthelial-cell secretion of the cytokine APRIL. Cell. 2007;26:812–826. doi: 10.1016/j.immuni.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 38.Belnoue E, Pihlgren M, McGaha TL, Tougne C, Rochat A-F, Bossen C, et al. APRIL is critical for plasmablast survival in the bone marrow and poorly expressed by early-life bone marrow stromal cells. Blood. 2008;111:2755–2764. doi: 10.1182/blood-2007-09-110858. [DOI] [PubMed] [Google Scholar]
- 39.Castigli E, Scott S, Dedeoglu F, Bryce P, Jabara H, Bhan AK, et al. Impaired IgA class switching in APRIL-deficient mice. Proc Natl Acad Sci U S A. 2004;101:3903–3908. doi: 10.1073/pnas.0307348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacob N, Yang H, Pricop L, Liu Y, Gao X, Zheng SG, et al. Accelerated pathological and clinical nephritis in systemic lupus erythematosus-prone New Zealand Mixed 2328 mice doubly deficient in TNF receptor 1 and TNF receptor 2 via a Th17-associated pathway. J Immunol. 2009;182:2532–2541. doi: 10.4049/jimmunol.0802948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scholz JL, Crowley JE, Tomayko MM, Steinel N, O'Neill PJ, Quinn WJ, III, et al. BLyS inhibition eliminates primary B cells but leaves natural and acquired humoral immunity intact. Proc Natl Acad Sci U S A. 2008;105:15517–15522. doi: 10.1073/pnas.0807841105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Varfolomeev E, Kischkel F, Martin F, Seshasayee D, Wang H, Lawrence D, et al. APRIL-deficient mice have normal immune system development. Mol Cell Biol. 2004;24:997–1006. doi: 10.1128/MCB.24.3.997-1006.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dall'Era M, Chakravarty E, Wallace D, Genovese M, Weisman M, Kavanaugh A, et al. Reduced B lymphocyte and immunoglobulin levels after atacicept treatment in patients with systemic lupus erythematosus: results of a multicenter, phase Ib, double-blind, placebo-controlled, dose-escalating trial. Arthritis Rheum. 2007;56:4142–4150. doi: 10.1002/art.23047. [DOI] [PubMed] [Google Scholar]
- 44.Stein JV, López-Fraga M, Elustondo FA, Carvalho-Pinto CE, Rodríguez D, Gómez-Caro R, et al. APRIL modulates B and T cell immunity. J Clin Invest. 2002;109:1587–1598. doi: 10.1172/JCI15034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pers J-O, Daridon C, Devauchelle V, Jousse S, Saraux A, Jamin C, et al. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann NY Acad Sci. 2005;1050:34–39. doi: 10.1196/annals.1313.004. [DOI] [PubMed] [Google Scholar]
- 46.Toubi E, Kessel A, Rosner I, Rozenbaum M, Paran D, Shoenfeld Y. The reduction of serum B-lymphocyte activating factor levels following quinacrine add-on therapy in systemic lupus erythematosus. Scand J Immunol. 2006;63:299–303. doi: 10.1111/j.1365-3083.2006.01737.x. [DOI] [PubMed] [Google Scholar]
- 47.Stohl W, Metyas S, Tan S-M, Cheema GS, Oamar B, Roschke V, et al. Inverse association between circulating APRIL levels and serologic and clinical disease activity in patients with systemic lupus erythematosus. Ann Rheum Dis. 2004;63:1096–1103. doi: 10.1136/ard.2003.018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huard B, Schneider P, Mauri D, Tschopp J, French LE. T cell costimulation by the TNF ligand BAFF. J Immunol. 2001;167:6225–6231. doi: 10.4049/jimmunol.167.11.6225. [DOI] [PubMed] [Google Scholar]
- 49.Ng LG, Sutherland APR, Newton R, Qian F, Cachero TG, Scott ML, et al. B cell-activating factor belonging to the TNF family (BAFF)-R is the principal BAFF receptor facilitating BAFF costimulation of circulating T and B cells. J Immunol. 2004;173:807–817. doi: 10.4049/jimmunol.173.2.807. [DOI] [PubMed] [Google Scholar]